

Abstract

Cationic Lewis acids (LAs) are gaining interest as targets for frustrated Lewis pair (FLP)-mediated catalysis. Unlike neutral boranes, which are the most prevalent LAs for FLP hydrogenations, the Lewis acidity of cations can be tuned through modulation of the counteranion; however, detailed studies on such anion effects are currently lacking in the literature. Herein, we present experimental and computational studies which probe the mechanism of H2 activation using iPr3SnOTf (1-OTf) in conjunction with a coordinating (quinuclidine; qui) and noncoordinating (2,4,6-collidine; col) base and compare its reactivity with {iPr3Sn·base}{Al[OC(CF3)3]4} (base = qui/col) systems which lack a coordinating anion to investigate the active species responsible for H2 activation and hence resolve any mechanistic roles for OTf– in the iPr3SnOTf-mediated pathway.
Keywords: frustrated Lewis pair, hydrogen activation, anion, tin, stannylium
Introduction
Frustrated Lewis pairs (FLPs), where bulky Lewis acid (LA) and Lewis base (LB) partners are sterically precluded from forming strong classical adducts, are now well-established systems for small-molecule activation with main group compounds.1,2 Perhaps most notably, H2 heterolysis can be effected by FLPs to form H+ and H– equivalents, which can be delivered to unsaturated bonds to form a reduced compound and the original FLP. Accordingly, FLP-catalyzed hydrogenations of organic compounds containing the C=X (X = C, N, and O) bonds have received widespread attention and have been thoroughly studied.3 The LAs used in these reactions are predominantly triarylboranes BAr3, particularly B(C6F5)3 and similar derivatives4 thereof, where the electronic and steric profiles have been tuned by the variation of aryl substituents (although these are predominantly confined to H, F, and Cl),5 which can afford a systematic manipulation of BAr3 Lewis acidity. Accordingly, novel BAr3 have recently been developed that can catalyze the hydrogenations of substrate classes previously inaccessible by FLPs or display the catalytic activity in the presence of alcohols/moisture, yet accessing such heteroleptic boranes requires laborious multistep syntheses.5e,6 Although some aspects of the H2 activation mechanism between the BAr3 and N- or P-centered bases are still being debated,7 it is generally assumed that facile H2 heterolysis is associated with the initial formation of a noncovalent LA–LB “encounter complex”, which subsequently reacts with H2 in a cooperative manner (i.e., a concerted mechanism via a single transition state).8 Important exceptions are “frustrated radical pairs”, which activate substrates via single electron pathways.9 Recently, the range of LAs displaying FLP-like reactivity has expanded to encompass a number of cationic species (or neutral species that act as surrogates for the active cationic LA) based on B(III),10 Si(IV),11 C(IV),12 N(III),13 P(V),14 and Sb(V).15,16
Tuning the LA strength, and hence the reactivity, of these LAs may also be achieved through ligand design within the formally cationic fragment, akin to BAr3 compounds; however, their anions also provide an additional locus for variation and hence a comparatively simple route to optimize the Lewis acidity of the cationic species. For instance, replacement of [TfO]− for [B(C6F5)4]− in phosphonium or stibonium LAs can switch on otherwise absent reactivity (see Figure 1), which has been rationalized only by reference to the relative sequestration of the cation by more strongly coordinating anions, resulting in weaker LAs. A detailed analysis of any intimate effects the anion may exert during a reaction (e.g. by its coordination within a transition state, which could perturb its energy) have not been previously undertaken for cationic LAs.
Figure 1.

Examples of cationic LAs where reactivity was promoted by using a weakly coordinating counteranion (top) and the stannylium-based LA/base systems investigated for H2 activation in this study (bottom). RF = C(CF3)3.
We reported previously that iPr3SnOTf (1-OTf; Tf = O2SCF3), which acts as a surrogate for the stannylium ion [iPr3Sn]+ ([1]+), activates H2 in conjunction with LBs (e.g., 2,4,6-collidine; col) and catalyzes the hydrogenation of imines and ketones and the reductive amination of carbonyls.17 Despite the diversity of hydrogenation substrates accessible by 1-OTf, which can operate in the presence of moisture and strong LBs, productive hydrogenation using 1-OTf/col is rather slow and requires forcing conditions (>120 °C, 10 bar H2). A thorough understanding of the factors affecting 1-OTf/LB catalytic hydrogenations would clearly aid the development of superior R3SnX LA hydrogenation catalysts, although it should be borne in mind that this will require a more complicated treatment, cf. B-based FLPs. This is predominantly due to the presence of the (hemi-labile) TfO anion and the fact that the [R3Sn]+ species are known to readily bind two anionic/neutral donors (as opposed to only one for BAr3); such five-coordinate adducts may also be important off-cycle entities in catalysis. A previous computational study by Pati et al. suggested that the weakly associated complexes of 1-OTf and either DABCO (1,4-diazabicyclo[2.2.2]octane) or col cleave H2 via an FLP-type concerted mechanism18 analogous to that seen in the FLP reactivity of the electrophilic [(C6F5)3P-X]+ species.14a−14d Pati et al. assumed that 1-OTf was the only active LA despite the experimental evidence for the formation of [1·(LB)]+ adducts (e.g. 119Sn NMR shifts) in 1-OTf/LB mixtures;17a accordingly, any role of [1·(LB)]+ species was not investigated, nor the potential impact of charged hypervalent Sn species upon reductions mediated by 1-OTf, all of which could arise from the displacement of labile [TfO]− during the reaction pathway (see Scheme 1). Clearly, ascertaining the correct catalytic speciation in these more complex systems is crucial if targeted improvements in activity/selectivity are to proceed in a logical and predictive manner. Herein, we report a comprehensive study into the mechanism of H2 activation using 1-OTf/LB pairs, using complementary experimental and theoretical methods, which reveal the noninnocent nature of [TfO]− in influencing both kinetic and thermodynamic factors. These results provide fundamental insight into how heavier-element cationic Lewis pairs activate H2, which will help guide the design of new cationic p-block LA catalysts by encouraging further research into the role and tunability of the anion in reactions featuring such LAs.
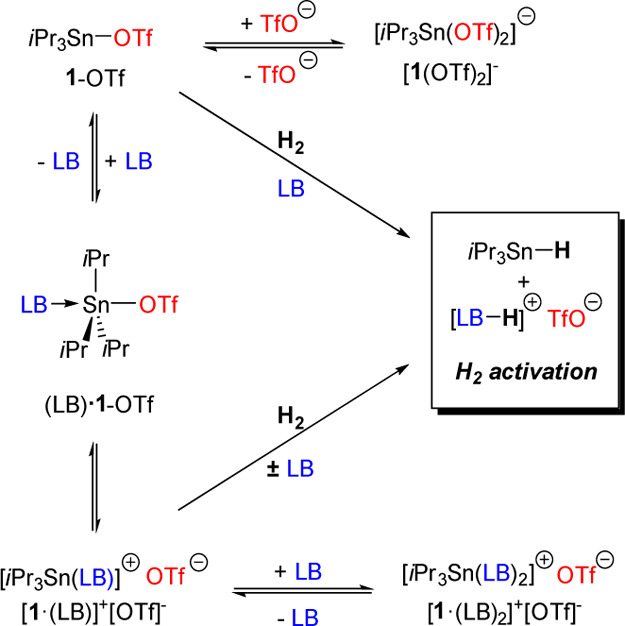
Scheme 1. Possible Sn-Containing Species in 1-OTf + LB Combinations and Mechanistic Pathways for H2 Activation.
Results and Discussion
Identifying Ground-State Speciation in 1-OTf/Quinuclidine Lewis Pairs
Because DABCO contains two donor atoms, which in theory could both interact with LAs and accordingly complicate the identification of equilibria speciation in 1-OTf/LB pairs, we chose instead to study the interaction of 1-OTf with quinuclidine (qui) because of its similar basicity [pKa(MeCN): [DABCO-H]+ 18.3;19 [qui-H]+ 19.5]20 and steric profile. 1-OTf exhibits a broad peak in its 119Sn{1H} NMR spectrum at δ = 166 ppm in 1,2-difluorobenzene (DFB),21 which moves to 34 ppm and sharpens upon addition of 1 equiv of qui at room temperature (RT), suggesting a spontaneous association to form (qui)·1-OTf and/or formation of the ion pair [1·(qui)]+[OTf]−.
Gratifyingly, slow cooling of this solution yielded single crystals that were analyzed by X-ray diffraction; the solved structure (Figure 2) reveals the former species: a five-coordinate adduct in which OTf– and the qui coordinate trans across the [1]+ core, with a trigonal bipyramidal geometry for the Sn atom [bond angles (°): C–Sn–N = 91.41(9), 93.73(9), and 93.76(9); O1–Sn–N = 177.40(7)]. Although this (qui)·1-OTf adduct represents the ground-state structure in the solid phase, it is possible that [1·(qui)]+[OTf]− is accessible at RT in solution. In order to spectroscopically identify [1·(qui)]+ as a distinct moiety, it was necessary to characterize it in the absence of any exchange equilibria, which was achieved by exchanging [OTf]− for the much more weakly coordinating aluminate {Al[OC(CF3)3]4}− ([Al(ORF)4]−); this was chosen because it is particularly robust to strongly Lewis and Brønsted acidic media.22 Thus, the reaction of K[Al(ORF)4], 1-OTf, and qui (1:1:1) in DFB resulted in the precipitation of KOTf, with the 19F NMR spectrum of the filtrate showing only a single signal for the aluminate anion, demonstrating clean anion metathesis (Scheme 2). Layering this solution with pentane yielded crystals suitable for X-ray diffraction studies; the structural solution to these data is represented in Figure 3 and corresponds to the salt [1·(qui)]+[Al(ORF)4]−.23
Figure 2.
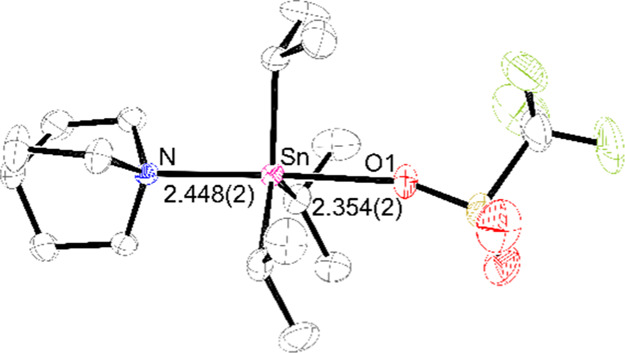
Oak Ridge Thermal Ellipsoid Plot (ORTEP) diagram of (qui)·1-OTf. Thermal ellipsoids set at 50% probability. C atoms in black, F atoms in green, N atom in blue, O atoms in red, S atom in yellow, and Sn atom in pink. The Sn–N and Sn–O bond lengths are given in Å with estimated standard deviations (ESDs) in parentheses. H atoms are omitted for clarity.
Scheme 2. Syntheses of [1·(LB)]+[Al(ORF)4]−; LB = qui or col (Collidine, 2,4,6-Trimethylpyridine).

Figure 3.
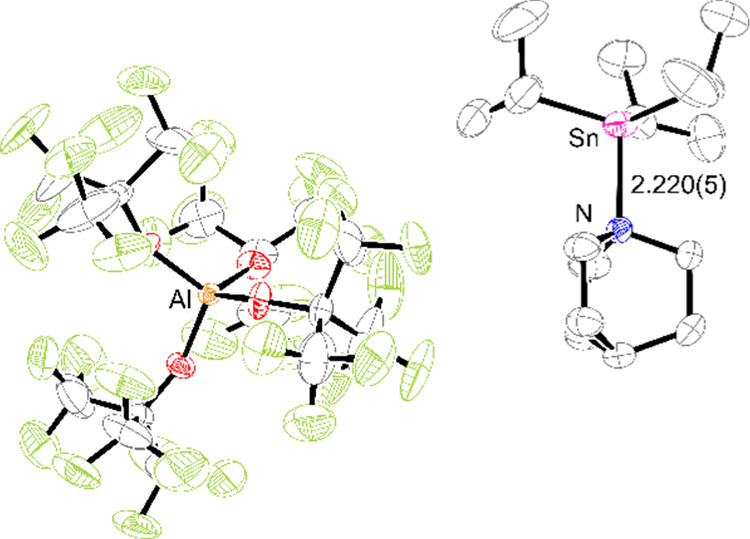
ORTEP diagram of [1·(qui)]+[Al(ORF)4]−. Thermal ellipsoids set at 50% probability. Al atom in orange, C atoms in black, F atoms in green, N atom in blue, O atoms in red, and Sn atom in pink. The Sn–N bond length is given in Å with the ESD in parentheses. The H atoms are omitted for clarity.
As anticipated, the [Al(ORF)4]− ion does not display any close contacts to the [1]+ unit and therefore does not coordinate to the LA Sn atom. Consequently, the [1·(qui)]+ assembly adopts a distorted tetrahedral geometry around the Sn center [N–Sn–C bond angles (°) = 103.7(2), 102.8(2), 102.9(3)]. The Sn–N bond distance is considerably shorter than that observed for the qui·1-OTf adduct [2.448(2) Å], which is consistent with a Sn–N weakening in the latter because of a trans-labilizing effect of the OTf– and/or increased steric encumbrance. The 1H NMR spectroscopic analysis of the DFB solutions of [1·(qui)]+[Al(ORF)4]− showed that the observed chemical shifts and couplings for the iPr groups are similar to those for 1-OTf in DFB; the relative integration of these with the signals for qui confirms a 1:1 ratio between [iPr3Sn]+ and qui. The 119Sn{1H} NMR spectrum, however, conclusively shows the formation of a new stannylium adduct with a sharp resonance at δ = 113 ppm, which is upfield from 1-OTf (δ = 166 ppm), but notably downfield of that seen for 1-OTf/qui mixtures (δ = 34 ppm). Taken together, these spectroscopic data confirm that the gross solid-state structure of [1·(qui)]+[Al(ORF)4]− is retained in the solution. Curiously, the 119Sn{1H} NMR spectroscopic data for the adduct formed from 1-OTf and qui in situ, or by redissolution of (qui)·1-OTf crystals obtained thereof, are inconsistent with those of either authentic 1-OTf or [1·(qui)]+. This suggests that neither compound is present exclusively. It is plausible that a rapid equilibrium containing 1-OTf, (qui)·1-OTf, and [1·(qui)]+ exists in the solution. To understand better the speciation, we assessed the relative energies of these species computationally (Scheme 3), with our density functional theory (DFT) calculations24 predicting the formation of a ternary complex (qui)·1-OTf to be clearly favored thermodynamically with respect to qui + 1-OTf, in contrast with the previously obtained results for the related DABCO + 1-OTf system.18,25 Additionally, dissociation of the adduct (qui)·1-OTf into [1·(qui)]+ + [OTf]− is also found to be thermodynamically feasible, with the latter computed to be 4.8 kcal/mol more stable than the original qui + 1-OTf reference state.
Scheme 3. Possible Reactant-State Species in the Solution Phase for the 1-OTf + qui Lewis Pair; Computed Relative Stabilities (in kcal/mol at 298 K) Are Shown in Parentheses24.

We also probed two other possible adducts: one where the dissociated [OTf]− binds to 1-OTf and the other where a further qui molecule coordinates to [1·(qui)]+; of these two ternary adducts, [1(OTf)2]− is stabilized notably with respect to the dissociated form, whereas [1·(qui)2]+ is predicted to be an unstable species (see Scheme 3). The optimized structures of possible binary and ternary adducts are depicted in Figure 4, and the computed bond distances suggest considerably weakened Sn–ligand bonds in the ternary species. The notable difference between the computationally and X-ray crystallographically determined Sn–N/O bond lengths in the (qui)·1-OTf complex is likely due to the intermolecular forces in the crystal structure, which appreciably influences the character of the Sn–N/O dative bonds. Of note, one of the Sn–N bonds in [1·(qui)2]+ is particularly elongated, which results in asymmetry across the equatorial plane and is likely due to steric effects.26 Having predicted that 1-OTf, (qui)·1-OTf, and [1·(qui)]+ are thermally accessible in a solution at RT, we sought to identify each species experimentally by manipulating the equilibria between them. Upon portion-wise addition of qui to 1-OTf in DFB, the 1H NMR iPr methine resonance (δ = 2.15 ppm) moves upfield to a limiting value of δ = 1.94 ppm (5.5 equiv qui), after which further qui results in no further change. Similarly, the 119Sn{1H} NMR resonance moves upfield from that observed for pure 1-OTf (δ = 166 → 0 ppm; see the Supporting Information). Only a single resonance attributable to the [1]+ fragment was observed for any qui:1-OTf ratio in the 119Sn{1H} NMR spectra (the same is true for the iPr resonances in the 1H NMR spectra), indicating that the observed shifts are weighted averages of the multiple species, all of which are in rapid exchange.
Figure 4.
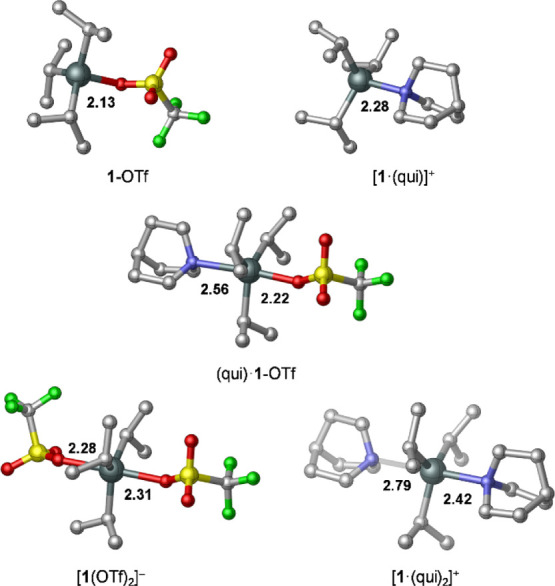
Optimized structures of possible binary and ternary adducts in the solution phase for the 1-OTf + qui Lewis pair. Selected bond distances are given in Å. All hydrogen atoms are omitted for clarity.
A variable temperature 119Sn{1H} NMR experiment of a 1:1 mixture of 1-OTf and qui (CD2Cl2)27 was undertaken to slow the exchange between the adducts (Figure 5, spectra A1 and A2),28 which revealed the broad RT resonance (δ = 45 ppm) to split into three distinct peaks (i–iii) below 263 K. Resonance i (δ = 119 ppm, 233 K) is very similar to that seen for authentic [1·(qui)]+[Al(ORF)4]− (δ = 113 ppm, 293 K; Figure 5, spectrum B), and accordingly, it is assigned to the cation fragment therein. The existence of the latter implies that dissociation of [OTf]− from (qui)·1-OTf has occurred, which is computationally predicted to bind spontaneously to 1-OTf and form [1·(OTf)2]− (ΔG = −3.2 kcal/mol).29 To verify this, an equimolar solution of [nBu4N][OTf] and 1-OTf (DFB, 293 K) was prepared, which displayed a broad peak at δ = 62.5 ppm in the 119Sn{1H} spectrum (Figure 5, spectrum C); this is significantly upfield from the chemical shift of either 1-OTf or [1·(qui)]+ and is almost identical to that observed for ii (62.9 ppm, 233 K), so it is attributed to [1(OTf)2]−. Notably at temperatures below 203 K, only one peak at δ = 14 ppm is observed (Figure 5, spectrum A3); DFT calculations predict the 1:1 adduct between qui and 1-OTf to be the most stable species at low temperatures (for details, see the Supporting Information, Section 6.1), and accordingly, we ascribe resonance iii to (qui)·1-OTf.
Figure 5.

(A1–A3): Stacked 119Sn{1H} NMR spectra of a 1:1 mixture of 1-OTf + qui in CD2Cl2 at selected temperatures. (B) 119Sn{1H} NMR spectrum of authentic [1·(qui)][Al(ORF)4] in DFB. (C) 119Sn{1H} NMR spectrum of 1-OTf + [nBu4N][OTf] in DFB.
Mechanism for the Activation of H2
Admission of 4 bar H2 to [1·(qui)][Al(ORF)4] in DFB led to no reaction by 1H or 119Sn{1H} NMR spectroscopy at RT after several days, nor after heating at 100 °C for 10 h. Furthermore, this salt did not scramble HD to H2, D2, and HD, which would detect rapidly reversible heterolysis. These results exclude a simple mechanism by which the Sn–N bond can elongate (with or without complete cleavage) to activate H2 in a manner akin to that reported for the related classical adduct [iPr3Si–PtBu3][B(C6F5)4].11a In support of this conclusion, computations predict the activation energy for H2 cleavage via this mechanism to be prohibitively high (ΔG⧧ = 36.4 kcal/mol, see Supporting Information Section 6.4 for the transition-state structure).
Because 1-OTf and [1·(qui)]+ are both present at RT in solutions of 1-OTf/qui LP, either could feasibly engage with qui to mediate H2 cleavage; accordingly, we sought to elucidate whether they could act as LAs for the FLP-mediated H2 activation. [1·(qui)][Al(ORF)4] is a valid model compound to study the reactivity of [1·(qui)]+ as a LA fragment, so using [1·(qui)][Al(ORF)4] + qui (hereafter referred to as the “[1·(qui)]+/qui” LP) allows the investigation of H2 activation, where [OTf]− cannot be involved at any point in the reaction.
When one equivalent of qui is added to [1·(qui)][Al(ORF)4], the evidence of H2 activation is observed immediately after H2 admission by the appearance of NMR spectroscopic resonances attributable to 1-H [SnH: δ (1H) = 5.10 ppm, 1J(1H–117Sn) = 1404 Hz, 1J(1H–119Sn) = 1470 Hz; δ (119Sn{1H}) = −47.3 ppm] and [qui-H]+ [qui-H+: δ (1H) = 13.84 ppm] (Scheme 4).17a The 27Al and 19F NMR spectra remain unchanged, indicating that [Al(ORF)4]− remains intact and does not participate in H2 activation, as expected. Based on these observations, we propose the mechanism shown in Scheme 5, which is supported computationally.30 H2 is cleaved via a termolecular transition state as illustrated in Figure 6, which requires the addition of free qui, akin to H2 activation mediated by Ar3B and N/P-centered FLPs. Here, the qui coordinated in [1·(qui)]+ acts as a leaving group, while the external qui is the LB for H2 activation.
Scheme 4. H2 Activation by [1·(qui)]+ Only Occurs When Free qui LB Is Present; RF = C(CF3)3.

Scheme 5. Proposed Mechanism for H2 Activation with the [1·(qui)]+/qui Lewis Pair via a Ternary TS; [Al(ORF)4] Counteranion Omitted; RF = C(CF3)3.

Figure 6.
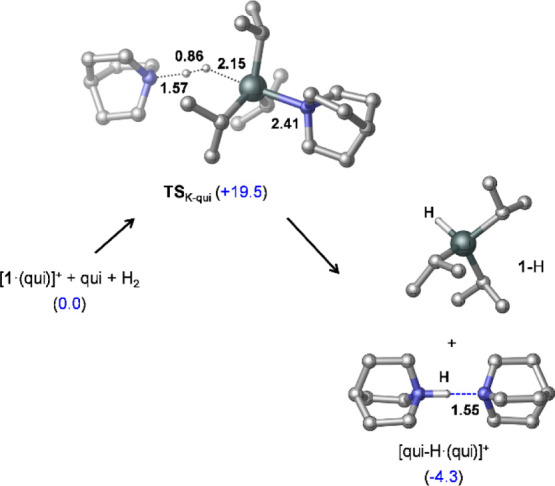
Transition-state TSK-qui identified computationally for H2 activation with [1·(qui)]+/qui and the species corresponding to the most stable product state (1-H + [qui-H·(qui)]+). Relative stabilities (in kcal/mol) are shown in parentheses, and selected bond distances are given in Å. All hydrogen atoms except those from H2 are omitted for clarity.
Computations reveal that H2 splitting and dissociation of the departing qui molecule take place concertedly, yet in an asynchronous manner (after TSK-qui, the dissociation of qui lags behind the H2 cleavage). Thus, a weakly bound termolecular intermediate [qui-H]+·(1-H)·(qui) is produced in this process (lying at 9.4 kcal/mol in free energy, see Supporting Information Section 6.2 for the ternary intermediate structure); however, the separation of 1-H and the formation of the H-bonded [qui-H·(qui)]+ species result in a significant stabilization on the product side (the reaction is exergonic by 4.3 kcal/mol). Computations predict the free energy barrier of this H2 cleavage reaction to be 19.5 kcal/mol, which is consistent with the observed rate at the applied reaction conditions.
Interestingly, while the qui-mediated hydrogenolysis of [1·(qui)][Al(ORF)4] with H2 is quantitative after 48 h (RT), the analogous reaction with 1-OTf reaches only ca. 50% conversion to 1-H, albeit much more quickly (2 h). Figure 7 shows the time evolution of the respective 1H NMR spectra for these reactions to illustrate their differing rates. Initially, as H2 activation by 1-OTf/qui proceeds, the 119Sn{1H} NMR spectra show the expected resonance at δ = −47 ppm for 1-H growing in intensity and a peak at δ = 40 ppm corresponding to the aforementioned fast equilibrium between 1-OTf, (qui)·1-OTf, [1·(qui)]+[OTf]−, and [1(OTf)2]− species; the latter moves upfield and sharpens as the reaction approaches completion, reaching a limiting value of δ = 19 ppm, which we attribute to the generation of [1(OTf)2]−.
Figure 7.

1H NMR spectra at selected time points (shown in parentheses in h) during the activation of H2 with 1-OTf/qui and [1·(qui)]+/qui LPs at RT. The initial rate of H2 activation with 1-OTf/qui is much faster than [1·(qui)]+/qui; however, the former reaches only ca. 50% conversion to 1-H. Contrastingly, the comparable reaction with [1·(qui)]+/qui LP is slower, although it eventually forms 1-H quantitatively. * denotes the resonances associated with 1-H.
This result implies that as H2 activation proceeds, [qui-H]+[OTf]− accumulates, and [OTf]− is concomitantly sequestered by 1-OTf to produce inactive [1(OTf)2]− (Scheme 3); this association quenches the electrophilicity of 1-OTf because the five-coordinate anionic adduct is not Lewis acidic enough to participate in H2 heterolysis. Therefore, the attainable conversion of H2 cleavage is impaired by the increasing concentration of [1(OTf2)]−. In support of this hypothesis, the incremental addition of [nBu4N]+[OTf]− to 1-OTf in DFB reproduces the upfield movement of the 119Sn{1H} NMR shift for the 1-OTf/qui/H2 reaction exactly, confirming that the binding of [OTf]− to 1-OTf occurs in situ during H2 activation (see the Supporting Information). Conversely, during H2 activation by [1·(qui)]+/qui, the noninteracting counteranion in the [qui-H]+[Al(ORF)4]− product cannot coordinate and quench the [1·(qui)]+ LA, so the reaction can proceed to quantitative conversion.
It is notable that for this latter LP system, the concentration of uncoordinated qui should remain constant throughout the reaction with H2 (Scheme 5); indeed, it is found that the [1·(qui)]+/qui LP is actually catalytic with respect to qui, and H2 activation proceeds with substoichiometric amounts of added qui, albeit at correspondingly reduced rates. For the 1-OTf/qui LP, however, the concentration of free qui is reduced by its association with 1-OTf and decreases further as the reaction proceeds. The concentration of free qui is therefore expected to be lower at all times for H2 cleavage reactions employing 1-OTf/qui versus [1·(qui)]+/qui; this difference makes a direct quantitative kinetic comparison extremely difficult. Nevertheless, the initial H2 activation rate is clearly higher for the 1-OTf/qui LP in spite of the lower concentration of free qui available, which supports the conclusion that H2 activation is inherently faster at the beginning of the reaction for 1-OTf/qui than [1·(qui)]+/qui. In order to rationalize our observations regarding the rates and yields of H2 heterolysis induced by these two qui-dependent systems, we additionally examined the basic mechanistic features of the latter reaction, computationally (Figure 8).
Figure 8.
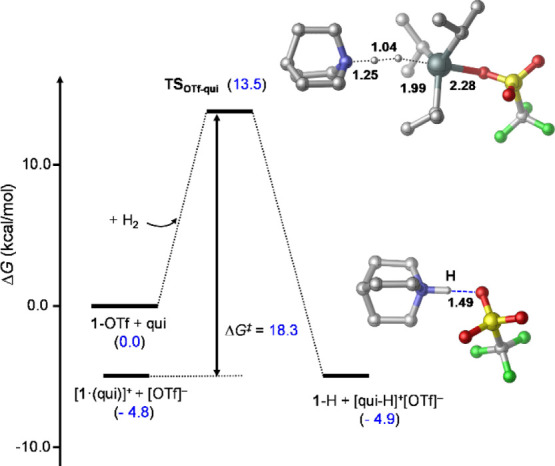
Transition-state TSOTf-qui identified computationally for H2 activation with 1-OTf/qui and the species corresponding to the most stable product state (1-H + [qui-H]+[OTf]−). Relative stabilities (in kcal/mol) are shown in parentheses, and selected bond distances are given in Å.33
Consistent with our experiment results, the transition state identified for the H2 splitting process (TSOTf-qui) possesses a significantly lower barrier with respect to the 1-OTf + qui + H2 state31 (13.5 kcal/mol) when compared to that obtained for the [1·(qui)]+ + qui + H2 reaction (19.5 kcal/mol). When the ground state of the reactants in the former case is factored in, corresponding to [1·(qui)]+ + [OTf]− + H2, the overall kinetic barrier is actually 18.3 kcal/mol (see Scheme 3 and Figure 8). These computed barriers are in agreement with the measured reaction times.32 Our computational analysis suggests that the differing energies of TSOTf-qui and TSK-qui relative to their corresponding reactant states could be associated with solvent effects (for a more detailed analysis of solvation effects, see Supporting Information Section 6.6). The reactant state of
involves only neutral components, whereas the H2 heterolysis yields two ionic species. The charge separation is notable already in TSOTf-qui, which has a relatively late character, so it is stabilized more by solvent polarity than the reactant state. On the other hand, no charge variation occurs in the reaction
Interestingly, on the product side of the reaction with the 1-OTf/qui Lewis pair, the 1-H + [qui-H]+[OTf]− state is predicted to be the most stable form lying at −4.9 kcal/mol in free energy, which is close to that of the reactant state (−4.8 kcal/mol) and suggests that, even in the absence of subsequent capture of [OTf]− by 1-OTf to form [1(OTf)2]− (predicted to be exergonic by 3.2 kcal/mol; vide supra), the reaction would in fact still only reach approximately 50% conversion.
Employing Collidine as the LB Partner
To provide further context, we also studied the analogous system using col as a LB partner for [1]+-based LAs because it was found to be the optimally performing base surveyed for catalytic hydrogenations using 1-OTf and also to resolve any reactivity effects because of its different steric profile versus qui.17a,17b All attempts to isolate samples (col)·1-OTf or [1·(col)]+[OTf]−, from mixtures of the LPs by analogy with qui experiments, were unsuccessful. This indicates that the association between 1-OTf and col is weaker compared with qui, which is consistent given that col exhibits a lower basicity [pKa(MeCN) col-H+ = 14.98]34 and that its N center is hindered by the flanking 2,6-Me substituents. The datively bound adduct (col)·1-OTf, analogous to (qui)·1-OTf, could be identified computationally; however, an appreciable Sn–N bonding interaction is sterically prevented with only a weak col···1-OTf association evident from DFT calculations (see Supporting Information Section 6.7 for a ternary complex structure). A mixture of 1-OTf and col (1:1, DFB) displays a broad signal in the RT 119Sn{1H} NMR spectrum at δ = 142 ppm, which is significantly downfield from that observed for 1-OTf/qui (δ = 34 ppm) and moves upon warming to a limiting value at 353 K that is identical to authentic 1-OTf. When the solution is cooled, the peak moves upfield (δ = 116 ppm at 243 K), indicating that col is binding to 1-OTf more strongly.35 Our computations suggest that the small change in chemical shift (24 ppm) can be ascribed to the [1·(col)]+ complex formed in low concentrations. Although several different species might be contributing to the observed average chemical shift, these could not be resolved; nonetheless, it is clear that the association between 1-OTf and col is significantly less favored than for qui.
Fortunately, [1·(col)]+[Al(ORF)4]− can be prepared by an analogous route to [1·(qui)]+[Al(ORF)4]− (Scheme 2), as a yellow oil that unfortunately resisted attempts at crystallization. Its 119Sn{1H} NMR spectrum exhibits a broad resonance at δ = 128 ppm, which is slightly downfield from the qui counterpart (δ = 113 ppm). Similar to [1·(qui)]+, [1·(col)]+[Al(ORF)4]− does not react with H2 unless additional col is added, resulting in the appearance of resonances attributable to 1-H + [col-H]+ by 1H and 119Sn{1H} NMR spectroscopy. This strongly suggests that [1·(qui/col)]+ LAs activate H2 with their corresponding LB partners via the same mechanism. Notably, however, an elevated pressure (10 bar H2) is required for H2 activation to proceed effectively by [1·(col)]+/col, and the rate to reach quantitative conversion is considerably slower (28 days) under analogous equimolar conditions. These latter results are consistent with previous observations17a,17b and are commensurate with the lower basicity of col relative to qui. For H2 activation with the 1-OTf/col LP, once again only partial conversion to 1-H and [col-H]+ is observed (ca. 50% in 4 days at RT) by 1H NMR spectroscopy because of the formation of [1(OTf)2]−; however, the initial reaction rate is faster when 1-OTf is the LA compared with [1·(col)]+.
To shed light on the similarities and differences between the qui and col systems thus far, the energetics of H2 activation for the [1·(col)]+/col and 1-OTf/col LPs were assessed computationally; the key structures and their relative stabilities are shown in Figures 9 and 10. The principal mechanistic features of these reactions are quite similar to those of the analogous reactions with qui as the LB (concerted asynchronous H2 splitting and base dissociation processes, formation of H-bonded species with the protonated base), yet there are profound differences in the computed energetics. For instance, H2 activation with [1·(col)]+/col is predicted to be more favored thermodynamically as compared to [1·(qui)]+/qui, which appears counterintuitive in light of the relative basicities; the computed proton affinities (kcal/mol) are −160.9 and −155.4 for qui and col, respectively.
Figure 9.

Transition-state TSK-col identified computationally for H2 activation with [1·(col)]+/col and the species corresponding to the most stable product state (1-H + [col-H·(col)]+). Relative stabilities (in kcal/mol) are shown in parentheses, and selected bond distances are given in Å.
Figure 10.
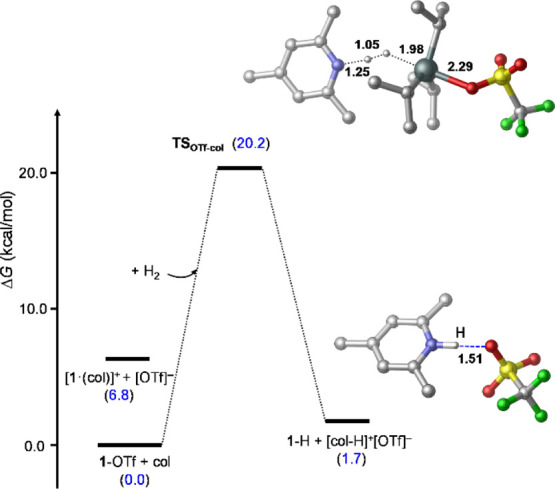
Transition-state TSOTf-col identified computationally for H2 activation with 1-OTf/col and the species corresponding to the most stable product state (1-H + [col-H]+[OTf]−). Relative stabilities (in kcal/mol) are shown in parentheses, and selected bond distances are given in Å.
However, the strained nature of the [1·(col)]+ binary complex implies substantial reactant-state destabilization with a dissociation free energy into [1]+ + col of only 17.6 kcal/mol, so the reaction becomes more exergonic versus qui (for a detailed energy decomposition analysis, see Supporting Information Section 6.8). Destabilizing steric effects are even more enhanced in the transition state for H2 splitting (TSK-col in Figure 9), which in combination with the reduced basicity of col results in a higher barrier compared to that obtained for [1·(qui)]+/qui + H2 (25.7 vs 19.5 kcal/mol, respectively).
For the 1-OTf/col system, the computational results predict the 1-OTf + col state to be clearly favored over the ionic [1·(col)]+[OTf]− alternative (Figure 10), and the H2 activation reaction is found to be less favored (both kinetically and thermodynamically) than the analogous reaction with 1-OTf/qui. The latter trend for the predicted energetics of these two reactions follows the difference in the proton affinity of the two bases, which is also in line with our experimental observations. Importantly, H2 activation by 1-OTf/col is predicted to have a much lower activation energy (20.2 kcal/mol) than the [1·(col)]+/col LP (25.7 kcal/mol), which is consistent with the observed reaction rates. We attribute this to the stabilization of TSOTf-col by the polar solvent relative to the neutral ground state (1-OTf + col + H2), and the destabilizing steric effects in TSK-col, which are weaker in TSOTf-col.
Relevance to Catalytic Hydrogenations Using 1-OTf/col
Our previous observations in the catalytic hydrogenation of imine and carbonyl compounds by 1-OTf + col (our optimum LB for catalysis) showed that the reduction steps involved the reaction of 1-H with protonated imines and (carbonyl)·1-OTf species (i.e., activated substrates), respectively. These were much more rapid than H2 activation, and notably, 1-H was observed neither by 1H nor by 119Sn{1H} NMR spectroscopy at any point during catalysis; hence, it was proposed that the H2 cleavage was the rate-determining step.17a,17b The current results show that [1(OTf)2]− forms as H2 heterolysis by 1-OTf/col proceeds and because of the sequestration of active LA 1-OTf, it would therefore be expected to affect the reaction rate deleteriously in catalytic reactions. It should be noted, however, that in catalytic reactions, the direct H2 cleavage products 1-H and [col-H]+[OTf]− are almost immediately consumed during proton and hydride transfers to the substrate; because H2 cleavage is the rate-determining step, the accumulation of [1(OTf)2]− will be suppressed and/or it will form reversibly (Scheme 6). The latter postulate is reinforced by the calculated small free energy difference between the formation of 1-H + [col-H][OTf] from 1-OTf + col + H2 (+1.7 kcal/mol) and binding of 1-OTf with [OTf]− (−3.2 kcal/mol); this implies that all of these species would coexist in a readily perturbed equilibrium, as 1-H + [col-H][OTf] is consumed in substrate hydrogenation. Indeed, [1(OTf)2]− is not observed by 119Sn{1H} NMR spectroscopy during catalytic reactions,17a,17b verifying that this species plays an insignificant role under the conditions of hydrogenation catalysis. Taking the results together, we propose that lowering the activation energy of H2 cleavage is the most important factor to improve the rate of hydrogenation by triorganostannylium-based catalysts. Because [OTf]− explicitly interacts with [1]+ in the reactants and the lowest energy TS possible for H2 heterolysis by 1-OTf/col, this anion affects their stabilities; variation of this anion should consequently affect the activation energy as long as it remains associated in such a manner.
Scheme 6. Simplified Catalytic Cycle for Hydrogenations Mediated by 1-OTf/col. The Reduction Step Is Rapid Relative to the H2 Cleavage, Making 1-OTf + Col the Resting State under Catalytic Conditions; Because [OTf]− Does Not Accumulate in Situ, 1-OTf Is Not Sequestered (as [1(OTf)2]−) during the Catalytic Hydrogenations; X = NR″, O.

Conclusions
In conclusion, we have elucidated that 1-OTf is the LA involved in the lowest energy TS for H2 activation with both coordinating (qui) and noncoordinating (col) LBs. 1-OTf and qui associate to form the dative adduct (qui)·1-OTf, which was characterized by single-crystal X-ray crystallography; solution-phase NMR studies and DFT calculations reveal that this adduct is in equilibrium with 1-OTf, [1·(qui)]+ and [1(OTf)2]− at RT. By contrast, 1-OTf and col show a far lesser degree of association because of the greater steric protection of the LB. Ionic salts [1·(LB)]+[Al(ORF)4]− permitted the examination of the reactivity of an alternative LA adduct in the absence of a coordinating anion, where it was found that an additional extraneous LB was necessary to promote H2 activation. This indicates that these reactions proceed via a (LB)·H2·[1·(LB)]+ TS, which is supported by computational results that also predict analogous mechanisms for H2 heterolysis by 1-OTf/LB pairs. Although the [1·(LB)]+/LB systems do activate H2, their initial reaction rates are significantly slower than the corresponding 1-OTf/LB pairs, which is attributed to differing degrees of charge separation along the respective reaction pathways; in the latter case, this is more pronounced, resulting in the relative stabilization of the TS by highly polar solvent media (e.g., DFB), and hence a lower barrier to the H2 cleavage. The activation energies for H2 cleavage for both 1-OTf/LB and [1·(LB)]+/LB pairs are found to be significantly higher when LB = col, which is due to its lower basicity and the higher steric strain in the TS, versus qui. A surprising finding is that while [1·(LB)]+/LB LPs activate H2 quantitatively, the catalytically relevant 1-OTf/LB pairs achieve only ∼50% conversion. This is rationalized by the liberation of [OTf]− during H2 activation, which is subsequently sequestered by 1-OTf to form [1(OTf)2]−, as well as by the modest thermodynamic driving force of H2 activation.
Taken together, these results reveal that the triflate ion in 1-OTf/LB LPs exerts a considerable effect on both the thermodynamics (the relative stabilities of ground-state species prior to H2 activation and the products thereafter) and kinetics (the TS energy barriers) of H2 activation. Our present work highlights the importance of how the counteranion can impact the reactivity of cationic LAs beyond a simple modulation of Lewis acidity, and we anticipate that these findings will translate to the reactivity of other systems. Encouragingly, the ability to vary the anion partner of LA cations offers a design principle unavailable to neutral LAs. We expect that this will encourage further research into anion tunability for the tailoring of LA reactivity, especially toward the reactions of important small molecules.
Acknowledgments
We would like to thank Pete Haycock for providing support and assistance during the collection of NMR data. In addition, we would like to thank Andrea Hamza for her technical assistance in the handling of the many conformers studied herein.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c02023.
Author Contributions
The manuscript was written through contributions of all authors.
We wish to thank Imperial College for a President’s Ph.D. Scholarship award (R.C.T.-R.); the EPSRC for PhD (J.S.S. and A.D.P.) and grant funding (EP/N026004), including a Doctoral Prize Fellowship (EP/N509486/1, D.J.S.); and the Royal Society for a University Research Fellowship (A.E.A.; UF/160395). This work was also supported by the NKFIH (K-115660). Computational resources provided by the NIIF Supercomputer Center are acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- For key publications see:; a Welch G. C.; Juan R. R. S.; Masuda J. D.; Stephan D. W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]; b Welch G. C.; Stephan D. W. Facile heterolytic cleavage of dihydrogen by phosphines and boranes. J. Am. Chem. Soc. 2007, 129, 1880–1881. 10.1021/ja067961j. [DOI] [PubMed] [Google Scholar]; c Chase P. A.; Welch G. C.; Jurca T.; Stephan D. W. Metal-Free Catalytic Hydrogenation. Angew. Chem., Int. Ed. 2007, 46, 8050–8053. 10.1002/anie.200702908. [DOI] [PubMed] [Google Scholar]; d Chase P. A.; Jurca T.; Stephan D. W. Lewis acid-catalyzed hydrogenation: B(C6F5)3-mediated reduction of imines and nitriles with H2. Chem. Commun. 2008, 1701–1703. 10.1039/b718598g. [DOI] [PubMed] [Google Scholar]; e Chen D.; Klankermayer J. Metal-free catalytic hydrogenation of imines with tris(perfluorophenyl)borane. Chem. Commun. 2008, 2130–2131. 10.1039/b801806e. [DOI] [PubMed] [Google Scholar]; f Ashley A. E.; Thompson A. L.; O’Hare D. Non-metal Mediated Homogeneous Hydrogenation of CO2 to CH3OH. Angew. Chem., Int. Ed. 2009, 48, 9839–9843. 10.1002/anie.200905466. [DOI] [PubMed] [Google Scholar]; g Chernichenko K.; Madarász Á.; Pápai I.; Nieger M.; Leskelä M.; Repo T. A frustrated-Lewis-pair approach to catalytic reduction of alkynes to cis-alkenes. Nat. Chem. 2013, 5, 718–723. 10.1038/nchem.1693. [DOI] [PubMed] [Google Scholar]; h Légaré M.-A.; Courtemanche M.-A.; Rochette E.; Fontaine F.-G. Metal-free catalytic C-H bond activation and borylation of heteroarenes. Science 2015, 349, 513–516. 10.1126/science.aab3591. [DOI] [PubMed] [Google Scholar]
- For overviews of FLPs see:; a Erker G.; Stephan D. W.. Frustrated Lewis Pairs; Topics in Current Chemistry; Springer: Berlin, 2013; Vols I and II. [Google Scholar]; b Stephan D. W. Frustrated Lewis pairs: From concept to catalysis. Acc. Chem. Res. 2015, 48, 306–316. 10.1021/ar500375j. [DOI] [PubMed] [Google Scholar]; c Stephan D. W.; Erker G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]; d Stephan D. W. Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. 10.1021/jacs.5b06794. [DOI] [PubMed] [Google Scholar]
- For reviews of FLP-catalyzed hydrogenations see:; a Stephan D. W.; Greenberg S.; Graham T. W.; Chase P.; Hastie J. J.; Geier S. J.; Farrell J. M.; Brown C. C.; Heiden Z. M.; Welch G. C.; Ullrich M. Metal-Free Catalytic Hydrogenation of Polar Substrates by Frustrated Lewis Pairs. Inorg. Chem. 2011, 50, 12338–12348. 10.1021/ic200663v. [DOI] [PubMed] [Google Scholar]; b Paradies J. Metal-Free Hydrogenation of Unsaturated Hydrocarbons Employing Molecular Hydrogen. Angew. Chem., Int. Ed. 2014, 53, 3552–3557. 10.1002/anie.201309253. [DOI] [PubMed] [Google Scholar]; c Hounjet L. J.; Stephan D. W. Hydrogenation by Frustrated Lewis Pairs: Main Group Alternatives to Transition Metal Catalysts?. Org. Process Res. Dev. 2014, 18, 385–391. 10.1021/op400315m. [DOI] [Google Scholar]; d Scott D. J.; Fuchter M. J.; Ashley A. E. Designing effective “frustrated Lewis pair” hydrogenation catalysts. Chem. Soc. Rev. 2017, 46, 5689–5700. 10.1039/c7cs00154a. [DOI] [PubMed] [Google Scholar]; e Lam J.; Szkop K. M.; Mosaferi E.; Stephan D. W. FLP catalysis: main group hydrogenations of organic unsaturated substrates. Chem. Soc. Rev. 2019, 48, 3592–3612. 10.1039/c8cs00277k. [DOI] [PubMed] [Google Scholar]; f Paradies J. Mechanisms in Frustrated Lewis Pair-Catalyzed Reactions. Eur. J. Org. Chem. 2019, 2019, 283–294. 10.1002/ejoc.201800944. [DOI] [Google Scholar]
- a Scott D. J.; Fuchter M. J.; Ashley A. E. Nonmetal Catalyzed Hydrogenation of Carbonyl Compounds. J. Am. Chem. Soc. 2014, 136, 15813–15816. 10.1021/ja5088979. [DOI] [PubMed] [Google Scholar]; b Mahdi T.; Stephan D. W. Enabling Catalytic Ketone Hydrogenation by Frustrated Lewis Pairs. J. Am. Chem. Soc. 2014, 136, 15809–15812. 10.1021/ja508829x. [DOI] [PubMed] [Google Scholar]; c Scott D. J.; Simmons T. R.; Lawrence E. J.; Wildgoose G. G.; Fuchter M. J.; Ashley A. E. Facile Protocol for Water-Tolerant “Frustrated Lewis Pair”-Catalyzed Hydrogenation. ACS Catal. 2015, 5, 5540–5544. 10.1021/acscatal.5b01417. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mahdi T.; Stephan D. W. Facile Protocol for Catalytic Frustrated Lewis Pair Hydrogenation and Reductive Deoxygenation of Ketones and Aldehydes. Angew. Chem., Int. Ed. 2015, 54, 8511–8514. 10.1002/anie.201503087. [DOI] [PubMed] [Google Scholar]
- a Erős G.; Mehdi H.; Pápai I.; Rokob T. A.; Király P.; Tárkányi G.; Soós T. Expanding the Scope of Metal-Free Catalytic Hydrogenation through Frustrated Lewis Pair Design. Angew. Chem., Int. Ed. 2010, 49, 6559–6563. 10.1002/ange.201001518. [DOI] [PubMed] [Google Scholar]; b Ashley A. E.; Herrington T. J.; Wildgoose G. G.; Zaher H.; Thompson A. L.; Rees N. H.; Krämer T.; O’Hare D. Separating Electrophilicity and Lewis Acidity: The Synthesis, Characterization, and Electrochemistry of the Electron Deficient Tris (aryl)boranes B(C6F5)3–n(C6Cl5)n (n = 1–3). J. Am. Chem. Soc. 2011, 133, 14727–14740. 10.1021/ja205037t. [DOI] [PubMed] [Google Scholar]; c Erős G.; Nagy K.; Mehdi H.; Pápai I.; Nagy P.; Király P.; Tárkányi G.; Soós T. Catalytic Hydrogenation with Frustrated Lewis Pairs: Selectivity Achieved by Size-Exclusion Design of Lewis Acids. Chem.—Eur. J. 2012, 18, 574–585. 10.1002/chem.201102438. [DOI] [PubMed] [Google Scholar]; d Sivaev I. B.; Bregadze V. I. Lewis acidity of boron compounds. Coord. Chem. Rev. 2014, 270–271, 75–88. 10.1016/j.ccr.2013.10.017. [DOI] [Google Scholar]; e Scott D. J.; Fuchter M. J.; Ashley A. E. Metal-Free Hydrogenation Catalyzed by an Air-Stable Borane: Use of Solvent as a Frustrated Lewis Base. Angew. Chem., Int. Ed. 2014, 53, 10218–10222. 10.1002/anie.201405531. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lawson J. R.; Melen R. L. Tris(pentafluorophenyl)borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. 10.1021/acs.inorgchem.6b02911. [DOI] [PubMed] [Google Scholar]; g Paradies J. From structure to novel reactivity in frustrated Lewis pairs. Coord. Chem. Rev. 2019, 380, 170–183. 10.1016/j.ccr.2018.09.014. [DOI] [Google Scholar]; h Carden J. L.; Dasgupta A.; Melen R. L. Halogenated triarylboranes: synthesis, properties and applications in catalysis. Chem. Soc. Rev. 2020, 49, 1706–1725. 10.1039/c9cs00769e. [DOI] [PubMed] [Google Scholar]
- a Fasano V.; Ingleson M. Recent Advances in Water-Tolerance in Frustrated Lewis Pair Chemistry. Synthesis 2018, 50, 1783–1795. 10.1055/s-0037-1609843. [DOI] [Google Scholar]; b Gyömöre Á.; Bakos M.; Földes T.; Pápai I.; Domján A.; Soós T. Moisture-Tolerant Frustrated Lewis Pair Catalyst for Hydrogenation of Aldehydes and Ketones. ACS Catal. 2015, 5, 5366–5372. 10.1021/acscatal.5b01299. [DOI] [Google Scholar]; c Dorkó É.; Szabó M.; Kótai B.; Pápai I.; Domján A.; Soós T. Expanding the Boundaries of Water-Tolerant Frustrated Lewis Pair Hydrogenation: Enhanced Back Strain in the Lewis Acid Enables the Reductive Amination of Carbonyls. Angew. Chem., Int. Ed. 2017, 56, 9512–9516. 10.1002/anie.201703591. [DOI] [PubMed] [Google Scholar]; d Bakos M.; Gyömöre Áy.; Domján A.; Soós T. Auto-Tandem Catalysis with Frustrated Lewis Pairs for Reductive Etherification of Aldehydes and Ketones. Angew. Chem., Int. Ed. 2017, 56, 5217–5221. 10.1002/anie.201700231. [DOI] [PubMed] [Google Scholar]; e Sitte N. A.; Bursch M.; Grimme S.; Paradies J. Frustrated Lewis Pair Catalyzed Hydrogenation of Amides: Halides as Active Lewis Base in the Metal-Free Hydrogen Activation. J. Am. Chem. Soc. 2019, 141, 159–162. 10.1021/jacs.8b12997. [DOI] [PubMed] [Google Scholar]; f Hoshimoto Y.; Kinoshita T.; Hazra S.; Ohashi M.; Ogoshi S. Main-Group-Catalyzed Reductive Alkylation of Multiply Substituted Amines with Aldehydes Using H2. J. Am. Chem. Soc. 2018, 140, 7292–7300. 10.1021/jacs.8b03626. [DOI] [PubMed] [Google Scholar]
- For a recent overview of unsettled mechanistic issues, see:Daru J.; Bakó I.; Stirling A.; Pápai I. Mechanism of Heterolytic Hydrogen Splitting by Frustrated Lewis Pairs: Comparison of Static and Dynamic Models. ACS Catal. 2019, 9, 6049–6057. 10.1021/acscatal.9b01137. [DOI] [Google Scholar]
- For a review on relevant computational studies, see:Rokob T. A.; Pápai I. Hydrogen Activation by Frustrated Lewis Pairs: Insights from Computational Studies. Top. Curr. Chem. 2013, 332, 157–211. 10.1007/128_2012_399. [DOI] [PubMed] [Google Scholar]
- We excluded the possibility of radical-mediated H2 cleavage by Lewis pairs comprising [iPr3Sn]+ fragments in our previous studies; see ref (17). For examples of “frustrated radical pairs” see:; a Liu L. L.; Cao L. L.; Shao Y.; Stephan D. W. Single Electron Delivery to Lewis Pairs: An Avenue to Anions by Small Molecule Activation. J. Am. Chem. Soc. 2017, 139, 10062–10071. 10.1021/jacs.7b05120. [DOI] [PubMed] [Google Scholar]; b Liu L.; Cao L. L.; Shao Y.; Ménard G.; Stephan D. W. A Radical Mechanism for Frustrated Lewis Pair Reactivity. Inorg. Chem. 2017, 3, 259–267. 10.1016/j.chempr.2017.05.022. [DOI] [Google Scholar]; c Liu L. L.; Cao L. L.; Zhu D.; Zhou J.; Stephan D. W. Homolytic cleavage of peroxide bonds via a single electron transfer of a frustrated Lewis pair. Chem. Commun. 2018, 54, 7431–7434. 10.1039/c8cc03522a. [DOI] [PubMed] [Google Scholar]; d Merk A.; Großekappenberg H.; Schmidtmann M.; Luecke M. P.; Lorent C.; Driess M.; Oestreich M.; Klare H. F. T.; Müller T. Single-Electron Transfer Reactions in Frustrated and Conventional Silylium Ion/Phosphane Lewis Pairs. Angew. Chem., Int. Ed. 2018, 57, 15267–15271. 10.1002/anie.201808922. [DOI] [PubMed] [Google Scholar]; e Liu L. L.; Stephan D. W. Radicals derived from Lewis acid/base pairs. Chem. Soc. Rev. 2019, 48, 3454–3463. 10.1039/c8cs00940f. [DOI] [PubMed] [Google Scholar]; f Bennett E. L.; Lawrence E. J.; Blagg R. J.; Mullen A. S.; MacMillan F.; Ehlers A. W.; Scott D. J.; Sapsford J. S.; Ashley A. E.; Wildgoose G. G.; Slootweg J. C. A New Mode of Chemical Reactivity for Metal-Free Hydrogen Activation by Lewis Acidic Boranes. Angew. Chem., Int. Ed. 2019, 58, 8362–8366. 10.1002/anie.201900861. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Soltani Y.; Dasgupta A.; Gazis T. A.; Ould D. M. C.; Richards E.; Slater B.; Stefkova K.; Vladimirov V. Y.; Wilkins L. C.; Willcox D.; Melen R. L. Radical Reactivity of Frustrated Lewis Pairs with Diaryl Esters. Cell Reports Phys. Sci. 2020, 1, 100016. 10.1016/j.xcrp.2020.100016. [DOI] [Google Scholar]
- a Eisenberger P.; Bailey A. M.; Crudden C. M. Taking the F out of FLP: Simple lewis acid-base pairs for mild reductions with neutral boranes via borenium ion catalysis. J. Am. Chem. Soc. 2012, 134, 17384–17387. 10.1021/ja307374j. [DOI] [PubMed] [Google Scholar]; b Farrell J. M.; Hatnean J. A.; Stephan D. W. Activation of Hydrogen and Hydrogenation Catalysis by a Borenium Cation. J. Am. Chem. Soc. 2012, 134, 15728–15731. 10.1021/ja307995f. [DOI] [PubMed] [Google Scholar]; c Eisenberger P.; Bestvater B. P.; Keske E. C.; Crudden C. M. Hydrogenations at Room Temperature and Atmospheric Pressure with Mesoionic Carbene-Stabilized Borenium Catalysts. Angew. Chem., Int. Ed. 2015, 54, 2467–2471. 10.1002/anie.201409250. [DOI] [PubMed] [Google Scholar]; d Clark E. R.; Grosso A. D.; Ingleson M. J. The Hydride-Ion Affinity of Borenium Cations and Their Propensity to Activate H2 in Frustrated Lewis Pairs. Chem.—Eur. J. 2013, 19, 2462–2466. 10.1002/chem.201203318. [DOI] [PubMed] [Google Scholar]; e Lam J.; Günther B. A. R.; Farrell J. M.; Eisenberger P.; Bestvater B. P.; Newman P. D.; Melen R. L.; Crudden C. M.; Stephan D. W. Chiral carbene–borane adducts: precursors for borenium catalysts for asymmetric FLP hydrogenations. Dalton Trans. 2016, 45, 15303–15316. 10.1039/c6dt02202b. [DOI] [PubMed] [Google Scholar]; f Mercea D. M.; Howlett M. G.; Piascik A. D.; Scott D. J.; Steven A.; Ashley A. E.; Fuchter M. J. Enantioselective reduction of N-alkyl ketimines with frustrated Lewis pair catalysis using chiral borenium ions. Chem. Commun. 2019, 55, 7077–7080. 10.1039/c9cc02900a. [DOI] [PubMed] [Google Scholar]
- a Herrington T. J.; Ward B. J.; Doyle L. R.; McDermott J.; White A. J. P.; Hunt P. A.; Ashley A. E. Bypassing a highly unstable frustrated Lewis pair: dihydrogen cleavage by a thermally robust silylium–phosphine adduct. Chem. Commun. 2014, 50, 12753–12756. 10.1039/c4cc05905k. [DOI] [PubMed] [Google Scholar]; b Oestreich M.; Hermeke J.; Mohr J. A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. 10.1039/c4cs00451e. [DOI] [PubMed] [Google Scholar]; c Weicker S. A.; Stephan D. W. Activation of Carbon Dioxide by Silyl Triflate-Based Frustrated Lewis Pairs. Chem.—Eur. J. 2015, 21, 13027–13034. 10.1002/chem.201501904. [DOI] [PubMed] [Google Scholar]; d Mallov I.; Ruddy A. J.; Zhu H.; Grimme S.; Stephan D. W. C–F Bond Activation by Silylium Cation/Phosphine Frustrated Lewis Pairs: Mono-Hydrodefluorination of PhCF3, PhCF2H and Ph2CF2. Chem.—Eur. J. 2017, 23, 17692–17696. 10.1002/chem.201705276. [DOI] [PubMed] [Google Scholar]
- a Clark E. R.; Ingleson M. J. N-Methylacridinium Salts: Carbon Lewis Acids in Frustrated Lewis Pairs for σ-Bond Activation and Catalytic Reductions. Angew. Chem., Int. Ed. 2014, 53, 11306–11309. 10.1002/anie.201406122. [DOI] [PubMed] [Google Scholar]; b Mosaferi E.; Ripsman D.; Stephan D. W. The air-stable carbocation salt [(MeOC6H4)CPh2][BF4] in Lewis acid catalyzed hydrothiolation of alkenes. Chem. Commun 2016, 52, 8291–8293. 10.1039/c6cc03970g. [DOI] [PubMed] [Google Scholar]; c Fasano V.; Radcliffe J. E.; Curless L. D.; Ingleson M. J. N-Methyl-Benzothiazolium Salts as Carbon Lewis Acids for Si–H Sigma-Bond Activation and Catalytic (De)hydrosilylation. Chem.—Eur. J. 2017, 23, 187–193. 10.1002/chem.201604613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhou J.; Liu L. L.; Cao L. L.; Stephan D. W. An umpolung of Lewis acidity/basicity at nitrogen by deprotonation of a cyclic (amino)(aryl)nitrenium cation. Chem. Commun. 2018, 54, 4390–4393. 10.1039/c8cc01331d. [DOI] [PubMed] [Google Scholar]; b Zhou J.; Liu L. L.; Cao L. L.; Stephan D. W. Nitrogen-Based Lewis Acids: Synthesis and Reactivity of a Cyclic (Alkyl)(Amino)Nitrenium Cation. Angew. Chem., Int. Ed. 2018, 57, 3322–3326. 10.1002/anie.201713118. [DOI] [PubMed] [Google Scholar]; c Waked A. E.; Ostadsharif Memar R.; Stephan D. W. Nitrogen-Based Lewis Acids Derived from Phosphonium Diazo Cations. Angew. Chem., Int. Ed. 2018, 57, 11934–11938. 10.1002/anie.201804183. [DOI] [PubMed] [Google Scholar]
- a Caputo C. B.; Hounjet L. J.; Dobrovetsky R.; Stephan D. W. Lewis Acidity of Organofluorophosphonium Salts: Hydrodefluorination by a Saturated Acceptor. Science 2013, 341, 1374–1377. 10.1126/science.1241764. [DOI] [PubMed] [Google Scholar]; b Pérez M.; Hounjet L. J.; Caputo C. B.; Dobrovetsky R.; Stephan D. W. Olefin Isomerization and Hydrosilylation Catalysis by Lewis Acidic Organofluorophosphonium Salts. J. Am. Chem. Soc. 2013, 135, 18308–18310. 10.1021/ja410379x. [DOI] [PubMed] [Google Scholar]; c Stein T. V.; Peréz M.; Dobrovetsky R.; Winkelhaus D.; Caputo C. B.; Stephan D. W. Electrophilic Fluorophosphonium Cations in Frustrated Lewis Pair Hydrogen Activation and Catalytic Hydrogenation of Olefins. Angew. Chem., Int. Ed. 2015, 54, 10178–10182. 10.1002/anie.201504109. [DOI] [PubMed] [Google Scholar]; d Fasano V.; LaFortune J. H. W.; Bayne J. M.; Ingleson M. J.; Stephan D. W. Air- and water-stable Lewis acids: synthesis and reactivity of P-trifluoromethyl electrophilic phosphonium cations. Chem. Commun. 2018, 54, 662–665. 10.1039/c7cc09128a. [DOI] [PubMed] [Google Scholar]; e Postle S.; Podgorny V.; Stephan D. W. Electrophilic phosphonium cations (EPCs) with perchlorinated-aryl substituents: towards air-stable phosphorus-based Lewis acid catalysts. Dalton Trans. 2016, 45, 14651–14657. 10.1039/c6dt01339b. [DOI] [PubMed] [Google Scholar]; f Barrado A. G.; Bayne J. M.; Johnstone T. C.; Lehmann C. W.; Stephan D. W.; Alcarazo M. Dicationic phosphonium salts: Lewis acid initiators for the Mukaiyama-aldol reaction. Dalton Trans. 2017, 46, 16216–16227. 10.1039/c7dt03197a. [DOI] [PubMed] [Google Scholar]; g Bayne J. M.; Fasano V.; Szkop K. M.; Ingleson M. J.; Stephan D. W. Phosphorous(V) Lewis acids: water/base tolerant P3-trimethylated trications. Chem. Commun. 2018, 54, 12467–12470. 10.1039/c8cc06564k. [DOI] [PubMed] [Google Scholar]
- a Pan B.; Gabbaï F. P. [Sb(C6F5)4][B(C6F5)4]: An Air Stable, Lewis Acidic Stibonium Salt That Activates Strong Element-Fluorine Bonds. J. Am. Chem. Soc. 2014, 136, 9564–9567. 10.1021/ja505214m. [DOI] [PubMed] [Google Scholar]; b Tofan D.; Gabbaï F. P. Fluorinated antimony(V) derivatives: strong Lewis acidic properties and application to the complexation of formaldehyde in aqueous solutions. Chem. Sci. 2016, 7, 6768–6778. 10.1039/c6sc02558g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review of non-borane FLPs see:Weicker S. A.; Stephan D. W. Main Group Lewis Acids in Frustrated Lewis Pair Chemistry: Beyond Electrophilic Boranes. Bull. Chem. Soc. Jpn. 2015, 88, 1003–1016. 10.1246/bcsj.20150131. [DOI] [Google Scholar]
- a Scott D. J.; Phillips N. A.; Sapsford J. S.; Deacy A. C.; Fuchter M. J.; Ashley A. E. Versatile Catalytic Hydrogenation Using A Simple Tin(IV) Lewis Acid. Angew. Chem., Int. Ed. 2016, 55, 14738–14742. 10.1002/anie.201606639. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sapsford J. S.; Scott D. J.; Allcock N. J.; Fuchter M. J.; Tighe C. J.; Ashley A. E. Direct Reductive Amination of Carbonyl Compounds Catalyzed by a Moisture Tolerant Tin(IV) Lewis Acid. Adv. Synth. Catal. 2018, 360, 1066–1071. 10.1002/adsc.201701418. [DOI] [PMC free article] [PubMed] [Google Scholar]; . For research using the (PhCH2)3SnOTf LA see:; c Cooper R. T.; Sapsford J. S.; Turnell-Ritson R. C.; Hyon D.-H.; White A. J. P.; Ashley A. E. Hydrogen activation using a novel tribenzyltin Lewis acid. Philos. Trans. R. Soc., A 2017, 375, 20170008. 10.1098/rsta.2017.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Das S.; Mondal S.; Pati S. K. Mechanistic Insights into Hydrogen Activation by Frustrated N/Sn Lewis Pairs. Chem.—Eur. J. 2018, 24, 2575–2579. 10.1002/chem.201705861. [DOI] [PubMed] [Google Scholar]; b Das S.; Pati S. K. Unravelling the mechanism of tin-based frustrated Lewis pair catalysed hydrogenation of carbonyl compounds. Catal. Sci. Technol. 2018, 8, 5178–5189. 10.1039/c8cy01227j. [DOI] [Google Scholar]
- Busca G. Bases and Basic Materials in Chemical and Environmental Processes. Liquid versus Solid Basicity. Chem. Rev. 2010, 110, 2217–2249. 10.1021/cr9000989. [DOI] [PubMed] [Google Scholar]
- Glasovac Z.; Eckert-Maksić M.; Maksić Z. B. Basicity of organic bases and superbases in acetonitrile by the polarized continuum model and DFT calculations. New J. Chem. 2009, 33, 588–597. 10.1039/b814812k. [DOI] [Google Scholar]
- In the absence of donors 1-OTf is only sparingly soluble in 1,2-dichlorobenzene at room temperature, which is the optimum solvent for hydrogenation catalysis. To ensure that all of the solutions studied were homogeneous, the solvent was changed to the more polar 1,2-difluorobenzene, in which 1-OTf is fully soluble (all H2 activation reactions using 1-OTf/LB pairs described herein proceed in both DCB and DFB with no reactivity differences).
- a Krossing I. The Facile Preparation of Weakly Coordinating Anions: Structure and Characterisation of Silverpolyfluoroalkoxyaluminates AgAl(ORF)4, Calculation of the Alkoxide Ion Affinity. Chem.—Eur. J. 2001, 7, 490–502. . [DOI] [PubMed] [Google Scholar]; b Krossing I.; Raabe I. Noncoordinating anions—Fact or fiction? A survey of likely candidates. Angew. Chem., Int. Ed. 2004, 43, 2066–2090. 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]; c Krossing I.; Reisinger A. Perfluorinated Alkoxyaluminate Salts of Cationic Brønsted Acids: Synthesis, Structure, and Characterization of [H(OEt2)2][Al{OC(CF3)3}4] and[H(THF)2][Al{OC(CF3)3}4]. Eur. J. Inorg. Chem. 2005, 2005, 1979–1989. 10.1002/ejic.200400436. [DOI] [Google Scholar]
- [1·(LB)]+[Al(ORF)4]– salts were found to be extremely sensitive to moisture upon isolation, decomposing rapidly to (iPr3Sn)2O – (1)2O – and [qui-H]+[Al(ORF)4]– (observed by 1H and 119Sn{1H} NMR spectroscopy). Consequently, this affected the activity of [1·(LB)]+ [Al(ORF)4]– in reactions with H2, and frustratingly the resonances of (1)2O obscured those of 1-H in the 1H NMR spectra. To minimise the amount of contaminating (1)2O, and maintain the reproducibility of experiments using [1·(LB)]+ [Al(ORF)4]–, the salts were prepared freshly in situ in DFB and used immediately for all reactions reported herein. For characterisation of (1)2O using NMR spectroscopy, see:Lockhart T. P.; Manders W. F.; Brinckman F. E. Spin Coupling Through Oxygen. Influence Of Structure And Solvent On 2J(119Sn,117Sn) In The 119Sn NMR Of Hexaorganodistannoxanes. J. Organomet. Chem. 1985, 286, 153–158. 10.1016/0022-328x(85)88002-5. [DOI] [Google Scholar]
- The DFT calculations were carried out using the ωB97X-D functional along with the Def2SVP and Def2TZVPP basis sets. The reported relative stabilities were obtained from solution phase Gibbs free energies. To model the global solvation effects, the SMD implicit solvation model was employed. For further computational details, see the Supporting Information Section S5.
- This seems to contradict the results obtained previously for DABCO/1-OTf.18 Pati et al. found the formation of the datively bound (DABCO)·1-OTf complex to be only marginally exergonic with respect to the 1-OTf + DABCO state. The orientation of the iPr groups in the most stable form of (qui)·1-OTf is different from that reported for the analogous (DABCO)·1-OTf complex (ref (18a)), which may explain the difference found in the relative stabilities of the two complexes.
- Only one set of (unobscured) qui resonances are observed in the 1H NMR spectrum (δ = 2.98 and 1.67 ppm) for a 1:1 mixture of [1·(qui)][Al(ORF)4] and qui, in DFB. The corresponding resonances in [1·(qui)]+ are appreciably downfield at δ = 3.16 and 1.86 ppm, respectively. This is consistent with qui being bound to the electron deficient [1]+ fragment in the former, while in the mixture a weighted average of chemical shifts between coordinated and uncoordinated qui is seen, implying a fast exchange equilibrium. Although [1·(qui)2]+ is calculated to be unstable relative to [1·(qui)]+ + qui (4.7 kcal/mol), this could be considered as an activation energy to degenerate exchange between bound and unbound qui, which will be readily surmountable at room temperature.
- The melting point of DFB (−35 °C; 239 K) limits the utility of this solvent for low-temperature NMR studies. Accordingly CD2Cl2 was used instead.
- Although broadening of the 119Sn{1H} NMR resonance is observed at 243 K in DFB, the solvent freezes below this temperature (mp = 239 K). The data are included in the Supporting Information.
- Spontaneous dissociation of [OTf]− from 1-OTf to form [1]+ is thought to be highly unlikely due to the very reactive and unstable nature of stannylium ions, especially in weak donor solvents such as DFB or CH2Cl2.
- [1·(qui)]+ and [Al(ORF)4]– are predicted to be separated in DFB (dissociation energy = −11.1 kcal/mol); [Al(ORF)4]– was therefore omitted from mechanistic calculations (for more details see Supporting Information Section S6.3).
- In principle, the [1·(qui)]+/[OTf]− pair may also induce FLP-type H2 activation, however, the barrier of this process is predicted to be much higher (31.2 kcal/mol) than that of the 1-OTf/qui pair (see Supporting Information, Section S6.5 for transition state structure).
- We note, however, that the level of agreement we find here should not be considered as a measure for the accuracy of the applied methodology.
- A direct comparison between the two systems [1·qui]+/qui and 1-OTf/qui is not straightforward because there is neither a common reference state, nor common reaction coordinate. We have attempted to make the best comparison possible using the two free energy profiles starting with their own reference states, including the H2 activation transition states, and product states.
- Kaljurand I.; Kütt A.; Sooväli L.; Rodima T.; Mäemets V.; Leito I.; Koppel I. A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- Further cooling of CD2Cl2 solutions beyond this temperature was uninformative due to precipitation of 1-OTf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.