

Abstract
S100A4 is a low-molecular-mass (12 kDa) EF-hand Ca2+-binding S100 protein that is expressed in a broad range of normal tissue and cell types. S100A4 can be secreted from some cells to act in an autocrine or paracrine fashion on target cells and tissues. S100A4 has been reported in the extracellular fluids of subjects with several inflammatory diseases, including asthma. Airway smooth muscle plays a critical role in airway inflammation by synthesizing and secreting inflammatory cytokines. We hypothesized that S100A4 may play an immunomodulatory role in airway smooth muscle. Trachealis smooth muscle tissues were stimulated with recombinant His-S100A4, and the effects on inflammatory responses were evaluated. S100A4 induced the activation of Akt and NF-κB and stimulated eotaxin secretion. It also increased the expression of RAGE and endogenous S100A4 in airway tissues. Stimulation of airway smooth muscle tissues with IL-13 or TNF-α induced the secretion of S100A4 from the tissues and promoted the expression of endogenous receptors for advanced glycation end products (RAGE) and S100A4. The role of RAGE in mediating the responses to S100A4A was evaluated by expressing a mutant nonfunctional RAGE (RAGEΔcyto) in tracheal muscle tissues and by treating tissues with a RAGE inhibitor. S100A4 did not activate NF-κB or Akt in tissues that were expressing RAGEΔcyto or treated with a RAGE inhibitor, indicating that S100A4 mediates its effects by acting on RAGE. Our results demonstrate that inflammatory mediators stimulate the synthesis and secretion of S100A4 in airway smooth muscle tissues and that extracellular S100A4 acts via RAGE to mediate airway smooth muscle inflammation.
Keywords: airway smooth muscle, inflammation, RAGE, S100A4
INTRODUCTION
S100A4 is a member of the family of low-molecular-mass (12 kDa) EF hand Ca2+-binding S100 proteins (1, 12). It was first identified in tumor cells and is highly expressed in metastatic cancer cells (13–15). S100A4 is also expressed in a broad range of normal tissue and cell types, including fibroblasts, smooth muscle cells, endothelial cells, and bone marrow-derived cells (1, 3, 14). S100A4 binds to a number of intracellular targets that regulate cytoskeletal dynamics, cell motility, and proliferation. Some S100 family proteins, including S100A4 are also secreted or released from cells and can act extracellularly to regulate the activities of target cells in an autocrine or paracrine fashion (1, 21, 23, 33, 35).
S100A4 has been identified as a candidate gene in allergy and has been reported in the extracellular fluids of subjects with several inflammatory diseases including rheumatoid arthritis, allergic dermatitis, asthma, and seasonal allergic rhinitis (1, 5, 18, 22, 28, 31). There is evidence that S100A4 is elevated in the sputum of asthmatic subjects (18) and in the nasal fluids of allergic patients during pollen season (5). Elevated S100A4 has also been found in the lungs and in the bronchoalveolar lavage fluid (BALF) of a mouse model of allergic asthma (5, 18). The treatment of these mice with S100A4-neutralizing antibodies reduced inflammatory cell accumulation and inflammatory mediators in the lungs (5, 18). In addition, mice with a genetic deletion of S100A4 are protected from allergic dermatitis (5).
Airway smooth muscle plays a critical role in airway inflammation and immunomodulation by synthesizing and secreting inflammatory cytokines (9, 19, 34, 36). Cytokines such as IL-4 and IL-13 and other extracellular stimuli can induce the expression of a synthetic inflammatory phenotype in airway smooth muscle characterized by the activation of the serine/threonine kinase Akt/PKB and the synthesis and secretion of eotaxin and other immunomodulatory cytokines by the muscle tissue (10, 19, 26, 36). The activation of synthetic pathways by these inflammatory mediators concurrently suppresses the expression of smooth muscle differentiation marker proteins such as smooth muscle myosin heavy chain (SmMHC) (10, 19, 26, 36, 37). Akt and inflammatory signaling pathways in airway smooth muscle cells can be activated by a variety of inflammatory stimuli as well as a number of nonhormonal extracellular stimuli (9, 10, 26, 34). However, the possibility that S100A4 plays an immunomodulatory role in airway smooth muscle has not been evaluated.
S100A4 is secreted extracellularly by a number of cell types and has been found to activate several cell surface receptors, including receptors for advanced glycation end products (RAGE), Toll-like receptor 4 (TLR4), epidermal growth factor receptor (EGFR), interleukin-10 (IL10R), and annexin-A2 (1, 11, 12, 23). RAGE receptors are expressed in many tissue types at low levels and have been demonstrated in lung tissues (4, 32, 38). In the lungs, RAGE receptors expressed on structural cells have been shown to play a critical role in allergic airway inflammation in murine models (27, 32).
In this study, we sought to determine whether extracellular S100A4 acts as an immunomodulatory regulator in airway smooth muscle tissues. Our results demonstrate that the inflammatory mediators IL-13 and TNF-α trigger the synthesis and secretion of S100A4 in airway smooth muscle tissues, and that extracellular S100A4 acts on RAGE receptors expressed on the muscle tissues to stimulate inflammatory responses.
MATERIALS AND METHODS
Reagents and antibodies.
Sources of the antibodies are as follows: rabbit anti-human polyclonal phospho-Ser473 Akt, rabbit anti-human polyclonal S100A4 (Invitrogen); mouse anti-human monoclonal Akt, rabbit anti-human polyclonal phospho-Ser536 NF-κB p65, rabbit anti-human polyclonal phospho-Tyr641 STAT6 (Cell Signaling); mouse anti-human monoclonal NF-κB p65, mouse anti-human monoclonal STAT6 (BD Biosciences Pharmingen); rabbit anti-human polyclonal RAGE (Abcam); mouse anti-human monoclonal smooth muscle myosin heavy chain (SmMHC), mouse anti-human α-actinin (Sigma); horseradish peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG (Amersham Biosciences), IRDye 800CW goat anti-rabbit IgG and IR Dye 680LT goat anti-mouse IgG (Li-Cor). Validation of the specificity of all antibodies has been shown in the results or has been demonstrated in previous studies (10, 36, 44).
Other materials and reagents used were as follows: canine recombinant IL-13 and human His-S100A4 (R&D Systems) and human eotaxin1 ELISA kit (R&D Systems); S100A4 ELISA kit ver. 2 (CircuLex, MBL Medical & Biological Laboratory Co., Ltd), plasmids encoding human RAGE, pcDNA3-RAGE (Addgene plasmid no. 71435), and pcDNA3-RAGEΔcyto (Addgene plasmid no. 71436) (20); pCMV6-XL4 human untagged S100A4 (no. SC109749, Origene); and RAGE Antagonist, FPS-ZM1, from Sigma-Aldrich (cat. no. 553030).
Quantitative RT-PCR reagents were as follows: TRIzol reagent (Invitrogen), iScrip cDNA synthesis kit (Bio-Rad), TaqMan Fast Advanced Master Mix, and primers and probes for S100A4 and β-actin were from Thermo Fisher Scientific. Primers and probes for RAGE were from Integrated DNA Technologies.
Preparation of smooth muscle tissues.
All procedures involving animals were in accordance with procedures approved by the Institutional Animal Care and Use Committee (IACUC) of Indiana University School of Medicine under the National Research Council’s Guide for the Care and Use of Laboratory Animals. Mongrel dogs (20–25 kg, either sex) were procured by the Indiana University Laboratory Animal Resource Center (LARC) at the Indiana University School of Medicine from LBL Kennels (Reelsville, IN). Animals were euthanized by LARC personnel in accordance with procedures approved by the IACUC) of Indiana University School of Medicine by intravenous injection of Fatal Plus (pentobarbital sodium: 390 mg/mL; propylene glycol: 0.01 mg/mL; ethyl alcohol: 0.29 mg/mL; benzyl alcohol (preservative): 0.2 mg/mL) at a dose of ~0.3 mL/kg. After euthanization, a tracheal segment was immediately removed and placed in physiological saline solution (PSS) aerated with 95% O2–5% CO2 to maintain a pH at 7.4. Multiple strips of trachealis muscle tissue were dissected from each trachea after removal of the epithelium and connective tissue layers. Prior to each experimental protocol, tracheal smooth muscle tissue strips were tied to wires under tension and subjected to an initial equilibration procedure to assure uniform tension and viability. Tissue strips were then subjected to stimulation with inflammatory mediators IL-13, TNF-α or S100A4, plasmid transfection procedures, and/or other experimental procedures.
Transfection of plasmids into tracheal smooth muscle tissues.
Plasmids encoding RAGE, RAGEΔcyto, untagged S100A4 (WT S100A4) were introduced into smooth muscle tissue strips by the method of reversible permeabilization, as previously described (36, 37). After the initial equilibration period, muscle tissues were attached to metal wires and incubated successively in each of the following solutions: Solution 1, which contained 10 mM EGTA, 5 mM Na2ATP, 120 mM KCl, 2 mM MgCl2, and 20 mM N-tris (hydoxymethyl) methyl-2-aminoethanesulphonic acid (TES) (at 4°C, pH 7.1,100% O2 for 120 min); Solution 2, which contained 0.1 mM EGTA, 5 mM Na2ATP, 120 mM KCl, 2 mM MgCl2, and 20 mM TES and 30 µg/mL plasmids (at 4°C, pH 7.1, overnight); Solution 3, which contained 0.1 mM EGTA, 5 mM Na2ATP, 120 mM, 10 mM MgCl2 and 20 mM TES (at 4°C, pH 7.1, 100% O2 for 30 min); and Solution 4, which contained 110 mM NaCl, 3.4 mM KCl, 0.8 mM MgSO4, 25.8 mM NaHCO3, 1.2 mM KH2PO4 (at 22°C, pH 7.4 for 60 min and aerated with 95% O2 and 5% CO2). After 30 min in Solution 4, CaCl2 was added gradually to reach a final concentration of 2.4 mM. After the introduction of plasmids, muscle tissue strips were maintained in a CO2 incubator at 37°C for 2–3 days in Dulbecco’s modified Eagle’s medium (DMEM) to allow for expression of the recombinant proteins. Five mM Na2ATP, 100 U/mL penicillin, 100 µg/mL streptomycin, and 10 μg/mL plasmids were added to the medium during the incubation period. Sham-treated tissues were subjected to identical procedures except that no plasmids were used.
Immunoblots.
Frozen muscle tissue strips were pulverized in liquid nitrogen using a mortar and pestle and pulverized tissues were mixed with lysis buffer (1% Triton X-100, 0.4% SDS, 20 mM Tris-HCl, 2 mM EDTA, and phosphatase and protease inhibitors) for protein extraction. Proteins were separated by 8% or 15% SDS-PAGE and transferred to a nitrocellulose membrane for immunoblotting. Proteins were visualized by infrared fluorescence using a Li-Cor Odyssey Fc imaging system.
Measurement of eotaxin secretion from muscle tissues.
Tracheal muscle tissue strips were incubated with 5 μg/mL S100A4 or 50 ng/mL IL-13 for 20 h. The tissue incubation medium from each sample was collected and concentrated to 100 µL using centrifugal filters. The concentration of eotaxin1 in each sample was determined by ELISA using human CCL11/eotaxin antibody (R&D Systems) and quantified using a standard curve (36). All samples and standards were measured in duplicate.
Analysis of S100A4 secretion from tissues.
Tissues were incubated with 50 ng/mL IL-13 or 50 ng/mL TNF-α for 20 h in DMEM, and the incubation medium from each tissue was collected. The concentration of S100A4 in each sample of incubation medium was determined by ELISA using anti-human S100A4 monoclonal antibody and quantified using a standard curve. All samples and standards were measured in duplicate.
S100A4 secreted into the incubation medium was also measured by immunoblotting. The incubation medium from tissues was collected and concentrated to 100 µL using a centrifugal filter. Proteins in the medium were separated by 15% SDS-PAGE and transferred to a nitrocellulose membrane for immunoblotting.
Quantitative RT-PCR.
mRNA for S100A4 and RAGE was measured using quantitative RT-PCR (qRT-PCR). Total RNA was isolated from airway smooth muscle tissues using TRIzol, and 0.5–1 µg of total RNA from each sample was reverse transcribed to cDNA using an iScrip cDNA synthesis kit. Quantitative RT-PCR was performed on an Applied Biosystems QuantStudio3 PCR machine using TaqMan fast advanced master mix. The preoptimized primers and probes for canine S100A4 and β-actin were as follows: S100A4 (assay ID: Cf02701114_m1) and β-actin (assay ID: Cf04931159_m1). Primers and probe for canine RAGE were as follows: forward primer: 5′-AGGACCAGGGAACCTACAG-3′, probe 5′-ACGACACTGACACCACGGCTTT-3′, and reverse primer: 5′-CCTGCCCTTCCACATAGC-3′. The changes in mRNA levels for target genes were quantified using relative quantitative analysis. The ∆Ct for mRNA expression of target genes was calculated relative to the Ct (threshold cycle) of β-actin. Relative mRNA expression of target genes was calculated using the 2−∆∆Ct method (25).
Statistical analysis.
Differences among treatment groups were analyzed for statistical significance using one‐way ANOVA with or without repeated measures and Tukey's multiple comparison test or by using two‐tailed paired or unpaired Student's t‐tests. Differences were considered statistically significant when P < 0.05. Data are expressed as means ± SE, and n represents the number of muscle tissue strips used for each measurement.
RESULTS
Stimulation of airway smooth muscle tissues with S100A4 activates inflammatory signaling pathways and eotaxin secretion.
The effect of extracellular S100A4 on proinflammatory signaling pathways was determined by incubating tracheal smooth muscle tissues with 5 µg/mL recombinant His-S100A4 for 30 min in PSS. Stimulation with S100A4 resulted in a significant increase in Akt phosphorylation at Ser473, an indicator of Akt activation, and a significant increase in NF-κB p65 phosphorylation at its activation site, Ser536 (30) (Fig. 1, A and B). The effect of S100A4 on Akt phosphorylation was qualitatively similar to the effect of stimulation for a comparable time period with the inflammatory mediator, IL-13 (Fig. 1A). Both S100A4 and IL-13 also stimulated a significant increase in eotaxin secretion by the tracheal smooth muscle tissues (Fig. 1C).
Fig. 1.

S100A4 stimulates inflammatory signaling pathways in airway smooth muscle (ASM) tissues. A and B: stimulation of tracheal smooth muscle tissues with recombinant His-S100A4 or IL-13 significantly increases Akt phosphorylation (A) (one-way ANOVA with repeated measures, n = 6, *P < 0.01) and NF-κB Ser536 phosphorylation (B) (two-tailed paired Student’s t test, n = 6, *P < 0.0007) relative to unstimulated (US) tissues C: eotaxin secretion was significantly increased by stimulation with S100A4 or IL-13 in ASM tissues (one-way ANOVA with repeated measures, n = 5, *P < 0.05). D: expression of smooth muscle myosin heavy chain (SmMHC) was significantly inhibited in tissues stimulated with S100A4 or IL-13 (one-way ANOVA with repeated measures, n = 8, *P < 0.001).
The stimulation of airway smooth muscle tissues with the Th2 cytokines IL-13 and IL-4 suppresses the expression of smooth muscle differentiation marker proteins, including the phenotype-specific protein, smooth muscle myosin heavy chain (SmMHC) (10, 19, 36, 37). Therefore, we evaluated the effect of S100A4 on SmMHC expression. Stimulation with S100A4 caused a significant decrease in the expression of SmMHC that was comparable to that caused by IL-13 or IL-4 (Fig. 1D).
These results demonstrate that the extracellular stimulation of airway smooth muscle tissues with S100A4 activates inflammatory signaling pathways and induces inflammatory responses by the muscle tissues.
The inflammatory effects of S100A4 on airway smooth muscle tissues are mediated by RAGE receptors.
We investigated whether S100A4 regulates inflammatory signaling pathways in airway smooth muscle tissues by acting on RAGE receptors. Airway smooth muscle tissues were transfected with plasmids encoding the recombinant mutant nonfunctional RAGE (RAGEΔcyto). The RAGE construct RAGEΔcyto can act as a receptor for extracellular ligands, but it is missing the cytoplasmic domain that activates its downstream effectors such as NF-κB (2, 20, 29). After transfection with RAGEΔcyto or full-length wild-type (WT) RAGE, tissues robustly expressed both of the recombinant RAGE proteins (Fig. 2A). Tissues expressing WT RAGE, RAGEΔcyto, and sham-treated tissues were stimulated with His-S100A4 for 30 min or left unstimulated. Basal NF-κB Ser536 phosphorylation and Akt Ser473 phosphorylation were not significantly different among the three treatment groups. NF-κB Ser536 phosphorylation and Akt Ser473 phosphorylation both increased significantly in response to S100A4 in sham-treated tissues and in tissues expressing WT RAGE (Fig. 2, B and C). The expression of RAGEΔcyto significantly inhibited S100A4-stimulated NF-κB Ser536 phosphorylation and Akt Ser473 phosphorylation in the airway smooth muscle tissues, demonstrating that S100A4 acts through RAGE receptors to activate both NF-κB and Akt (Fig. 2, B and C).
Fig. 2.

S100A4 regulates inflammatory signaling pathways by acting through receptors for advanced glycation end products (RAGE) in airway smooth muscle tissues. A: RAGE recombinant protein expression was confirmed by immunoblotting using RAGE antibody. More RAGE protein was observed in tissues treated with plasmids for wild-type (WT)-RAGE or RAGEΔcyto than in sham-treated tissues. B and C: NF-κB Ser536 phosphorylation and Akt Ser473 phosphorylation were significantly increased in response to His-S100A4 stimulation in tissues expressing WT-RAGE and in sham-treated tissues. The expression of RAGEΔcyto significantly inhibited S100A4-stimulated NF-κB Ser536 phosphorylation and Akt Ser473 phosphorylation (n = 6; *P < 0.01). D: expression of RAGEΔcyto prevents the suppression of smooth muscle myosin heavy chain (SmMHC) protein expression in response to S100A4 in airway smooth muscle tissues (n = 5; P > 0.05). Stimulation with His-S100A4 significantly inhibits SmMHC expression in sham-treated tissues (n = 5; *P < 0.001) or tissues expressing WT-RAGE (n = 5; *P < 0.05). All values are means ± SE (one-way ANOVA with repeated measures).
We also evaluated the effect of the expression of WT RAGE and RAGEΔcyto on SmMHC expression by stimulating tissues with His-S100A4 overnight after transfection (Fig. 2D). Stimulation with His-S100A4 resulted in a significant decrease in the expression of SmMHC in Sham-treated tissues and in tissues expressing WT RAGE, but His-S100A4 did not stimulate a significant reduction in SmMHC expression in tissues expressing RAGEΔcyto. The expression of WT RAGE also caused a significant reduction in SmMHC expression in unstimulated tissues, but the expression of RAGEΔcyto had no significant effect on SmMHC expression in the absence of stimulation with His-S100A4. Thus, expression of the inactive RAGE receptor prevented changes in phenotypic expression induced by S100A4, which is further evidence that S100A4 mediates its effects by acting on RAGE.
Inhibition of RAGE prevents the inflammatory responses of airway smooth muscle to S100A4.
The role of RAGE in mediating the effects of S100A4 on airway smooth muscle tissues was further evaluated by treating tissues with the RAGE antagonist FPS-ZM1 for 2 h and then stimulating them with His-S100A4. Treatment with the RAGE inhibitor significantly inhibited S100A4-stimulated NF-κB Ser536 phosphorylation and Akt Ser473 phosphorylation (Fig. 3, A and B). The effect of inhibiting RAGE on SmMHC expression and eotaxin secretion were evaluated by incubating tissues for 20 h with the RAGE inhibitor (Fig. 3, C and D). Treatment of tissues with the RAGE inhibitor prevented the reduction in SmMHC expression induced by stimulation with His-S100A4 and significantly inhibited S100A4-stimulated eotaxin secretion from tissues.
Fig. 3.

The inhibition of receptors for advanced glycation end products (RAGE) suppresses inflammatory responses to S100A4 in airway smooth muscle tissues. Airway smooth muscle tissues were treated for 2 h with 100 μM of the RAGE antagonist, FPS-ZM1 [RAGE inhibitor (Inh)] and their responses to stimulation with His-S100A4 were determined. A and B: stimulation with S100A4 significantly increased NF-κB Ser536 phosphorylation and Akt Ser473 phosphorylation. Treatment with RAGE inhibitor significantly inhibited S100A4-stimulated NF-κB Ser536 phosphorylation (n = 5, *P < 0.01) and Akt Ser473 phosphorylation (n = 5, *P < 0.001). C: smooth muscle myosin heavy chain (SmMHC) protein was significantly inhibited by stimulation with His-S100A4. Treatment of tissues with RAGE Inh prevented the reduction in SmMHC expression induced by stimulation with His-S100A4 (n = 6, *P < 0.001). D: stimulation of tissues with His-S100A4 significantly increased eotaxin secretion as measured by ELISA. Treatment of tissues with RAGE Inh prevented the increase in eotaxin secretion stimulated by His-S100A4 (n = 4, *P < 0.01). All values are means ± SE (one-way ANOVA with repeated measures).
These results demonstrate that the pharmacological inhibition of RAGE prevents inflammatory responses stimulated by S100A4 in airway smooth muscle tissues.
Stimulation of airway smooth muscle tissues with IL-13 or TNF-α induces the secretion of S100A4.
We sought to determine whether airway smooth muscle tissues secrete S100A4 in response to stimulation with inflammatory mediators. Airway tissues were incubated with 50 ng/mL IL-13 or 50 ng/mL TNF-α for 20 h, and the incubation medium was analyzed for S100A4 using ELISA and also by immunoblotting S100A4 in the incubation medium (Fig. 4). Stimulation with either IL-13 or TNF-α significantly increased S100A4 secretion from airway smooth muscle tissues, as determined by either method. The time course for the secretion of S100A4 was analyzed by immunoblotting S100A4 in the incubation medium of tissues stimulated with IL-13 or TNF-α for periods ranging from 0 to 25 h (Fig. 4C). An increase in S100A4 in the tissue incubation medium could be detected after 5 h of stimulation with either IL-13 or TNF-α and reached a maximum after 20–25 h of incubation.
Fig. 4.
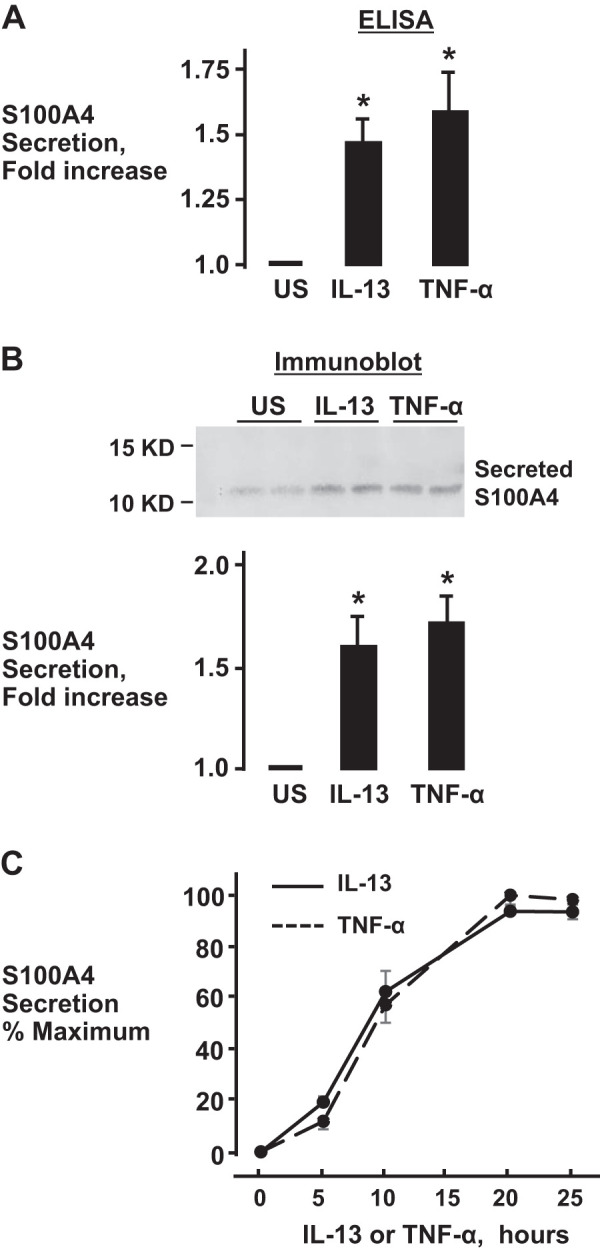
The inflammatory cytokines IL-13 and TNF-α induce the secretion of S100A4 by airway smooth muscle tissues. IL-13 and TNF-α significantly increased the secretion of S100A4 from tissues compared with unstimulated tissues. S100A4 secretion was measured by ELISA (one-way ANOVA, n = 9, *P < 0.05) (A) or by immunoblotting (one-way ANOVA with repeated measures, n = 7, *P < 0.01) (B). C: time course for secreted S100A4 in the incubation medium during stimulation with IL-13 or TNF-α. All values are means ± SE.
Stimulation of airway smooth muscle tissues with IL-13, TNF-α, or S100A4 potentiates the expression of endogenous S100A4 and RAGE.
Tissues were incubated for 20 h with 50 ng/mL IL-13, 50 ng/mL TNF-α, or 5 µg/mL His-S100A4 (Fig. 5). The expression of both RAGE protein and endogenous S100A4 protein was significantly increased in tissues stimulated with IL-13, TNF-α, or His-S100A4, as compared with unstimulated tissues (Fig. 5, A and B). Incubation of tissues with IL-13, TNF-α, or His-S100A4 also significantly increased mRNA levels for both S100A4 and RAGE, confirming that the increases in the expression of S100A4 and RAGE protein resulted from an increase in transcription (Fig. 5C). The increased expression of S100A4 or RAGE protein was evident by 10 h of stimulation with IL-13, TNF-α, or His-S100A4 and continued to increase for up to 20–25 h (Fig. 5D).
Fig. 5.

Stimulation of airway smooth muscle with the inflammatory cytokines IL-13, TNF-α, or S100A4 promotes the expression of S100A4 and receptors for advanced glycation end products (RAGE). A: immunoblot of S100A4 and RAGE in airway smooth muscle tissue extracts after stimulation with IL-13, TNF-α, or His-S100A4 for 20 h. B and C: incubation with IL-13, TNF-α, or His-S100A4 significantly increased the expression of S100A4 and RAGE protein (n = 7, *P < 0.001) (B) and mRNA (n = 6, *P < 0.001) (C) in airway smooth muscle tissues. D: time course for protein expression of RAGE and S100A4 in airway smooth muscle tissues during stimulation with IL-13, TNF-α, or His-S100A4. Values are means ± SE. Statistical analysis performed using one-way ANOVA with repeated measures.
An increase in the expression of endogenous S100A4 in airway smooth muscle tissues stimulates S100A4 secretion and activates inflammatory signaling pathways.
We sought to determine whether the increased expression of endogenous S100A4 in airway smooth muscle tissues activates inflammatory responses by the tissues by increasing S100A4 secretion. Smooth muscle tissues were transfected with plasmids encoding WT-S100A4 and incubated for 2 days to allow for the expression of recombinant WT-S100A4. The secretion of S100A4 was then measured in the incubation medium by immunoblotting. The transfection of tissues with plasmids encoding WT-S100A4 resulted in a significant increase in the expression of intracellular S100A4 protein relative to sham-treated tissues (Fig. 6A). There was a significant increase in S100A4 secretion in tissues that were overexpressing S100A4 (Fig. 6B). S100A4 overexpression also resulted in a significant increase in the activation of NF-κB and Akt and a significant decrease in the expression of SmMHC (Fig. 6C). These results demonstrate that an increase in S100A4 expression potentiates the secretion of S100A4 and stimulates the activation of inflammatory signaling pathways in airway smooth muscle tissues.
Fig. 6.

S100A4 overexpression in airway smooth muscle tissues increases S100A4 secretion and the activation of inflammatory signaling pathways. A: S100A4 overexpression in tissues expressing recombinant S100A4 (no epitope tag) was confirmed by immunoblotting. More S100A4 was detected in tissues transfected with plasmids compared with sham-treated tissues (n = 5, *P = 0.005). B: S100A4 in the incubation medium was detected by immunoblotting. The overexpression of S100A4 significantly increased the secretion of S100A4 from airway smooth muscle tissues (n = 4, *P = 0.035). C: overexpression of S100A4 significantly increases NF-κB Ser536 phosphorylation (n = 7; *P = 0.006) and Akt Ser473 phosphorylation (n = 6; *P = 0.002) compared with sham-treated tissues. Smooth muscle myosin heavy chain (SmMHC) expression was significantly lower in tissues overexpressing S100A4 compared with sham-treated tissues (n = 4; *P = 0.006). Values are means ± SE. Statistical analysis performed by paired Student’s t test.
DISCUSSION
The results of this study provide the first demonstration that the S100 family protein S100A4 acts as an inflammatory mediator in airway tissues (Fig. 7). The inflammatory effects of S100A4 on airway smooth muscle tissues are similar to those of IL-13 and TNF-α (10, 19, 36): S100A4 activates Akt and NF-κB, enhances secretion of the chemokine eotaxin, and suppresses expression of the smooth muscle phenotype-specific protein SmMHC. Our results also provide the first evidence that the inflammatory effects of S100A4 on airway smooth muscle tissues are mediated by RAGE receptors expressed on the muscle tissues. We find that S100A4 acts in an autocrine fashion to promote airway smooth muscle inflammation: the inflammatory cytokines IL-13 and TNF-α stimulate the expression of S100A4 in the airway muscle tissues and increase its secretion into the extracellular fluid, where S100A4 can then act on resident airway tissues to promote inflammation. These results suggest that S100A4 may be a potent inflammatory stimulus in asthma and allergic inflammation of the airways and that it acts to potentiate the effects of inflammatory cytokines.
Fig. 7.

S100A4 plays an autocrine role in promoting inflammation in airway smooth muscle tissues. Extracellular S100A4 acts on receptors for advanced glycation end products (RAGE) in airway smooth muscle to activate proinflammatory pathways mediated by Akt and NF-κB. The activation of Akt and NF-κB promotes eotaxin secretion. Akt suppresses the expression of smooth muscle myosin heavy chain (SmMHC) by inhibiting the translocation of serum response factor (SRF) to the nucleus. Activated NF-κB increases the transcription and expression of S100A4 and RAGE. Increased expression of intracellular S100A4 promotes S100A4 secretion from airway smooth muscle tissues and the activation of inflammatory signaling pathways.
Our results show that S100A4 activates proinflammatory signaling pathways mediated by Akt and NF-κB by acting as a ligand for RAGE receptors in airway smooth muscle tissue (Fig. 1). We expressed a mutant nonfunctional RAGE construct, RAGEΔcyto in the airway smooth muscle tissues. RAGEΔcyto has a deletion of most of its cytoplasmic domain and cannot activate downstream signaling pathways mediated by NF-κB and Akt (2, 20, 29) (Fig. 2). The expression of inactive RAGE receptors completely inhibited intracellular signaling stimulated by S100A4, suggesting that RAGE is the primary receptor for S100A4 in this tissue. We also evaluated the effects of treating airway smooth muscle tissues with a small molecule inhibitor of RAGE (Fig. 3). The inhibition of endogenous RAGE receptors also blocked inflammatory responses to stimulation by S100A4 and prevented the transition of airway smooth muscles to a synthetic inflammatory phenotype. Although RAGE receptors are known to be expressed in the lungs (4, 27, 32), their expression in airway smooth muscle tissues has not previously been demonstrated. Our study thus provides the first demonstration that RAGE receptors regulate inflammatory signaling pathways in airway smooth muscle and that S100A4 mediates its inflammatory effects by activating RAGE receptors expressed on the airway smooth muscle cells. These results are consistent with in vivo studies in murine models of asthma that have suggested a role for RAGE in allergic lung inflammation (27, 32).
Our study showed that treatment of airway smooth muscle tissues with either IL-13 or TNF-α stimulates the expression of endogenous S100A4 and the secretion of S100A4 from the muscle tissues (Figs. 4 and 5). When we overexpressed recombinant WT-S100A4 in the muscle tissues, the secretion of S100A4 by airway smooth muscle tissues was increased (Fig. 6). This suggests that the overexpression of S100A4 that is induced by the inflammatory stimuli IL-13 and TNF-α promotes its secretion.
Our evidence that S100A4 acts as an inflammatory stimulus in airway smooth muscle tissues is consistent with other studies suggesting a role for S100A4 in promoting airway inflammation. In a murine model of allergic asthma (18), treatment of asthmatic mice with S100A4-neutralizing antibodies inhibited inflammatory cell recruitment and reduced inflammatory mediators in the lungs of the mice. In another study, S100A4-deficient mice did not develop symptoms of allergic dermatitis and had impaired recruitment of macrophages to sites of inflammation in vivo (5).
S100A4 has direct effects on inflammatory cells and can regulate the motility of leukocytes (1, 12, 24); however, our results demonstrate that S100A4 can directly trigger proinflammatory responses by airway smooth muscle, and that S100A4 can regulate its own secretion from airway muscle cells. Thus, S100A4 could regulate lung inflammation through its proinflammatory effects on resident airway cells and other structural cells in the lungs. There is evidence for S100A4-mediated inflammatory effects on other structural cells: S100A4 can stimulate a RAGE-mediated inflammatory signaling cascade in chondrocytes (41). S100A4 can also stimulate a phenotypic transition from a rhomboid to a synthetic phenotype by activating RAGE in cultured porcine arterial smooth muscle cells (8).
Other members of the S100A protein family have been proposed to play roles in asthma and inflammatory diseases (7, 16, 42). Both the heterodimer S100A8/S100A9 and S100A12 can be secreted into the extracellular space by myeloid cells. Studies of airway cells in culture have indicated that S100A8/S100A9 can inhibit airway smooth muscle migration and proliferation, which led the authors to propose that it may counteract airway remodeling (39, 40, 43). Both proinflammatory and anti-inflammatory roles for S100A12 in airway inflammation have been proposed. On one hand, S100A12 has been proposed to potentiate both asthma and innate immunity by promoting monocyte recruitment and airway inflammation (42); on the other hand, S100A12 suppressed airway inflammation and airway hyperreactivity in a murine model of allergic airway inflammation (6, 17). Our evidence clearly demonstrates that S100A4 functions as a proinflammatory stimulus in airway smooth muscle tissues and that airway smooth muscle could be a source for the S100A4 in the extracellular fluids of asthmatic subjects and animal models of airway inflammation (5, 18).
In summary, our results demonstrate that inflammatory mediators associated with airway inflammation can trigger the release of S100A4 by airway smooth muscle tissues and that extracellular S100A4 acts on RAGE receptors on the muscle cells to activate proinflammatory pathways. These results suggest that S100A4 plays a pivotal role in regulating the inflammation of airway smooth muscle tissues and that it could contribute significantly to inflammatory diseases of the airways. S100A4 may provide a useful therapeutic target for the amelioration of airway inflammation, and elevated S100A4 may serve as a marker for inflammatory airways disease.
GRANTS
These studies were supported by National Heart, Lung, and Blood Institute Grant HL029289.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.W., W.Z., and S.J.G. conceived and designed research; Y.W., W.Z., and S.J.G. performed experiments; Y.W., W.Z., and S.J.G. analyzed data; Y.W., W.Z., and S.J.G. interpreted results of experiments; Y.W., W.Z., and S.J.G. prepared figures; Y.W., W.Z., and S.J.G. drafted manuscript; Y.W., W.Z., and S.J.G. edited and revised manuscript; Y.W., W.Z., and S.J.G. approved final version of manuscript.
REFERENCES
- 1.Ambartsumian N, Klingelhöfer J, Grigorian M. The multifaceted S100A4 protein in cancer and inflammation. Methods Mol Biol 1929: 339–365, 2019. doi: 10.1007/978-1-4939-9030-6_22. [DOI] [PubMed] [Google Scholar]
- 2.Angelo MF, Aguirre A, Avilés Reyes RX, Villarreal A, Lukin J, Melendez M, Vanasco V, Barker P, Alvarez S, Epstein A, Jerusalinsky D, Ramos AJ. The proinflammatory RAGE/NF-κB pathway is involved in neuronal damage and reactive gliosis in a model of sleep apnea by intermittent hypoxia. PLoS One 9: e107901, 2014. doi: 10.1371/journal.pone.0107901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boye K, Maelandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol 176: 528–535, 2010. doi: 10.2353/ajpath.2010.090526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 143: 1699–1712, 1993. [PMC free article] [PubMed] [Google Scholar]
- 5.Bruhn S, Fang Y, Barrenäs F, Gustafsson M, Zhang H, Konstantinell A, Krönke A, Sönnichsen B, Bresnick A, Dulyaninova N, Wang H, Zhao Y, Klingelhöfer J, Ambartsumian N, Beck MK, Nestor C, Bona E, Xiang Z, Benson M. A generally applicable translational strategy identifies S100A4 as a candidate gene in allergy. Sci Transl Med 6: 218ra4, 2014. doi: 10.1126/scitranslmed.3007410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camoretti-Mercado B, Karrar E, Nuñez L, Bowman MA. S100A12 and the airway smooth muscle: beyond inflammation and constriction. J Allergy Ther 3, Suppl 1: S1–S7, 2012. doi: 10.4172/2155-6121.S1-007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camoretti-Mercado B, Pauer SH, Yong HM, Smith DC, Deshpande DA, An SS, Liggett SB. Pleiotropic effects of bitter taste receptors on [Ca2+]i mobilization, hyperpolarization, and relaxation of human airway smooth muscle cells. PLoS One 10: e0131582, 2015. doi: 10.1371/journal.pone.0131582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaabane C, Heizmann CW, Bochaton-Piallat ML. Extracellular S100A4 induces smooth muscle cell phenotypic transition mediated by RAGE. Biochim Biophys Acta 1853: 2144–2157, 2015. doi: 10.1016/j.bbamcr.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 9.Damera G, Tliba O, Panettieri RA Jr. Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther 22: 353–359, 2009. doi: 10.1016/j.pupt.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desai LP, Wu Y, Tepper RS, Gunst SJ. Mechanical stimuli and IL-13 interact at integrin adhesion complexes to regulate expression of smooth muscle myosin heavy chain in airway smooth muscle tissue. Am J Physiol Lung Cell Mol Physiol 301: L275–L284, 2011. doi: 10.1152/ajplung.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donato R. RAGE: a single receptor for several ligands and different cellular responses: the case of certain S100 proteins. Curr Mol Med 7: 711–724, 2007. doi: 10.2174/156652407783220688. [DOI] [PubMed] [Google Scholar]
- 12.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 proteins. Curr Mol Med 13: 24–57, 2013. doi: 10.2174/156652413804486214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebralidze A, Tulchinsky E, Grigorian M, Afanasyeva A, Senin V, Revazova E, Lukanidin E. Isolation and characterization of a gene specifically expressed in different metastatic cells and whose deduced gene product has a high degree of homology to a Ca2+-binding protein family. Genes Dev 3: 1086–1093, 1989. doi: 10.1101/gad.3.7.1086. [DOI] [PubMed] [Google Scholar]
- 14.Garrett SC, Varney KM, Weber DJ, Bresnick AR. S100A4, a mediator of metastasis. J Biol Chem 281: 677–680, 2006. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 15.Gross SR, Sin CG, Barraclough R, Rudland PS. Joining S100 proteins and migration: for better or for worse, in sickness and in health. Cell Mol Life Sci 71: 1551–1579, 2014. doi: 10.1007/s00018-013-1400-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halayko AJ, Ghavami S. S100A8/A9: a mediator of severe asthma pathogenesis and morbidity? Can J Physiol Pharmacol 87: 743–755, 2009. doi: 10.1139/Y09-054. [DOI] [PubMed] [Google Scholar]
- 17.Hofmann Bowman MA, Heydemann A, Gawdzik J, Shilling RA, Camoretti-Mercado B. Transgenic expression of human S100A12 induces structural airway abnormalities and limited lung inflammation in a mouse model of allergic inflammation. Clin Exp Allergy 41: 878–889, 2011. doi: 10.1111/j.1365-2222.2011.03714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang X, Qu D, Liang Y, Huang Q, Li M, Hou C. Elevated S100A4 in asthmatics and an allergen-induced mouse asthma model. J Cell Biochem 120: 9667–9676, 2019. doi: 10.1002/jcb.28245. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, Gunst SJ. Phenotype transitions induced by mechanical stimuli in airway smooth muscle are regulated by differential interactions of parvin isoforms with paxillin and Akt. Am J Physiol Lung Cell Mol Physiol 318: L1036–L1055, 2020. doi: 10.1152/ajplung.00506.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem 274: 19919–19924, 1999. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 21.Jonasson L, Grauen Larsen H, Lundberg AK, Gullstrand B, Bengtsson AA, Schiopu A. Stress-induced release of the S100A8/A9 alarmin is elevated in coronary artery disease patients with impaired cortisol response. Sci Rep 7: 17545, 2017. doi: 10.1038/s41598-017-17586-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klingelhöfer J, Senolt L, Baslund B, Nielsen GH, Skibshøj I, Pavelka K, Neidhart M, Gay S, Ambartsumian N, Hansen BS, Petersen J, Lukanidin E, Grigorian M. Up-regulation of metastasis-promoting S100A4 (Mts-1) in rheumatoid arthritis: putative involvement in the pathogenesis of rheumatoid arthritis. Arthritis Rheum 56: 779–789, 2007. doi: 10.1002/art.22398. [DOI] [PubMed] [Google Scholar]
- 23.Leclerc E, Fritz G, Vetter SW, Heizmann CW. Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta 1793: 993–1007, 2009. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 24.Li ZH, Dulyaninova NG, House RP, Almo SC, Bresnick AR. S100A4 regulates macrophage chemotaxis. Mol Biol Cell 21: 2598–2610, 2010. doi: 10.1091/mbc.e09-07-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Lockett AD, Wu Y, Gunst SJ. Elastase alters contractility and promotes an inflammatory synthetic phenotype in airway smooth muscle tissues. Am J Physiol Lung Cell Mol Physiol 314: L626–L634, 2018. doi: 10.1152/ajplung.00334.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milutinovic PS, Alcorn JF, Englert JM, Crum LT, Oury TD. The receptor for advanced glycation end products is a central mediator of asthma pathogenesis. Am J Pathol 181: 1215–1225, 2012. doi: 10.1016/j.ajpath.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oslejsková L, Grigorian M, Gay S, Neidhart M, Senolt L. The metastasis associated protein S100A4: a potential novel link to inflammation and consequent aggressive behaviour of rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis 67: 1499–1504, 2008. doi: 10.1136/ard.2007.079905. [DOI] [PubMed] [Google Scholar]
- 29.Riuzzi F, Sorci G, Donato R. RAGE expression in rhabdomyosarcoma cells results in myogenic differentiation and reduced proliferation, migration, invasiveness, and tumor growth. Am J Pathol 171: 947–961, 2007. doi: 10.2353/ajpath.2007.070049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J Biol Chem 280: 34538–34547, 2005. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- 31.Šenolt L, Cerezo LA, Šumová B, Pecha O, Pleštilová L, Forejtová Š, Růžičková O, Hušáková M, Závada J, Pavelka K, Vencovský J, Mann H. High levels of metastasis-inducing S100A4 protein and treatment outcome in early rheumatoid arthritis: data from the PERAC cohort. Biomarkers 20: 47–51, 2015. doi: 10.3109/1354750X.2014.989544. [DOI] [PubMed] [Google Scholar]
- 32.Taniguchi A, Miyahara N, Waseda K, Kurimoto E, Fujii U, Tanimoto Y, Kataoka M, Yamamoto Y, Gelfand EW, Yamamoto H, Tanimoto M, Kanehiro A. Contrasting roles for the receptor for advanced glycation end-products on structural cells in allergic airway inflammation vs. airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol 309: L789–L800, 2015. doi: 10.1152/ajplung.00087.2015. [DOI] [PubMed] [Google Scholar]
- 33.Tardif MR, Chapeton-Montes JA, Posvandzic A, Pagé N, Gilbert C, Tessier PA. Secretion of S100A8, S100A9, and S100A12 by neutrophils involves reactive oxygen species and potassium efflux. J Immunol Res 2015: 296149, 2015. doi: 10.1155/2015/296149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tliba O, Panettieri RA Jr. Noncontractile functions of airway smooth muscle cells in asthma. Annu Rev Physiol 71: 509–535, 2009. doi: 10.1146/annurev.physiol.010908.163227. [DOI] [PubMed] [Google Scholar]
- 35.Van Crombruggen K, Vogl T, Pérez-Novo C, Holtappels G, Bachert C. Differential release and deposition of S100A8/A9 proteins in inflamed upper airway tissue. Eur Respir J 47: 264–274, 2016. doi: 10.1183/13993003.00159-2015. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y, Huang Y, Gunst SJ. Focal adhesion kinase (FAK) and mechanical stimulation negatively regulate the transition of airway smooth muscle tissues to a synthetic phenotype. Am J Physiol Lung Cell Mol Physiol 311: L893–L902, 2016. doi: 10.1152/ajplung.00299.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y, Huang Y, Herring BP, Gunst SJ. Integrin-linked kinase regulates smooth muscle differentiation marker gene expression in airway tissue. Am J Physiol Lung Cell Mol Physiol 295: L988–L997, 2008. doi: 10.1152/ajplung.90202.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie J, Méndez JD, Méndez-Valenzuela V, Aguilar-Hernández MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal 25: 2185–2197, 2013. doi: 10.1016/j.cellsig.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Xu YD, Wang Y, Yin LM, Park GH, Ulloa L, Yang YQ. S100A8 protein attenuates airway hyperresponsiveness by suppressing the contraction of airway smooth muscle. Biochem Biophys Res Commun 484: 184–188, 2017. doi: 10.1016/j.bbrc.2017.01.033. [DOI] [PubMed] [Google Scholar]
- 40.Xu YD, Wei Y, Wang Y, Yin LM, Park GH, Liu YY, Yang YQ. Exogenous S100A8 protein inhibits PDGF-induced migration of airway smooth muscle cells in a RAGE-dependent manner. Biochem Biophys Res Commun 472: 243–249, 2016. doi: 10.1016/j.bbrc.2016.02.098. [DOI] [PubMed] [Google Scholar]
- 41.Yammani RR, Carlson CS, Bresnick AR, Loeser RF. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum 54: 2901–2911, 2006. doi: 10.1002/art.22042. [DOI] [PubMed] [Google Scholar]
- 42.Yang Z, Yan WX, Cai H, Tedla N, Armishaw C, Di Girolamo N, Wang HW, Hampartzoumian T, Simpson JL, Gibson PG, Hunt J, Hart P, Hughes JM, Perry MA, Alewood PF, Geczy CL. S100A12 provokes mast cell activation: a potential amplification pathway in asthma and innate immunity. J Allergy Clin Immunol 119: 106–114, 2007. doi: 10.1016/j.jaci.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 43.Yin LM, Li HY, Zhang QH, Xu YD, Wang Y, Jiang YL, Wei Y, Liu YY, Yang YQ. Effects of S100A9 in a rat model of asthma and in isolated tracheal spirals. Biochem Biophys Res Commun 398: 547–552, 2010. doi: 10.1016/j.bbrc.2010.06.116. [DOI] [PubMed] [Google Scholar]
- 44.Zhang W, Gunst SJ. Non-muscle (NM) myosin heavy chain phosphorylation regulates the formation of NM myosin filaments, adhesome assembly and smooth muscle contraction. J Physiol 595: 4279–4300, 2017. doi: 10.1113/JP273906. [DOI] [PMC free article] [PubMed] [Google Scholar]