

Abstract
Substantial evidence underscores the clinical efficacy of inhibiting CYP17A1-mediated androgen biosynthesis by abiraterone for treatment of prostate oncology. Previous structural analysis and in vitro assays revealed inconsistencies surrounding the nature and potency of CYP17A1 inhibition by abiraterone. Here, we establish that abiraterone is a slow-, tight-binding inhibitor of CYP17A1, with initial weak binding preceding the subsequent slow isomerization to a high-affinity CYP17A1-abiraterone complex. The in vitro inhibition constant of the final high-affinity CYP17A1-abiraterone complex ( ( Ki* = 0.39 nM )yielded a binding free energy of -12.8 kcal/mol that was quantitatively consistent with the in silico prediction of −14.5 kcal/mol. Prolonged suppression of dehydroepiandrosterone (DHEA) concentrations observed in VCaP cells after abiraterone washout corroborated its protracted CYP17A1 engagement. Molecular dynamics simulations illuminated potential structural determinants underlying the rapid reversible binding characterizing the two-step induced-fit model. Given the extended residence time (42 hours) of abiraterone within the CYP17A1 active site, in silico simulations demonstrated sustained target engagement even when most abiraterone has been eliminated systemically. Subsequent pharmacokinetic-pharmacodynamic (PK-PD) modeling linking time-dependent CYP17A1 occupancy to in vitro steroidogenic dynamics predicted comparable suppression of downstream DHEA-sulfate at both 1000- and 500-mg doses of abiraterone acetate. This enabled mechanistic rationalization of a clinically reported PK-PD disconnect, in which equipotent reduction of downstream plasma DHEA-sulfate levels was achieved despite a lower systemic exposure of abiraterone. Our novel findings provide the impetus for re-evaluating the current dosing paradigm of abiraterone with the aim of preserving PD efficacy while mitigating its dose-dependent adverse effects and financial burden.
SIGNIFICANCE STATEMENT
With the advent of novel molecularly targeted anticancer modalities, it is becoming increasingly evident that optimal dose selection must necessarily be predicated on mechanistic characterization of the relationships between target exposure, drug-target interactions, and pharmacodynamic endpoints. Nevertheless, efficacy has always been perceived as being exclusively synonymous with affinity-based measurements of drug-target binding. This work demonstrates how elucidating the slow-, tight-binding inhibition of CYP17A1 by abiraterone via in vitro and in silico analyses was pivotal in establishing the role of kinetic selectivity in mediating time-dependent CYP17A1 engagement and eventually downstream efficacy outcomes.
Introduction
Prostate cancer is ranked as the second most frequent cancer and the fifth leading cause of death among male malignancies worldwide (Bray et al., 2018). Therapeutic interventions in prostate cancer are defined upon patient stratification into the clinical states continuum (Scher et al., 2016). Although localized prostate cancer remains amenable to curative interventions via radiation or surgery, androgen deprivation therapy (ADT) in the form of surgical or medical castration has become the standard of care in clinically advanced or disseminated disease. However, the initial response to ADT is often unsustainable, and patients inevitably experience progression to castration-resistant prostate cancer within 2 to 3 years (Pienta and Bradley, 2006; Harris et al., 2009). Metastases (mCRPC) are evident in ≥84% of diagnosed cases, and prognosis is poor in mCRPC. Substantial evidence alludes to the reactivation of androgen receptor (AR)-mediated signaling as a key driver in disease progression despite castrate serum testosterone (<50 ng/dl) levels (Mostaghel et al., 2007). As a result, secondary hormonal manipulation via targeted molecular therapies has gained traction in the therapeutic management of mCRPC.
Bifunctional CYP17A1 occupies a pivotal role in both adrenal and de novo intratumoral androgen biosynthesis in which it catalyzes sequential 17α-hydroxylation and C17,20-lyase reactions (Fig. 1A) (Porubek, 2013). The resulting product, dehydroepiandrosterone (DHEA), is a critical precursor for the downstream generation of potent AR ligands testosterone and dihydrotestosterone. Consequently, abiraterone [administered as prodrug abiraterone acetate (AA)] was first developed and approved in 2011 as a first-in-class CYP17A1 inhibitor to treat mCRPC (Vasaitis et al., 2011; Yin and Hu, 2014). More recently, in men with metastatic hormone-sensitive prostate cancer (mHSPC), AA in combination with ADT was shown to significantly increase overall survival as compared with ADT monotherapy, culminating in the expansion of the clinical indications of AA to include the population of patients with mHSPC (Fizazi et al., 2017).
Fig. 1.

Clinical utility of abiraterone and D4A in prostate cancer. (A) Endogenous androgen biosynthesis pathway showing the inhibitory effects of abiraterone/D4A on CYP17A1. (B) Schematic describing the two proposed mechanisms of slow-, tight-binding inhibition of CYP17A1 by abiraterone/D4A (C) Chemical structures of abiraterone and D4A in which the latter is formed via metabolism of abiraterone by 3β-hydroxysteroid dehydrogenase (3βHSD).
AA is currently indicated to be administered at a dose of 1000 mg daily either 1 hour before or 2 hours after food (Janssen, 2019). Given that no dose-limiting toxicities were detected up to 2000 mg in a phase I dose escalation trial, the current dose selection was justified based on a plateau in anticipated toxicities (i.e., upstream mineralocorticoid excess) observed at doses above 750 mg (Attard et al., 2008). Implicit in this classic maximal tolerated dose (MTD) paradigm are the principles that 1) a linear dose-efficacy relationship exists and 2) toxicities are direct manifestations of exacerbated pharmacology (Ji et al., 2018). However, in a recent phase II trial, comparisons of low-dose AA (250 mg with a low-fat meal) versus standard-dose AA (1000 mg fasting) demonstrated that despite trough abiraterone concentrations being significantly higher in the standard-dose group compared with the low-dose group, the extent of dehydroepiandrosterone-sulfate (DHEA-S) suppression was similar, verifying that the efficacy of CYP17A1 inhibition was preserved (Szmulewitz et al., 2018). This observed pharmacokinetic-pharmacodynamic (PK-PD) uncoupling underscores how targeted therapeutics such as AA may achieve optimal antitumor activity at doses significantly lower than required to elicit adverse outcomes. With the diminished utility of MTD-based strategies, rational dose selection for AA must instead be predicated on mechanistic and quantitative characterization of the relationships between target exposure, drug-target interactions, and PD endpoints (Minasian et al., 2014; Sachs et al., 2016).
However, the mechanism of CYP17A1 inhibition by abiraterone has not been fully characterized. Firstly, X-ray crystal structures and spectral ligand binding assays reported coordination of the C17 pyridine of abiraterone with CYP17A1 heme iron, indicating a potent but reversible type II interaction (DeVore and Scott, 2012). Garrido et al. (2014) postulated that abiraterone is a slowly reversible CYP17A1 inhibitor and demonstrated its tight-binding effect with jump dilution experiments. Furthermore, abiraterone was suggested to be a slow-binding inhibitor with enhanced inhibition upon preincubation with CYP17A1 (Jarman et al., 1998). Collectively, the apparent complexities in the inhibition kinetics of CYP17A1 by abiraterone allude to a potential slow-, tight-binding phenomenon (Fig. 1B). Consequently, both inhibitory potency (thermodynamic selectivity) and target occupancy (kinetic selectivity) by abiraterone against CYP17A1 must be explicitly considered when defining its in vivo drug activity.
Moreover, it was revealed that Δ4-abiraterone (D4A) (Fig. 1C), a downstream metabolite of abiraterone, inhibits multiple steroidogenic enzymes, including CYP17A1 (Li et al., 2015). The multitargeting effects of D4A, coupled with its direct AR antagonism, have underscored its potential clinical utility in prostate oncology. Considering the structural similarities between abiraterone and D4A, particularly the preservation of the pyridine ring, the inhibition kinetics of CYP17A1 by D4A are expected to be equally complex.
In this study, detailed in vitro biochemical and in silico molecular dynamics (MD) analyses allowed us to gain unprecedented insights into the kinetic and structural determinants of the abiraterone/D4A-CYP17A1 interaction. Utilizing a mechanistic PK-PD model, we subsequently demonstrated how the coupling between PK and the extended residence time of abiraterone within the CYP17A1 active site potentially plays a pivotal role in controlling time-dependent CYP17A1 occupancy and eventually downstream PD outcomes. Our findings could eventually guide the optimization of AA dosing across the continuum of prostate oncology.
Methods
Chemicals and Reagents.
Abiraterone and DHEA were purchased from Tokyo Chemical Industry (Tokyo, Japan), whereas D4A was synthesized in-house (Supplemental Fig. 1; Supplemental Methods). Prednisolone, progesterone, 17α-hydroxyprogesterone, 17α-hydroxypregnenolone, androstenedione, testosterone-2,3,4-13C3, ketoconazole, and hydroxylamine hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). NADPH-regenerating system consisting of NADPH A (NADP+ and glucose 6-phosphate) and NADPH B (glucose-6 phosphate dehydrogenase) was purchased from BD Gentest (Woburn, MA). Dulbecco’s Modified Eagle’s medium high glucose, fetal bovine serum and 10× trypsin-EDTA were purchased from Gibco Life Technologies (Waltham, MA). One molar stock solution of phosphate buffer saline was purchased from Vivantis (Subang Jaya, Malaysia). All other analytical reagents were of analytical grade.
Enzymes.
Human CYP17A1R bactosomes containing recombinant human CYP17A1 (rCYP17A1) and human cytochrome P450–reductase enzymes supplemented with purified human cytochrome b5 were purchased from Cypex (Dundee, UK).
Slow-Binding Inhibition Kinetics.
To characterize the slow-binding inhibition of CYP17A1 by abiraterone and D4A, preincubation time dependence of reaction velocity was measured (Morrison and Walsh, 1988). Given the dual catalytic pathways associated with CYP17A1, both inhibition of progesterone 17α-hydroxylation and 17α-hydroxypregnenolone C17,20-lyase reactions were evaluated. Incubations (n = 3) were performed in 96-well plates. Abiraterone or D4A (5–100 nM) was preincubated for 0–30 minutes at 37°C with rCYP17A1 (5 pmol/ml) and NADPH B in 50 mM potassium phosphate buffer (pH 7.4) containing 2 mM MgCl2. Reactions were initiated by the addition of NADPH A and either 50 μM progesterone or 5 μM 17α-hydroxypregnenolone (saturating substrate conditions; Supplemental Fig. 2) and were allowed to proceed over the previously optimized incubation time of 20 minutes (Supplemental Fig. 2; Supplemental Methods). Eighty-microliter aliquots were subsequently removed and quenched with an equal volume of ice-cold acetonitrile containing the internal standard (prednisolone and androstenedione were used in the 17α-hydroxylase and C17,20-lyase inhibition assays, respectively). The quenched samples were centrifuged at 2755 g, 4°C, for 30 minutes, and the supernatant fraction was subjected to liquid chromatography tandem mass spectrometry (LC/MS/MS) analysis to determine the formation of 17α-hydroxyprogesterone or DHEA oxime, which were monitored as measures of residual enzyme activity. Oxime derivatization of DHEA and androstenedione prior to LC/MS/MS analysis is further described in the Supplemental Methods and Supplemental Fig. 3.
Semilogarithmic plots illustrating the percentage of rCYP17A1 activity remaining 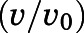 (relative to an [I] = 0 preincubation control sample) against preincubation time
(relative to an [I] = 0 preincubation control sample) against preincubation time 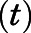 were generated for the determination of the observed first-order rate constant for the onset of inhibition (
were generated for the determination of the observed first-order rate constant for the onset of inhibition (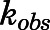 ) at various inhibitor concentrations (eq. 1) (Copeland, 2000, 2013).
) at various inhibitor concentrations (eq. 1) (Copeland, 2000, 2013).
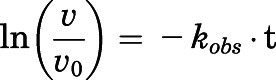 |
(1) |
Estimation of 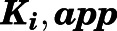 for the Initial Encounter Complex.
for the Initial Encounter Complex.
To obtain the apparent inhibition constant (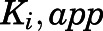 ) for the initial CYP17A1-inhibitor encounter complex, procedures were similar to that of the preincubation time-dependent inhibition assays, albeit without preincubation. The measured initial velocity
) for the initial CYP17A1-inhibitor encounter complex, procedures were similar to that of the preincubation time-dependent inhibition assays, albeit without preincubation. The measured initial velocity  across various concentrations of abiraterone and D4A (1–300 nM) was normalized against the velocity of the uninhibited reaction
across various concentrations of abiraterone and D4A (1–300 nM) was normalized against the velocity of the uninhibited reaction 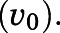 Data were fitted using standard four-parameter logistic models in GraphPad Prism 7.04 (San Diego, CA) as delineated in eq. 2 to generate
Data were fitted using standard four-parameter logistic models in GraphPad Prism 7.04 (San Diego, CA) as delineated in eq. 2 to generate 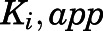 (Copeland, 2013).
(Copeland, 2013).
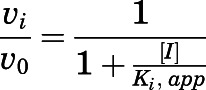 |
(2) |
Determination of 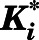 for the
for the  Complex.
Complex.
Using nonlinear regression in GraphPad Prism 7.04, the measured 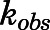 against
against  was analyzed via a two-step inhibition mechanism as shown in Fig. 1B, in which the initial rapid binding of inhibitor to enzyme
was analyzed via a two-step inhibition mechanism as shown in Fig. 1B, in which the initial rapid binding of inhibitor to enzyme 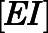 is accompanied by subsequent slower isomerization to the final enzyme-inhibitor
is accompanied by subsequent slower isomerization to the final enzyme-inhibitor 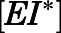 complex (Copeland, 2013). Equation 3a was used to obtain the forward
complex (Copeland, 2013). Equation 3a was used to obtain the forward 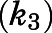 and reverse kinetic constants
and reverse kinetic constants 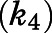 , describing the slow-binding inhibition. Given that
, describing the slow-binding inhibition. Given that 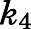 reflects the rate-limiting step for enzyme reactivation in the case of slow binding, the dissociation half-life for slow-binding inhibitors could be calculated using eq. 3b (Copeland, 2013).
reflects the rate-limiting step for enzyme reactivation in the case of slow binding, the dissociation half-life for slow-binding inhibitors could be calculated using eq. 3b (Copeland, 2013).
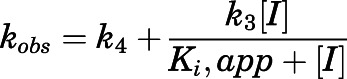 |
(3a) |
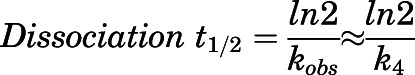 |
(3b) |
To determine the true values of 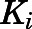 from experimental
from experimental 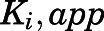 , reversible inhibition assays were performed, and Lineweaver-Burk transformations were used to discern the mode of inhibition (Supplemental Methods). Lastly, the inhibition constants
, reversible inhibition assays were performed, and Lineweaver-Burk transformations were used to discern the mode of inhibition (Supplemental Methods). Lastly, the inhibition constants 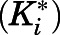 of the
of the  complex, representing the true target affinity of inhibitor to CYP17A1, were determined using eq. 4 (Copeland, 2013).
complex, representing the true target affinity of inhibitor to CYP17A1, were determined using eq. 4 (Copeland, 2013).
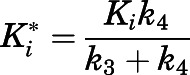 |
(4) |
Verifying the Functionality of the C17,20 Lyase Pathway in VCaP Cells.
VCaP prostate cancer cells (ATCC CRL-2876) were purchased from American Type Culture Collection (ATCC) (Manassas, VA) and authenticated by ATCC. VCaP cells were cultured in Dulbecco’s modified Eagle’s medium media supplemented with 10% v/v fetal bovine serum. For kinetic analyses, cells (passage numbers 8–16) were seeded in 24-well plates at a density of 0.4 million cells per well in a volume of 500 µl. Upon confluency, culture medium was first removed, and each well was washed with phosphate buffer saline to minimize residual metabolic waste. Cells were incubated with media containing 17α-hydroxypregnenolone (2.5–45 µM, 1% v/v methanol). After 30 minutes, 200-µl aliquots of medium were collected, spiked with internal standard, and subjected to a two-step liquid-liquid extraction using methyl-tert butyl ether before concentration under nitrogen gas (Supplemental Methods). The dried residues were derivatized via reconstitution with 50 µl of 100 mM hydroxylamine hydrochloride solution (50% v/v methanol/water) and heated at 60°C for 1 hour prior to LC/MS/MS measurement of DHEA oxime formation (Ming et al., 2014). Subsequent assessment of inhibitor functionality involved the simultaneous coincubation of inhibitors (abiraterone: 20 µM, 0.5% v/v methanol; ketoconazole: 25 µM, 0.5% v/v DMSO) or vehicle control (0.5% v/v methanol/DMSO), along with probe substrate (17α-hydroxypregnenolone: 30 µM, 0.6% v/v methanol) for 2 hours before analysis of DHEA oxime formation.
Functional Recovery Experiments in VCaP Cells.
Preincubation experiments utilizing VCaP cells were initiated by the addition of media containing abiraterone (40 µM, 1% v/v methanol), known reversible CYP17A1 inhibitor ketoconazole (40 µM, 1% v/v DMSO), along with vehicle controls (1% v/v methanol/DMSO). After 2 hours, inhibitors were removed, and fresh medium was added for the duration of the washout period (0, 30, or 60 minutes). Finally, medium containing the probe substrate 17α-hydroxypregnenolone (30 μM, 1% v/v methanol) was added for 30 minutes. Two-hundred-microliter aliquots of medium were collected and processed in the same manner as described in the previous section to measure DHEA oxime formation. At each time point (0, 30, or 60 minutes), unpaired one-tailed t tests were applied to evaluate whether the percentage recovery of maximal CYP17A1 activity (quantified by DHEA oxime formation in the respective vehicle controls) after abiraterone preincubation and washout was significantly suppressed in comparison with ketoconazole treatment (P value < 0.05).
LC/MS/MS Measurement of Metabolite Formation.
Samples were analyzed using an Agilent 1290 Infinity liquid chromatography system (Agilent Technologies, Santa Clara, CA) interfaced with an AB Sciex QTrap 5500 hybrid linear ion-trap quadrupole mass spectrometer equipped with a TurboIonSpray source (AB SCIEX, Framingham, MA). Injection volumes were 2 and 4 μl when quantifying 17α-hydroxyprogesterone and DHEA oxime production, respectively. AWaters ACQUITYTM ultra-high performance liquid chromatography ethylene bridged hybrid C18 column (1.7 μM, 2.1 × 50 mm; Waters, Milford) was used for chromatographic separation. The aqueous mobile phases comprised 0.1% v/v formic acid in water (A) and 0.1% v/v formic acid in acetonitrile (B). Mobile phases were delivered at 0.6 ml/min. The column and sample temperatures were maintained at 45°C and 4°C, respectively. The gradient program was as follows: linear gradient from 20% to 60% B (0–0.5 minutes), isocratic at 60% B (0.5–1.5 minutes), linear gradient from 60% to 98% B (1.50–1.51 minutes), isocratic at 98% B (1.51–2 minutes), linear gradient from 98% to 20% B (2–2.01 minutes), and isocratic at 20% B (2.01–2.5 minutes). All analyses were performed in electrospray ionization positive mode. The mass spectrometer source conditions were as follows: ion spray voltage +5500 V, source temperature 500°C, curtain gas 25 psi, GS1 (sheath gas) 30 psi, GS2 (drying gas) 30 psi, collision gas (nitrogen) medium. The compound-specific parameters are outlined in Supplemental Table 1 in which the peak areas of 17α-hydroxyprogesterone and DHEA oxime were quantified as the products formed via 17α-hydroxylation and the C17,20-lyase reaction, respectively. For cellular washout experiments, testosterone-2,3,4-13C3 was used as the internal standard. Chromatographic peak integration was performed using MultiQuant software (AB Sciex) to obtain the peak area of the analyte expressed as a ratio to the peak area of the internal standard (peak area ratio).
MD Simulations.
The X-ray crystal structure of abiraterone-bound CYP17A1 (Protein Data Bank 3RUK, chain D) was used as the template for MD simulations and PMF calculations. All MD simulations of the protein and the heme prosthetic group were performed using Groningen Machine for Chemical Simulations version 5.1.5 in conjunction with the Groningen Molecular Simulation 54A7 force field (Schmid et al., 2011; Abraham et al., 2015). The simple point charge water model was used to describe the solvent water. Simulations were performed under periodic boundary conditions in a rectangular box. A cutoff of 1.4 nm was applied to short-range nonbonded interactions, whereas long-range electrostatic interactions were calculated using the particle mesh Ewald algorithm (Darden et al., 1993; Essmann et al., 1995). Abiraterone was predicted to be neutral at pH 7.4 [ChemAxon, (Szegezdi and Csizmadia, 2007)]. Topology parameters for the neutral abiraterone were obtained using the Automated Topology Builder and Repository (Malde et al., 2011).
Abiraterone-bound CYP17A1 was placed in a cubic box of simple point charge water, including neutralizing counterions. A steepest-descents minimization, followed by a position restraint simulation for 100 picoseconds, was performed under a constant volume (NVT - number of particles, volume and temperature) ensemble. A constant pressure (NPT - number of particles, pressure and temperature) equilibration was performed for 100 picoseconds, using weak coupling to maintain pressure isotropically at 1.0 bar at a temperature of 310 K. A Parrinello-Rahman barostat was used to isotropically regulate pressure, along with a Nosé-Hoover thermostat to maintain temperature, ensuring that a true NPT ensemble was sampled (Parrinello and Rahman, 1981; Nosé, 1984; Hoover, 1985). Production MD simulations were conducted for 250 nanoseconds without any restraints.
PMF Calculations.
To calculate the binding free energy of abiraterone, PMF calculations of the abiraterone binding pathway from the CYP17A1 active site were performed along the central axis of the heme using the umbrella sampling method (Torrie and Valleau, 1977; Gordon et al., 2013). The CYP17A1 structure was placed in a rectangular box with dimensions adequate to fulfill the minimum image convention and provide space for “pulling” simulations to occur along the z-axis. The chosen reaction coordinate was defined as the axis passing through the heme, normal to the plane of the heme. A harmonic restraint with a force constant of 1000 kJ/(mol·nm2) was applied to the center of mass (COM) of abiraterone and moved along the reaction coordinate away from the core structure along the z-axis over 500 picoseconds with a pull rate of 10 nm ns−1. The pulling simulation was used to generate a set of 44 reference configurations (windows) separated by 0.05-nm intervals along the reaction coordinate. These windows were then used as starting configurations for umbrella sampling calculations. In each umbrella sampling simulation, the COM of abiraterone was harmonically restrained using a force constant of 1000 kJ/(mol·nm2) in the z-direction to allow sampling of a specific window along the reaction coordinate, where the motion of the abiraterone was not restrained in the xy-plane. MD simulations were performed for 20 nanoseconds with each window, providing a total simulation time of 880 nanoseconds for umbrella sampling.
The PMF profile was constructed using the weighted histogram analysis method (Kumar et al., 1992; Hub et al., 2010). The first 4 nanoseconds of each window were taken as equilibration and were therefore not included in the calculation of PMF values. The Kd was calculated from the PMF, W(z), using eq. 5 (Chen and Chung, 2012), where πr2 is the cross-sectional area of the binding path, NA is Avogadro’s number, and zmin and zmax are the z-positions of the abiraterone COM when fully bound to CYP17A1 and when in the bulk solvent, respectively. The radius R of the binding path was estimated as 0.5 nm based on the size of the heme porphyrin moiety.
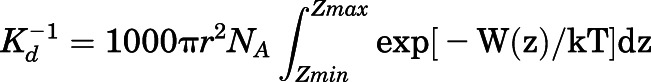 |
(5) |
∆G for the binding of abiraterone to CYP17A1 was calculated using eq. 6, where ∆G is the binding free energy, R is the universal gas constant, T is the temperature, and Kd is the equilibrium dissociation constant.
| (6) |
The volume of the binding pocket was estimated using the mdpocket program (Schmidtke et al., 2011). The pocket volume calculations were performed in an unliganded trajectory of CYP17A1-abiraterone simulation to estimate fluctuations in the presence of a bound ligand.
Development of a Mechanistic PK-PD Model Linking Abiraterone PK and CYP17A1-Abiraterone Binding Kinetics to Steroidogenic Flux.
We also sought to understand CYP17A1 target occupancy by abiraterone in the context of its in vivo PK (Dahl and Akerud, 2013). Considering the two-step slow-binding inhibition mechanism as shown in Fig. 1B, time-dependent changes in the concentration of each enzyme species (i.e., 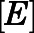 ,
, 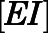 ,
, 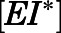 ) were simulated after a single 1000- or 500-mg dose of AA (Acharya et al., 2012). Dynamic in vivo target occupancy was determined by the summation of
) were simulated after a single 1000- or 500-mg dose of AA (Acharya et al., 2012). Dynamic in vivo target occupancy was determined by the summation of 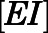 and
and 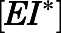 .
.
Equations 7, a–d describe the system of differential equations that was consecutively solved in parallel over very small intervals to simulate in vivo target occupancy (Vauquelin, 2017). The initial value of 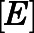 was defined as the steady-state concentration of CYP17A1 in vivo. CYP17A1 has an abundance of approximately 35,000 pmol/g tissue. Considering the weight (10 g) and volume of the adrenal glands (0.0085 l) gives a steady-state concentration of 41 µM. The flux in the concentration of free inhibitor
was defined as the steady-state concentration of CYP17A1 in vivo. CYP17A1 has an abundance of approximately 35,000 pmol/g tissue. Considering the weight (10 g) and volume of the adrenal glands (0.0085 l) gives a steady-state concentration of 41 µM. The flux in the concentration of free inhibitor  near the target was assumed to be equal to the plasma concentration-time profile of abiraterone after AA administration.
near the target was assumed to be equal to the plasma concentration-time profile of abiraterone after AA administration.
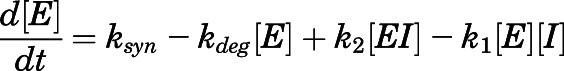 |
(7a) |
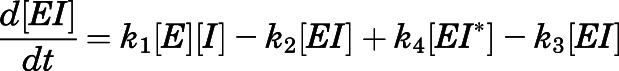 |
(7b) |
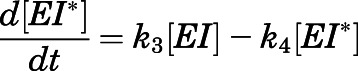 |
(7c) |
| (7d) |
Supplemental Table 2 presents a summary of the relevant input parameters. Values for microscopic rate constants 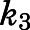 and
and 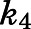 were derived from the preincubation time-dependent kinetic experiments. As highlighted in eq. 8a, the overall target affinity
were derived from the preincubation time-dependent kinetic experiments. As highlighted in eq. 8a, the overall target affinity 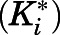 of abiraterone for CYP17A1 can be described as a composite of the various microscopic rate constants. With rearrangement, eq. 8b emphasizes the equivalence of
of abiraterone for CYP17A1 can be described as a composite of the various microscopic rate constants. With rearrangement, eq. 8b emphasizes the equivalence of 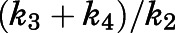 and the
and the 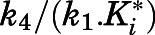 ratios (Vauquelin, 2017).
ratios (Vauquelin, 2017).
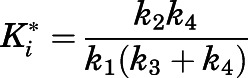 |
(8a) |
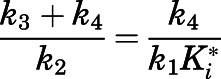 |
(8b) |
For induced-fit binding, slow dissociation and high affinity rely on the stability of the 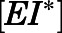 , a scenario necessitating that
, a scenario necessitating that 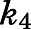 becomes the kinetic bottleneck of the dissociation process (Copeland et al., 2006; Copeland, 2013; Vauquelin, 2017). Hence, to qualify as an induced-fit binder, it follows that
becomes the kinetic bottleneck of the dissociation process (Copeland et al., 2006; Copeland, 2013; Vauquelin, 2017). Hence, to qualify as an induced-fit binder, it follows that 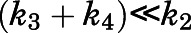 and
and 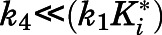 (Vauquelin, 2017). Consequently, the eventual estimates of
(Vauquelin, 2017). Consequently, the eventual estimates of 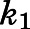 and
and 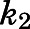 satisfied these inequality relationships as well as the
satisfied these inequality relationships as well as the 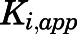 of the initial
of the initial 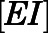 complex obtained from kinetic analyses.
complex obtained from kinetic analyses.
In subsequent PK-PD simulations, CYP17A1 engagement varying as a function of both abiraterone concentration and time was prospectively correlated with the extent of DHEA-sulfate suppression in a dynamic in vitro model of intracellular steroidogenesis as developed by Ahmed et al. (Ahmed et al., 2018) (Supplemental Methods). All analyses were performed in RStudio 1.1.456 using the deSolve package (R Core Team, 2013).
Correlating Abiraterone PK with Androgen Reduction.
A phase II trial by Szmulewitz et al. (2018) demonstrated comparable efficacy of low-dose AA (250 mg with a low-fat meal) to standard-dose AA (1000 mg fasting). Utilizing plasma abiraterone measurements collected after 8 days of therapy (before and 2, 3, and 4 hours after dosing) in the low-dose and high-dose groups, differences in PK parameters [i.e., area under the plasma concentration-time curve (AUC0–4 hours) and peak plasma concentrations (Cmax)] were evaluated and compared with AUC and Cmax values derived from two phase I trials (O’Donnell et al., 2004; Attard et al., 2008) studying the 500-mg dose of AA (fasted) in castrate patient cohorts. Group comparisons of log-transformed PK data were performed via one-way analysis of variance, followed by Dunnett’s multiple comparison test. Mean AUC and Cmax ratios as well as 95% confidence intervals (CIs) for pairwise comparisons were calculated by retransformation of the logarithmic results.
Results
Abiraterone and D4A Cause Slow- and Tight-Binding Inhibition of CYP17A1.
The binding of abiraterone and D4A to CYP17A1 was characterized by monitoring the formation of both 17α- hydroxyprogesterone and DHEA oxime as a function of preincubation time. Semilogarithmic plots illustrated time- and concentration-dependent decreases in residual CYP17A1 activity (relative to a [I] = 0 preincubation control sample) and enabled determination of the first-order rate constants for onset of inhibition (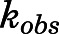 ) at various concentrations of abiraterone (Fig. 2, A and B) and D4A (Fig. 2, E and F) using eq. 1. The hyperbolic dependence of
) at various concentrations of abiraterone (Fig. 2, A and B) and D4A (Fig. 2, E and F) using eq. 1. The hyperbolic dependence of 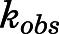 on abiraterone/D4A concentration as observed in the kinetic plots (Fig. 2, C, D, G, and H) is indicative of a two-step slow-binding inhibition mechanism (Fig. 1B) (Morrison and Walsh, 1988; Copeland, 2000). Concentration-dependent reduction of CYP17A1 activity in the absence of preincubation (y-intercept values in Fig. 2, A, B, E, and F) further substantiates a two-step model, in which the rapid, reversible binding of
on abiraterone/D4A concentration as observed in the kinetic plots (Fig. 2, C, D, G, and H) is indicative of a two-step slow-binding inhibition mechanism (Fig. 1B) (Morrison and Walsh, 1988; Copeland, 2000). Concentration-dependent reduction of CYP17A1 activity in the absence of preincubation (y-intercept values in Fig. 2, A, B, E, and F) further substantiates a two-step model, in which the rapid, reversible binding of 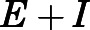 to form
to form  is manifested as an instantaneous effect on the initial reaction velocity (
is manifested as an instantaneous effect on the initial reaction velocity (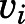 ). The apparent inhibition constant for the initial
). The apparent inhibition constant for the initial  complex (
complex (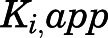 ) was determined by fitting the dependence of fractional velocity (
) was determined by fitting the dependence of fractional velocity (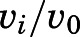 ) on inhibitor concentration using eq. 2 (Fig. 3, A, B, E, and F; Table 1). In the second step, the binding of abiraterone/D4A to CYP17A1 subsequently induces a time-dependent conformational change in CYP17A1 that results in a high-affinity
) on inhibitor concentration using eq. 2 (Fig. 3, A, B, E, and F; Table 1). In the second step, the binding of abiraterone/D4A to CYP17A1 subsequently induces a time-dependent conformational change in CYP17A1 that results in a high-affinity  complex (Fig. 1B). Nonlinear regression analyses based on eq. 3a allowed the forward (
complex (Fig. 1B). Nonlinear regression analyses based on eq. 3a allowed the forward (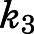 ) and reverse (
) and reverse (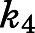 ) isomerization constants to be established (Table 1).
) isomerization constants to be established (Table 1).
Fig. 2.

Preincubation time-dependent inhibition of CYP17A1 by abiraterone and D4A. Semilogarithmic plots illustrate time- and concentration-dependent inhibition of CYP17A1 by (A and B) abiraterone or (E and F) D4A using progesterone and 17α-hydroxypregnenolone as probe substrates for 17α-hydroxyprogesterone and dehydroepiandrosterone formation, respectively. The relationship between the observed first-order rate constants for the onset of inhibition (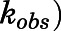 values determined from (A, B, E, and F) and inhibitor concentration was further investigated in (C, D, G, and H), and data were subsequently fitted to eq. 3a to calculate the forward (
values determined from (A, B, E, and F) and inhibitor concentration was further investigated in (C, D, G, and H), and data were subsequently fitted to eq. 3a to calculate the forward (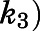 and reverse
and reverse 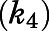 isomerization constants via nonlinear regression analysis. Goodness of fits (R2) for (C, D, G, and H) were determined to be 0.984, 0.984, 0.986, and 0.996, respectively. Each point in the semilogarithmic plots represents the mean ± S.D. of triplicate determinations.
isomerization constants via nonlinear regression analysis. Goodness of fits (R2) for (C, D, G, and H) were determined to be 0.984, 0.984, 0.986, and 0.996, respectively. Each point in the semilogarithmic plots represents the mean ± S.D. of triplicate determinations.
Fig. 3.

Reversible inhibition of CYP17A1 by abiraterone and D4A. Concentration-response plots represent the initial inhibited state (EI) of the CYP17A1-mediated 17α-hydroxylase and C17,20-lyase enzymatic reactions when slow-, tight-binding inhibitors (A and B) abiraterone and (E and F) D4A were introduced. The measured initial velocities (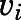 ) were used together with velocity of the uninhibited reaction (
) were used together with velocity of the uninhibited reaction (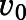 ) to calculate the fractional velocities across various concentrations of abiraterone and D4A (1–300 nM). Using eq. 2, apparent inhibition constants (
) to calculate the fractional velocities across various concentrations of abiraterone and D4A (1–300 nM). Using eq. 2, apparent inhibition constants (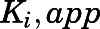 ) for the initial CYP17A1-inhibitor encounter complexes were first obtained from the midpoint of isotherm curves.
) for the initial CYP17A1-inhibitor encounter complexes were first obtained from the midpoint of isotherm curves. 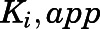 values were subsequently used in nonlinear regression analyses (eq. 3a) to determine the forward (
values were subsequently used in nonlinear regression analyses (eq. 3a) to determine the forward (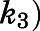 and reverse
and reverse 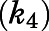 isomerization constants. Reversible inhibition experiments were also performed in the presence of multiple substrate (progesterone or 17α-hydroxypregnenolone) and (C and D) abiraterone or (G and H) D4A concentrations. To discern the mode of inhibition, Michaelis-Menten plots generated were subjected to double reciprocal transformations to yield Lineweaver-Burk graphs as presented in Supplemental Fig. 4. Based on the identified mode of inhibition, the inhibition constant for the initial EI complex (
isomerization constants. Reversible inhibition experiments were also performed in the presence of multiple substrate (progesterone or 17α-hydroxypregnenolone) and (C and D) abiraterone or (G and H) D4A concentrations. To discern the mode of inhibition, Michaelis-Menten plots generated were subjected to double reciprocal transformations to yield Lineweaver-Burk graphs as presented in Supplemental Fig. 4. Based on the identified mode of inhibition, the inhibition constant for the initial EI complex (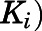 could eventually be derived from
could eventually be derived from 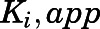 . Each point represents the mean ± S.D. of triplicate determinations.
. Each point represents the mean ± S.D. of triplicate determinations.
TABLE 1.
Summary of kinetic constants and dissociation t1/2 for the inhibition of CYP17A1 by abiraterone and D4A
| Inhibitor | CYP17A1 Activity |
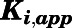 (nM)a (nM)a
|
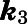 (min−1)b (min−1)b
|
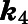 (min−1)b (min−1)b
|
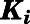 (nM)c (nM)c
|
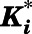 (nM) (nM) |
Dissociation t1/2 (h) |
|---|---|---|---|---|---|---|---|
| Abiraterone | 17α-Hydroxylase | 132 ± 2.02 | 0.0353 | 0.000275 | 50.7 | 0.39 | 42.0 |
| C17,20-Lyase | 118 ± 1.13 | 0.0224 | 0.000384 | 24.8 | 0.42 | 30.1 | |
| D4A | 17α-Hydroxylase | 76 ± 1.21 | 0.0218 | 0.000396 | 23.5 | 0.42 | 29.2 |
| C17,20-Lyase | 163 ± 3.02 | 0.0219 | 0.000121 | 57.4 | 0.32 | 95.5 |
From plot of 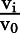 vs. [I]. Data represented as means ± S.D. of triplicate determinations.
vs. [I]. Data represented as means ± S.D. of triplicate determinations.
From plot of 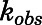 vs. [I].
vs. [I].
Value of Ki,app corrected for mixed-mode inhibition.
The Tight-Inhibited Complex (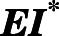 ) Demonstrates Significant Increases in Inhibitory Potency.
) Demonstrates Significant Increases in Inhibitory Potency.
Reversible inhibition of CYP17A1 by abiraterone (Fig. 3, C and D) and D4A (Fig. 3, G and H) was assessed. The intersection of lines above the x-axis in double reciprocal Lineweaver-Burk transformations revealed mixed-mode inhibition of CYP17A1 by both abiraterone (Supplemental Fig. 4, A and B) and D4A (Supplemental Fig. 4, C and D). Hence, the 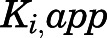 values determined for abiraterone and D4A were corrected to the true
values determined for abiraterone and D4A were corrected to the true 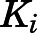 (Table 1). With acquisition of
(Table 1). With acquisition of 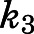 ,
, 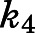 , and
, and 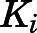 , the overall inhibition constants for the final
, the overall inhibition constants for the final  complex (
complex (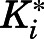 ) were calculated using eq. 4. For abiraterone and D4A, the isomerization of
) were calculated using eq. 4. For abiraterone and D4A, the isomerization of  to
to  was accompanied by significant increases in inhibitory potencies (
was accompanied by significant increases in inhibitory potencies (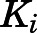 to
to 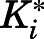 in Table 1), expounding the thermodynamic stabilization of the ground state of
in Table 1), expounding the thermodynamic stabilization of the ground state of  versus
versus  (Tonge, 2018). Using eq. 3b, which assumes that k4 is rate-determining, both abiraterone and D4A demonstrated prolonged residence time within the binding domain of CYP17A1 (Table 1). However, as reflected in Table 1, despite comparable inhibitory potencies across both 17α-hydroxylase and C17,20-lyase reactions for abiraterone and D4A, D4A exhibited longer residence time on CYP17A1 when the C17,20-lyase reaction was measured (95 hours vs. 29-42 hours), highlighting the discordance between affinity and residence time measurements.
(Tonge, 2018). Using eq. 3b, which assumes that k4 is rate-determining, both abiraterone and D4A demonstrated prolonged residence time within the binding domain of CYP17A1 (Table 1). However, as reflected in Table 1, despite comparable inhibitory potencies across both 17α-hydroxylase and C17,20-lyase reactions for abiraterone and D4A, D4A exhibited longer residence time on CYP17A1 when the C17,20-lyase reaction was measured (95 hours vs. 29-42 hours), highlighting the discordance between affinity and residence time measurements.
Binding Free Energy of the CYP17A1-Abiraterone Complex is Consistent with Kinetic Analyses.
Given that the X-ray crystal structure reported by DeVore and Scott (2012) has abiraterone bound to CYP17A1, subsequent MD simulations considered the CYP17A1-abiraterone interaction only. The potential of mean force (PMF) profile was generated to predict the binding affinity of abiraterone to CYP17A1. In the reported binding mode (referred to hereafter as pose A), the abiraterone steroid moiety orients near perpendicular to the plane of the heme. Based on the PMF profile of abiraterone unbinding pathway, a distinct energy well occurs at the reaction coordinate, z = 5 Å (Fig. 4A). The PMF calculations converged after 4 nanoseconds; further extension of simulations to 20 nanoseconds for each umbrella window did not appreciably change the PMF profile or binding free energy (SD = 0.5 kcal/mol). The binding mode associated with the energy well is essentially identical to that observed in the X-ray crystal structure, in which the N atom of abiraterone forms a coordinate bond with heme Fe (<2.6 Å). The computationally determined free energy of pose A was −14.5 kcal/mol and compares well to the experimental in vitro estimates (−12.5 and −12.8 kcal/mol for 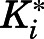 = 0.42 and 0.39 nM depending on substrate, Table 1). Consistent with the PMF profile, only one abiraterone binding pose A is identified using this approach (Fig. 4B).
= 0.42 and 0.39 nM depending on substrate, Table 1). Consistent with the PMF profile, only one abiraterone binding pose A is identified using this approach (Fig. 4B).
Fig. 4.

MD simulations investigating the binding of abiraterone to CYP17A1. (A) PMF profile for the unbinding of abiraterone from CYP17A1. (B) Long, unrestrained MD simulations demonstrated two dominant binding modes of abiraterone in CYP17A1 catalytic site. In pose A, the plane of the steroid moiety is near perpendicular to the plane of the heme. (C) In pose B, abiraterone is parallel to the plane of the heme. The key-binding region of the CYP17A1 active site is shown in magenta. The side chain atoms of important binding residues are shown as sticks, abiraterone as thick sticks, and heme as ball and sticks. C, O, and N atoms are shown in cyan, red, and blue, respectively.
Prolonged CYP17A1 Binding of Abiraterone Was Confirmed in Cellular Washout Experiments.
To ensure that any kinetic selectivity observed at the level of the purified recombinant CYP17A1 target could be qualitatively recapitulated in complex biologic systems, cellular washout experiments utilizing human prostate cancer cells (VCaP) were performed. Formation of DHEA at various concentrations of 17α-hydroxypregnenolone exhibited Michaelis-Menten kinetics, establishing the cellular functionality of CYP17A1 (Fig. 5A). In coincubation experiments, both abiraterone (20 µM) and ketoconazole (25 µM) produced depressions of the maximal response observed after 2 hours (quantified via DHEA formation in vehicle controls) (Fig. 5B), demonstrating inhibition of C17,20-lyase reaction by abiraterone and ketoconazole. Upon preincubation of abiraterone and subsequent washout, continued suppression of CYP17A1 activity was observed up to 60 minutes (Fig. 5C). In contrast, minimal prolongation of DHEA suppression was observed after washout of known reversible CYP17A1 inhibitor ketoconazole (Fig. 5C).
Fig. 5.

VCaP cell–based washout experiments to assess the phenotypic consequence of CYP17A1 engagement after inhibitor removal. (A) DHEA formation at increasing concentrations of 17α-hydroxypregnenolone conformed to saturable Michaelis-Menten kinetics (R2 = 0.974) (B) The effect of either abiraterone (20 µM) or ketoconazole (25 µM) coincubation on 17α-hydroxypregnenolone–mediated DHEA formation in VCaP cells. Cells were incubated for 2 hours at 37°C. (C) Time dependent reversal of CYP17A1 inhibition by abiraterone or ketoconazole as quantified via restoration of DHEA production. VCaP cells were preincubated for 2 hours without (control) or with abiraterone (40 µM) or ketoconazole (40 µM), washed, and further incubated with fresh medium for the indicated washout periods before final incubation with 30 μM 17α-hydroxypregnenolone for 30 minutes. DHEA oxime formation, representing residual CYP17A1 activity in (B) and recovery of CYP17A1 activity in (C) was expressed as the percentage of maximal DHEA oxime produced in the corresponding vehicle controls. In (C), an unpaired t test revealed overall significant difference between treatment arms across the 60-minute washout period (0 minutes: one-sided P value = 0.0158; 30 minutes: one-sided P value = 0.0071; 60 minutes: one-sided P value = 0.0281). Data in (A) represent the means ± S.D. from two experiments with triplicate determinations, whereas data in (B and C) represent the means ± S.D. from at least three experiments with triplicate determinations. *P < 0.05; **P < 0.01.
Identification of a Second Binding Mode Confirms the Two-Step Induced-Fit Binding Mechanism Observed in Kinetic Experiments.
In addition to the tight-binding mode, the kinetic experiments performed here identified a fast, reversible binding component of abiraterone to CYP17A1. Beyond the PMF calculations performed over a short simulation timescale, a long, unrestrained MD simulation (250 nanoseconds) was performed to explore additional binding event(s). MD simulations of the abiraterone-CYP17A1 complex demonstrated two dominant binding modes of abiraterone within the active site. In pose A, the steroid moiety of abiraterone is oriented near vertically above the plane of the heme between the F- and G- helices. This is similar to the chemical interactions and orientation reported in the X-ray crystal structure (DeVore and Scott, 2012) and pose A observed in the PMF calculations (Fig. 4B).
In the second binding mode, referred to as pose B, the plane of the steroid moiety is orientated parallel to the plane of the heme. The α-face of the steroid nucleus packs almost entirely along the I-helix, whereas in pose A, only a portion of the structure packs against the I-helix (Fig. 4C). In pose A, H-bonding occurs between the 3β-OH group of abiraterone and Asn202. In pose B, H-bonding occurs between the 3β-OH group and Arg 239 (2.2 Å) and Asp 298 (2.4 Å) of the G- and I-helices, respectively. We propose that the binding mode of abiraterone identified in pose B may represent the fast, reversible interaction that occurs prior to heme coordination.
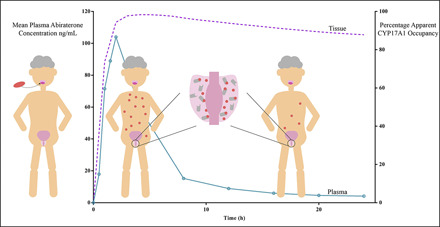
Simulated Target Occupancy after a Single In Vivo Dose of Abiraterone Exceeds Its PK Half-Life.
Simulations of apparent CYP17A1 occupancy after a single 1000- or 500-mg dose of AA were performed using numerical integration over a system of differential equations for the two-step slow-binding enzyme inhibition mechanism (Vauquelin, 2017). The terminal elimination 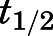 of abiraterone is approximately 16 hours (Fig. 6A). At the end of the dosing interval of 24 hours, when most abiraterone has been eliminated systemically, an on-target peak-to-trough occupancy ratio (in which trough is measured 24 hours after dosing) was calculated to be 1.1 for both 1000- and 500-mg doses, indicating approximately constant apparent target occupancy between two consecutive doses (Vauquelin, 2017). Notably, >80% apparent CYP17A1 engagement was preserved despite a dose reduction to 500 mg.
of abiraterone is approximately 16 hours (Fig. 6A). At the end of the dosing interval of 24 hours, when most abiraterone has been eliminated systemically, an on-target peak-to-trough occupancy ratio (in which trough is measured 24 hours after dosing) was calculated to be 1.1 for both 1000- and 500-mg doses, indicating approximately constant apparent target occupancy between two consecutive doses (Vauquelin, 2017). Notably, >80% apparent CYP17A1 engagement was preserved despite a dose reduction to 500 mg.
Fig. 6.

Mechanistic pharmacokinetic-pharmacodynamic simulations integrating abiraterone PK and CYP17A1-abiraterone binding kinetics into eventual predictions of efficacy outcomes. (A) Simulated apparent CYP17A1 occupancy over time plots after a single in vivo dose of 1000 or 500 mg of abiraterone acetate (AA). The clinical plasma concentration-time profiles of abiraterone are represented by open symbols, whereas the solid and dashed lines indicate the predicted percentage apparent CYP17A1 occupancy over time. Time-dependent changes in apparent CYP17A1 engagement were subsequently used to predict potential perturbations in the intracellular concentrations of (B) dehydroepiandrosterone-sulfate after multiple-dose administration of 1000 or 500 mg AA over 72 hours.
Translation of CYP17A1 Occupancy to PD Outcomes Rationalized a Clinically Observed PK-PD Disconnect.
Nevertheless, the translation of target occupancy to efficacy outcomes requires additional consideration of the amount of target that must be complexed with abiraterone to elicit the desired PD effect (i.e., target vulnerability). When multiple dosing of AA for 72 hours was used to perturb a baseline model of steroidogenesis, the model predicted comparable attenuations in intracellular DHEA-sulfate concentrations for the 1000- and 500-mg doses, calculated to be 3.26% and 4.68% at 72 hours of baseline measurements (without inhibition), respectively (Fig. 6B).
This PK-PD disconnect paralleled reported clinical findings from a phase II trial comparing low-dose AA (250 mg daily with a low-fat meal) versus standard-dose AA (1000 mg daily fasting). The systemic exposure (AUC0–4 hours) and Cmax of abiraterone were significantly higher in the standard-dose arm compared with the low-dose group (Fig. 7) (mean AUC0–4 hours ratio = 2.08, 95% CI: 1.02–4.24; mean Cmax ratio = 2.02, 95% CI: 1.01–4.05). Nevertheless, equipotent reduction of plasma DHEA-S levels was observed (Szmulewitz et al., 2018), confirming that the clinical efficacy of CYP17A1 inhibition was maintained even at lower systemic exposure of abiraterone.
Fig. 7.

Investigating potential correlations between abiraterone drug levels and the extent of DHEA-S suppression. Results from the phase II study by Szmulewitz et al. (2018) demonstrated how (A) the area under the plasma concentration-time curve of abiraterone from 0 to 4 hours (AUC0–4 hours), and (B) peak plasma concentrations (Cmax) remained significantly higher in the standard-dose arm (1000 mg AA, fasting, n = 20) compared with the low-dose arm (250 mg AA, fed, n = 20) (two-sided P value for AUC0–4 hours = 0.0417, and two-sided P value for Cmax = 0.0474). Combined analyses of phase I trials (Attard et al., 2008; O’Donnell et al., 2004, n = 10) in (A and B) revealed that administration of 500 mg of AA in a fasted state yielded AUC values that were greater than that measured in the low-dose arm (two-sided P value = 0.0038), whereas Cmax measurements were not significantly different. AUC and Cmax data were log-transformed and groups comparisons were performed using repeated-measures one-way ANOVA and Dunnett’s multiple comparisons tests.
Notably, as presented in Fig. 7B, comparable Cmax measurements (mean Cmax ratio = 0.98, 95% CI = 0.42–2.26) were observed between a 500-mg dose of AA administered under fasting conditions and 250 mg of AA administered with food. The mean AUC (AUC0–12 hours and AUC0–72 hours) associated with the 500-mg dose was expectedly greater than AUC0–4 hours, associated with the low-dose treatment arm (mean AUC ratio = 2.83, 95% CI = 1.42–5.62) (Fig. 7A). Based on PK measurements made up to 72 hours in a phase I dose escalation trial (Acharya et al., 2012), the calculated AUC0–4 hours is approximately 50% of the total AUC across different doses. Taken together, a 500-mg dose of AA is expected to yield similar exposure to 250 mg of AA administered with food, representing a viable alternative to utilizing the food effect to enhance the systemic exposure of abiraterone.
Discussion
The translational disconnect between preclinical and clinical pharmacology is a perennial challenge. Incorrect assumptions underlying various processes in the causal chain between drug exposure and pharmacological response often restrict the success of in vitro–to–in vivo extrapolation techniques. By elucidating the interaction between CYP17A1 and abiraterone/D4A, we demonstrate how inadequate characterization of target association-dissociation kinetics precludes an accurate determination of the in vivo time course of drug action.
For the first time, our results chronicle the profiling of abiraterone and its active metabolite D4A as slow-, tight-binding inhibitors of steroidogenic CYP17A1 adhering to a two-step induced-fit mechanism. As highlighted in Table 1, the conversion from the initial EI complex to the final EI* was accompanied by significant (>50-fold) increases in inhibitory potencies (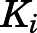 to
to 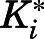 ) for both CYP17A1-mediated reactions. Hence, our results underscore how functional comparisons of compound inhibitory activity with the conventional assumption of rapid equilibrium may hinder detection of the time-dependent effects that are essential for holistic assessment of the true inhibitory potencies of nonclassic inhibitors. Notably, the Ki and Ki* values obtained largely reconcile the considerable incongruity observed in previous studies examining the nature and potency of CYP17A1 inhibition by abiraterone and D4A. Ki values quantifying the initial mixed-mode inhibition of progesterone 17α-hydroxylation by abiraterone (50.7 nM) and D4A (23.4 nM) are comparable to those obtained by Garrido et al. (2014) (Ki = 27 nM for abiraterone and 22 nM for D4A). Conversely, spectral ligand binding assays monitoring time- and concentration-dependent heme coordination have implicated abiraterone and D4A as tight-binding inhibitors with extremely high affinity for CYP17A1 (Garrido et al., 2014). This alternative approach measures the dissociation of I from
) for both CYP17A1-mediated reactions. Hence, our results underscore how functional comparisons of compound inhibitory activity with the conventional assumption of rapid equilibrium may hinder detection of the time-dependent effects that are essential for holistic assessment of the true inhibitory potencies of nonclassic inhibitors. Notably, the Ki and Ki* values obtained largely reconcile the considerable incongruity observed in previous studies examining the nature and potency of CYP17A1 inhibition by abiraterone and D4A. Ki values quantifying the initial mixed-mode inhibition of progesterone 17α-hydroxylation by abiraterone (50.7 nM) and D4A (23.4 nM) are comparable to those obtained by Garrido et al. (2014) (Ki = 27 nM for abiraterone and 22 nM for D4A). Conversely, spectral ligand binding assays monitoring time- and concentration-dependent heme coordination have implicated abiraterone and D4A as tight-binding inhibitors with extremely high affinity for CYP17A1 (Garrido et al., 2014). This alternative approach measures the dissociation of I from  (Kd = 2.6 nM for abiraterone and <1 nM for D4A) and is analogous to the Ki* established in the present work (Table 1). Independently, PMF calculations from MD simulations yielded binding affinity estimates of abiraterone with CYP17A1 (−14.5 kcal/mol) that also verified the experimentally measured binding affinity (−12.5 and −12.8 kcal/mol for
(Kd = 2.6 nM for abiraterone and <1 nM for D4A) and is analogous to the Ki* established in the present work (Table 1). Independently, PMF calculations from MD simulations yielded binding affinity estimates of abiraterone with CYP17A1 (−14.5 kcal/mol) that also verified the experimentally measured binding affinity (−12.5 and −12.8 kcal/mol for 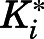 = 0.42 and 0.39 nM). The convergence of results from MD simulations and biochemical analyses confirms the formation of the
= 0.42 and 0.39 nM). The convergence of results from MD simulations and biochemical analyses confirms the formation of the  complex between abiraterone and CYP17A1.
complex between abiraterone and CYP17A1.
Our kinetic experiments have confirmed that the high-affinity interactions of abiraterone/D4A with CYP17A1 can be attributed to a two-step induced-fit mechanism. However, the conservation of the global conformation of CYP17A1 in the presence of abiraterone (Supplemental Fig. 5, A and B), D4A (Supplemental Fig. 5, C and D), and various ligands (Supplemental Fig. 6) (DeVore and Scott, 2012; Petrunak et al., 2014, 2017) suggest that abiraterone or D4A can bind in the active site in the same orientation as that observed in the X-ray crystal structure, which paradoxically renders the proposed induced-fit mechanism untenable. Nevertheless, molecular docking simulations are static by nature and are unable to simulate protein flexibility throughout binding (Salmaso and Moro, 2018). Indeed, cytochrome P450 enzymes with large binding sites have been demonstrated to undergo dramatic conformational changes to accommodate and optimize the binding of ligands with diverse structures and shapes (Ekroos and Sjogren, 2006; Nair et al., 2016; Sevrioukova and Poulos, 2017).
MD simulations demonstrated that the pyridine ring of abiraterone occupies an unfilled space of CYP17A1 between Val366 (on a K-L loop) and Val483 (β 4 loop) (DeVore and Scott, 2012), where its steroid nucleus binds near parallel to the plane of heme (pose B in Fig. 4C). This parallel pose has been comparatively observed for steroidal ligands binding to CYP19A1 [e.g., Protein Data Bank 3EQM, 5JKW; (Ghosh et al., 2018)].
To probe the molecular basis of the two binding poses A and B, we investigated and established conformational changes in the F-helix (Supplemental Table 3) and the region connecting the F- and G-helices (Nair et al., 2016), which are consistent with the secondary X-ray crystal structure of abiraterone-bound CYP17A1 (Supplemental Fig. 7). Our MD simulations further demonstrated fluctuations of ligand-bound CYP17A1 active site volume (496–1023 Å3) with flexibilities noted for the F-helix, K-L loop, and β4 loop region. Taken together, these conformational flexibilities possibly influence the binding modes of abiraterone.
In the X-ray crystal structure (pose A in Fig. 4A), abiraterone binds to Asn202 (F-helix) within CYP17A1 active site via H-bonding, whereas the roles of Arg239 and Asp298 were unclear (DeVore and Scott, 2012). It was observed during MD simulations that abiraterone adopts pose B when Arg239 (G-helix) and Asp298 (I-helix) are sufficiently close to form a salt bridge (∼3 Å), but abiraterone tends to assume pose A as the distance between these residues increases. Tyr201 was found to be the most flexible side chain in the CYP17A1 binding site, with a root mean square fluctuation value of 2.8 Å. The side chain dihedral angle of Tyr201, specifically χ1 (N-CA-CB-CG), repositions from a near gauche (∼70°) to a near trans (∼170°) conformation during simulations. This conformational rearrangement of Tyr201 favors the charge interaction between Arg239 and Asp298 and, hence, adoption of pose B. Thus, Tyr201, Arg239, and Asp298 play a crucial role in the reversible binding of abiraterone to CYP17A1. Collectively, MD simulations have provided insights into conformational adaptations within CYP17A1 that could justify the two predominant modes of abiraterone binding.
Apart from structural determinants, our findings underscore that equilibrium dissociation constants (thermodynamic selectivity) are not unequivocal indicators of the lifetime of drug-target complex (kinetic selectivity). This recognition has prompted the recent emergence of residence time (1/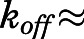 1/
1/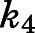 ) as the paramount indicator of PD efficacy (Copeland et al., 2006; Copeland, 2016; Tonge, 2018). Interpreting the prolonged residence time obtained for abiraterone in the context of previous dialysis and jump dilution studies demonstrates the validity of our estimates (Jarman et al., 1998; Garrido et al., 2014).
) as the paramount indicator of PD efficacy (Copeland et al., 2006; Copeland, 2016; Tonge, 2018). Interpreting the prolonged residence time obtained for abiraterone in the context of previous dialysis and jump dilution studies demonstrates the validity of our estimates (Jarman et al., 1998; Garrido et al., 2014).
Nevertheless, the potential contribution of kinetic selectivity to in vivo efficacy remains complex because of dynamic changes in drug concentration, target turnover, and the relationship between target occupancy and effect (i.e., target vulnerability) (Daryaee and Tonge, 2019). Selection of a 500-mg dose for comparison with the standard 1000-mg dose of AA was guided by results of a phase 1 single-dose study in castrate males in which target testosterone suppression was achieved and sustained for days 2–5 post-therapy (O’Donnell et al., 2004). Here, incorporating CYP17A1 turnover as well the plasma PK of abiraterone in relation to the lifetime of abiraterone-CYP17A1 complex, we first established that at both 1000- and 500-mg doses of AA, apparent protracted CYP17A1 engagement by abiraterone could be achieved in vivo (Fig. 6A). In subsequent PK-PD simulations, dynamic CYP17A1 engagement was correlated with in vitro androgen suppression. Interestingly, we further demonstrated comparable attenuations in intracellular DHEA-S levels associated with both 1000 and 500 mg AA doses (Fig. 6C). Taken together, our kinetic selectivity approach facilitated rationalization of a clinically observed PK-PD uncoupling highlighted by Szmulewitz et al. (2018) and advocates a lower dosing requirement of abiraterone than that mandated by the MTD approach. Given that a 500-mg dose of AA administered fasted was shown to yield comparable PK with 250 mg of AA administered with a low-fat meal (Fig. 7), dose reduction could represent a viable alternative to circumvent the high variability in oral bioavailability that often accompanies the food effect in drug PK.
With abiraterone slated for treating newly diagnosed high-risk mHSPC, its average duration of treatment could span up to 24 months (Fizazi et al., 2017). Therefore, the pharmacoeconomic implications of any potential dose reduction could be substantial, as abiraterone acetate has an approximate retail cost of USD 10,000 per month (Szmulewitz et al., 2018). Moreover, the inhibition of the 17α-hydroxylase reaction by abiraterone has been shown to produce dose-dependent mineralocorticoid-related toxicities that necessitates concomitant prednisone administration. Hence, potential dose reductions could also be instrumental in enhancing the safety of abiraterone therapy.
It is important to note that apparent occupancy-time profiles (Fig. 6A) were generated on the assumption of equivalent total tissue and plasma concentrations. This approach deviates from that advocated by the free drug hypothesis, in which drug molecules bind to plasma and tissue proteins, resulting in a reduction in free, pharmacologically active concentrations necessary for target engagement. Based on the free drug theory, with assumptions of steady-state PK and the dominance of passive diffusion processes, the unbound partition coefficient (Kp,uu) for a noneliminating tissue will equal unity (Cu,t/Cu,p = ∼1) (Zhang et al., 2019). Expectedly, when unbound plasma concentrations were used as a surrogate for unbound tissue concentrations to effect CYP17A1 engagement, the extent of CYP17A1 occupancy over time was significantly diminished (Supplemental Fig. 8A). Nevertheless, in the case of abiraterone, we believe that asymmetry of free drug concentrations between target tissues and plasma at steady state is a distinct possibility. Firstly, corroborating Mostaghel et al. (2017), our preliminary analyses demonstrated potential organic anion transporting polypeptide 1B3–mediated abiraterone uptake (Supplemental Fig. 8B). Coupled with evidence supporting increased expression of organic anion transporting polypeptide 1B3 in metastatic lesions from prostate cancer, intratumoral levels of abiraterone could be reasonably higher than that predicted by Kp,uu (Schulte and Ho, 2019). Additionally, another key assumption underpinning the free drug hypothesis is that interactions between the drug and the receptor are reversible. Consequently, concentration gradient can be the sole factor invoked to describe free drug permeation. However, with the tight-binding of abiraterone to CYP17A1, it is possible that with adequate abiraterone concentrations exceeding the target covalent binding threshold, a separate concentration gradient can be re-established with the excess free abiraterone (Zhang et al., 2019). This postulation is also supported by distribution studies performed in rats, in which the highest 14C-abiraterone concentrations detected within the adrenal gland were 15–35 times the corresponding blood concentration (European Medicines Agency, 2011). Taken together, there remains considerable uncertainties in occupancy determinations because of the inability to accurately estimate the molar ratio of intracellular free abiraterone versus CYP17A1. As such, the threshold and strength of correlation between target occupancy and effect as described by a target vulnerability function remains poorly defined. Consequently, direct quantification of in vivo target engagement via imaging techniques such as positron emission tomography becomes imperative to inform and correct estimates of apparent CYP17A1 occupancy.
In the case of D4A, residence time effects were also expected, given the prolonged CYP17A1 engagement observed in in vitro kinetic analyses. Nevertheless, prolongation of phenotypic response after D4A washout was not observed (data not shown). This apparent incongruity could be attributed to the susceptibility of the Δ4,3-keto structure of D4A to irreversible 5α-reduction and 5β-reduction within the tumor microenvironment (Li et al., 2016). In fact, direct incubation of D4A in LAPC4 prostate cancer cell line for 48 hours demonstrated substantial downstream conversion of D4A to the initial 5α-reduced metabolite (3-keto-5α-abiraterone), which is similarly present at higher concentrations than D4A in patients with mCRPC taking abiraterone (Li et al., 2016). Hence, it is possible that the elimination of D4A is not rate-limiting, diminishing the utility of a prolonged residence time in extending its PD durability (Dahl and Akerud, 2013).
In conclusion, this study provides compelling evidence that abiraterone is a slow-, tight-binding inhibitor of steroidogenic CYP17A1. Our in vitro enzyme kinetic analyses provided quantitative description of pertinent kinetic parameters characterizing the two-step binding that are entirely consistent with the results of MD simulations. Cellular washout experiments provided further corroborative evidence for the protracted residence time of abiraterone on CYP17A1. In addition, MD simulations offered insights into the fast, reversible step preceding tight-binding that was not apparent from either X-ray crystallography or molecular docking simulations. The long dissociation t1/2 of both abiraterone and D4A reflects protracted residence time on CYP17A1, which in turn creates the awareness that the incorporation of residence time along with PK parameters is imperative in the mechanistic PK-PD modeling of abiraterone to optimize mCRPC pharmacotherapy.
Acknowledgments
This research was undertaken with the assistance of resources from the National Computational Infrastructure, which is supported by the Australian government. The authors also acknowledge Flinders University’s High Performance Computing resources for this work.
Abbreviations
- AA
abiraterone acetate
- ADT
androgen deprivation therapy
- AR
androgen receptor
- ATCC
American Type Culture Collection
- AUC
area under the plasma concentration-time curve
- CI
confidence interval
- COM
center of mass
- D4A
∆4 abiraterone
- DHEA
dehydroepiandrosterone
- DHEA-S
dehydroepiandrosterone-sulfate
- EI
initial enzyme-inhibitor encounter complex
- EI*
the final high-affinity enzyme-inhibitor complex
- k1
rate constant for the initial binding of inhibitor to enzyme
- k2
rate constant for the dissociation of the initial EI complex
- k3
forward isomerization rate constant for the conversion of EI to EI*
- k4
reverse isomerization rate constant for the conversion of EI* to EI
- Ki
inhibition constant for the initial EI complex
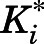
inhibition constant for the final EI* complex
- Ki,app
apparent inhibition constant for the initial EI complex
- Kobs
first-order rate constant for onset of inhibition
- LC/MS/MS
liquid chromatography tandem mass spectrometry
- mCRPC
metastatic castration-resistant prostate cancer
- MD
molecular dynamics
- mHSPC
metastatic hormone-sensitive prostate cancer
- MTD
maximal tolerated dose
- PD
pharmacodynamics
- PK
pharmacokinetics
- PMF
potential of mean force
- rCYP17A1
recombinant CYP17A1
- t1/2
half-life
Authorship Contributions
Participated in research design: Cheong, Nair, Neo, Tu, Lin, Fan, Miners, Chan.
Conducted experiments: Cheong, Nair, Neo, Tu, Lin, Fan, Miners, Chan.
Contributed new reagents or analytic tools: Tu, Chai.
Performed data analysis: Cheong, Nair, Neo, Tu, Lin, Fan, Szmulewitz, Peer, Figg, Miners, Chan.
Wrote or contributed to the writing of the manuscript: Cheong, Nair, Neo, Tu, Lin, Chiong, Esuvaranathan, Fan, Chai, Miners, Chan.
Footnotes
This work was supported by the Singapore Ministry of Education Tier 1 Academic Research Funding [Grant R-148-000-249-114] and the National University of Singapore President’s Graduate Fellowship to E.J.Y.C. and the National University of Singapore, Department of Pharmacy, Final Year Project Funding provided to R.W.Y.N. F.L. and H.F. gratefully acknowledge the financial support from the Biomedical Research Council of the Agency for Science, Technology and Research, Singapore.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. [Google Scholar]
- Acharya M, Bernard A, Gonzalez M, Jiao J, De Vries R, Tran N. (2012) Open-label, phase I, pharmacokinetic studies of abiraterone acetate in healthy men. Cancer Chemother Pharmacol 69:1583–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed KEM, Frøysa HG, Karlsen OD, Sagen JV, Mellgren G, Verhaegen S, Ropstad E, Goksøyr A, Kellmann R. (2018) LC-MS/MS based profiling and dynamic modelling of the steroidogenesis pathway in adrenocarcinoma H295R cells. Toxicology in Vitro 52:332–341. [DOI] [PubMed] [Google Scholar]
- Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, et al. (2008) Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 26:4563–4571. [DOI] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. [DOI] [PubMed] [Google Scholar]
- Chen R, Chung S-H. (2012) Binding modes and functional surface of anti-mammalian scorpion α-toxins to sodium channels. Biochemistry 51:7775–7782. [DOI] [PubMed] [Google Scholar]
- Copeland RA. (2013) Slow binding inhibitors, Evaluation of Enzyme Inhibitors in Drug Discovery pp 203–244, John Wiley & Sons, Inc, Hoboken NJ. [Google Scholar]
- Copeland RA. (2016) The drug-target residence time model: a 10-year retrospective. Nat Rev Drug Discov 15:87–95. [DOI] [PubMed] [Google Scholar]
- Copeland RA. (2000) Time-dependent inhibition, Enzymes: A Practical Introduction to Structure Mechanism, and Data Analysis pp 318–349, Wiley, New York. [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. (2006) Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov 5:730–739. [DOI] [PubMed] [Google Scholar]
- Dahl G, Akerud T. (2013) Pharmacokinetics and the drug-target residence time concept. Drug Discov Today 18:697–707. [DOI] [PubMed] [Google Scholar]
- Darden TA, York DM, Pedersen LG. (1993) Particle Mesh Ewald: An Nlog (N) Method for Ewald Sums in Large Systems. The Journal of Chemical Physics 98:10089–10092. [Google Scholar]
- Daryaee F, Tonge PJ. (2019) Pharmacokinetic-pharmacodynamic models that incorporate drug-target binding kinetics. Curr Opin Chem Biol 50:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVore NM, Scott EE. (2012) Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 482:116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekroos M, Sjögren T. (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci USA 103:13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. (1995) A smooth particle mesh Ewald method. J Chem Phys 103:8577–8593. [Google Scholar]
- European Medicines Agency (2011) Assessment report for Zytiga (abiraterone).
- European Medicines Agency (2017) EMA/816845/2017 - Assessment Report, Zytiga. https://www.ema.europa.eu/en/documents/variation-report/zytiga-h-c-2321-ii-0047-epar-assessment-report-variation_en.pdf.
- Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, Özgüroğlu M, Ye D, Feyerabend S, Protheroe A, et al. LATITUDE Investigators (2017) Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med 377:352–360. [DOI] [PubMed] [Google Scholar]
- Garrido M, Peng HM, Yoshimoto FK, Upadhyay SK, Bratoeff E, Auchus RJ. (2014) A-ring modified steroidal azoles retaining similar potent and slowly reversible CYP17A1 inhibition as abiraterone. J Steroid Biochem Mol Biol 143:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Egbuta C, Lo J. (2018) Testosterone complex and non-steroidal ligands of human aromatase. J Steroid Biochem Mol Biol 181:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Chen R, Chung S-H. (2013) Computational methods of studying the binding of toxins from venomous animals to biological ion channels: theory and applications. Physiol Rev 93:767–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WP, Mostaghel EA, Nelson PS, Montgomery B. (2009) Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol 6:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WG. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys Rev A Gen Phys 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- Hub JS, de Groot BL, van der Spoel D. (2010) g_wham—a free weighted histogram analysis Implementation including robust error and autocorrelation estimates. J Chem Theory Comput 6:3713–3720. [Google Scholar]
- Janssen (2019) ZYTIGA® (abiraterone acetate) [Highlights of Prescribing Information].
- Jarman M, Barrie SE, Llera JM. (1998) The 16,17-double bond is needed for irreversible inhibition of human cytochrome p45017alpha by abiraterone (17-(3-pyridyl)androsta-5, 16-dien-3beta-ol) and related steroidal inhibitors. J Med Chem 41:5375–5381. [DOI] [PubMed] [Google Scholar]
- Ji Y, Jin JY, Hyman DM, Kim G, Suri A. (2018) Challenges and opportunities in dose finding in oncology and immuno-oncology. Clin Transl Sci 11:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. (1992) THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem 13:1011–1021. [Google Scholar]
- Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin M-E, Auchus RJ, Sharifi N. (2016) Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature 533:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N. (2015) Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 523:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malde AK, Zuo L, Breeze M, Stroet M, Poger D, Nair PC, Oostenbrink C, Mark AE. (2011) An automated force field Topology builder (ATB) and repository: version 1.0. J Chem Theory Comput 7:4026–4037. [DOI] [PubMed] [Google Scholar]
- Minasian L, Rosen O, Auclair D, Rahman A, Pazdur R, Schilsky RL. (2014) Optimizing dosing of oncology drugs. Clin Pharmacol Ther 96:572–579. [DOI] [PubMed] [Google Scholar]
- Ming DS, Pham S, Deb S, Chin MY, Kharmate G, Adomat H, Beheshti EH, Locke J, Guns ET. (2014) Pomegranate extracts impact the androgen biosynthesis pathways in prostate cancer models in vitro and in vivo. J Steroid Biochem Mol Biol 143:19–28. [DOI] [PubMed] [Google Scholar]
- Morrison JF, Walsh CT. (1988) The behavior and significance of slow-binding enzyme inhibitors, in Advances in Enzymology and Related Areas of Molecular Biology (Meister A. 201–301, John Wiley & Sons, Inc., New York. [DOI] [PubMed] [Google Scholar]
- Mostaghel EA, Cho E, Zhang A, Alyamani M, Kaipainen A, Green S, Marck BT, Sharifi N, Wright JL, Gulati R, et al. (2017) Association of tissue abiraterone levels and SLCO genotype with intraprostatic steroids and pathologic response in men with high-risk localized prostate cancer. Clin Cancer Res 23:4592–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, et al. (2007) Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res 67:5033–5041. [DOI] [PubMed] [Google Scholar]
- Nair PC, McKinnon RA, Miners JO. (2016) Cytochrome P450 structure-function: insights from molecular dynamics simulations. Drug Metab Rev 48:434–452. [DOI] [PubMed] [Google Scholar]
- Nosé S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys 81:511–519. [Google Scholar]
- O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, et al. (2004) Hormonal impact of the 17α-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer 90:2317–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M, Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. [Google Scholar]
- Petrunak EM, DeVore NM, Porubsky PR, Scott EE. (2014) Structures of human steroidogenic cytochrome P450 17A1 with substrates. J Biol Chem 289:32952–32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrunak EM, Rogers SA, Aubé J, Scott EE. (2017) Structural and functional evaluation of clinically relevant inhibitors of steroidogenic cytochrome P450 17A1. Drug Metab Dispos 45:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pienta KJ, Bradley D. (2006) Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res 12:1665–1671. [DOI] [PubMed] [Google Scholar]
- Porubek D. (2013) CYP17A1: a biochemistry, chemistry, and clinical review. Curr Top Med Chem 13:1364–1384. [DOI] [PubMed] [Google Scholar]
- R Core Team (2013) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Sachs JR, Mayawala K, Gadamsetty S, Kang SP, de Alwis DP. (2016) Optimal dosing for targeted therapies in oncology: drug development cases leading by example. Clin Cancer Res 22:1318–1324. [DOI] [PubMed] [Google Scholar]
- Salmaso V, Moro S. (2018) Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: an overview. Front Pharmacol 9:923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, Antonarakis ES, Beer TM, Carducci MA, Chi KN, et al. Prostate Cancer Clinical Trials Working Group 3 (2016) Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the prostate cancer clinical trials working group 3. J Clin Oncol 34:1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF. (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J 40:843–856. [DOI] [PubMed] [Google Scholar]
- Schmidtke P, Bidon-Chanal A, Luque FJ, Barril X. (2011) MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 27:3276–3285. [DOI] [PubMed] [Google Scholar]
- Schulte RR, Ho RH. (2019) Organic anion transporting polypeptides: emerging roles in cancer pharmacology. Mol Pharmacol 95:490–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova IF, Poulos TL. (2017) Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc Natl Acad Sci USA 114:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi J, Csizmadia F. (2007) Calculating the pKa values of small and large molecules, in American Chemical Society Spring meeting. [Google Scholar]
- Szmulewitz RZ, Peer CJ, Ibraheem A, Martinez E, Kozloff MF, Carthon B, Harvey RD, Fishkin P, Yong WP, Chiong E, et al. (2018) Prospective international randomized phase II study of low-dose abiraterone with food versus standard dose abiraterone in castration-resistant prostate cancer. J Clin Oncol 36:1389–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonge PJ. (2018) Drug-target kinetics in drug discovery. ACS Chem Neurosci 9:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrie GM, Valleau JP. (1977) Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J Comput Phys 23:187–199. [Google Scholar]
- Vasaitis TS, Bruno RD, Njar VCO. (2011) CYP17 inhibitors for prostate cancer therapy. J Steroid Biochem Mol Biol 125:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G. (2017) Distinct in vivo target occupancy by bivalent- and induced-fit-like binding drugs. Br J Pharmacol 174:4233–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Hu Q. (2014) CYP17 inhibitors-abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat Rev Urol 11:32–42. [DOI] [PubMed] [Google Scholar]
- Zhang D, Hop CECA, Patilea-Vrana G, Gampa G, Seneviratne HK, Unadkat JD, Kenny JR, Nagapudi K, Di L, Zhou L, et al. (2019) Drug concentration asymmetry in tissues and plasma for small molecule-related therapeutic modalities. Drug Metab Dispos 47:1122–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]