

Abstract
The sphingosine kinase-1/sphingosine-1-phosphate pathway is linked with the cancer progression and survival of the chemotherapy-challenged cells. Sphingosine kinase-1 (SphK1) has emerged as an attractive drug target, but their inhibitors from natural sources are limited. In this study, we have chosen harmaline, one of the β-carboline alkaloids, and report its mechanism of binding to SphK1 and subsequent inhibition. Molecular docking combined with fluorescence binding studies revealed that harmaline binds to the substrate-binding pocket of SphK1 with an appreciable binding affinity and significantly inhibits the kinase activity of SphK1 with an IC50 value in the micromolar range. The cytotoxic effect of harmaline on non-small-cell lung cancer cells by MTT assay was found to be higher for H1299 compared to A549. Harmaline induces apoptosis in non-small-cell lung carcinoma cells (H1299 and A549), possibly via the intrinsic pathway. Our findings suggest that harmaline could be implicated as a scaffold for designing potent anticancer molecules with SphK1 inhibitory potential.
1. Introduction
Lung cancer has emerged as a leading cause of cancer deaths1 with 5 year survival rate lower than 15%.2 Smoking is the major cause of lung cancer with new causes that emerged as radon, arsenic, asbestos, and air pollution. They altogether synergistically increase the risk of lung cancer.3 It has two major histological subtypes: small-cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC), which accounts for more than 85% of all the cases among lung cancers.4 The most common subtypes of NSCLCs comprise large cell carcinoma, adenocarcinoma, and squamous cell carcinoma.5 The high mortality in NSCLC is associated with complexity in early diagnosis and its high rate of proliferation and metastasis.6,7 As a result, the current treatments for NSCLC are not fully successful.8 Hence, research in the area of development of effective diagnostics tools, anti-NSCLC molecules, and identification of biomarkers and novel molecular targets against non-small-cell lung cancer is much needed.9,10
Sphingosine kinase (SphK) is an evolutionarily conserved and unique class of lipid kinase11 that catalyzes the reversible phosphorylation of sphingosine to produce sphingosine-1-phosphate (S1P).12 SphKs have two isoforms, i.e., SphK1 and SphK2, which differ in subcellular location and function.13 SphK1 promotes cell proliferation and survival, while SphK2 induces cell apoptosis. The absence of both isoforms causes fatal effects, whereas the presence of one functional isoform circumvents the deleterious outcomes, as observed in knockout studies with mice.14,15 SphK1 is activated by a range of agonists,16 including growth factors, cytokines, or oncogenes via phosphorylation of serine 225,17 which causes it to delocalize to the plasma membrane.18
The S1P produced by SphKs is an important lipid mediator involved in multiple signaling pathways associated with mitogenesis, cell survival, cell migration, inflammation, and angiogenesis and acts extracellularly via interaction with cell-surface G-protein-coupled receptors (S1PR1–S1PR5) or through intracellular targets such as HDACs and TRAF2.19,20 The intracellular levels of S1P are tightly regulated by sphingolipid rheostat, which is essential for maintaining homeostasis in the cellular environment. The three core sphingolipid metabolites, viz., ceramide, sphingosine, and S1P, are the key players of sphingolipid rheostat, which dictates the metabolic fate of the cell and thus plays an important role in cell growth, motility, differentiation, and apoptosis.20,21 S1P promotes cell proliferation, while ceramide and sphingosine mediate cell cycle arrest and apoptosis. Thus, S1P plays a key role in regulating tumor cell growth, survival, invasion, and angiogenesis. Henceforth, modulation of the S1P levels in the cellular environment has become an important strategy for the treatment of cancer and other S1P-related pathologies.21,22
SphKs regulate S1P levels in cells and maintain a balance between prosurvival and apoptotic signaling. Aberrant expression of SphK1 leans the balance toward the prosurvival pathway.22,23 SphK1 has emerged as a potential anticancer target24 since it contributes to cancer progression as it is overexpressed in lung,25 colon,26 breast,27,28 and prostate29 cancer cells. SphK1 is also linked to the chemoresistance of the cancer cells as its overexpression inhibited chemotherapy-driven apoptosis in breast cancer cells (MCF-7) on treatment with anthracyclines.30 Likewise, it is responsible for the chemoresistance in many other cancer types, for example, in acute myeloid (HL-60)31 and prostate cancer cells (LNCaP).32 SphK1 causes tumor progression, invasion, metastasis, and chemo- and radioresistance in NSCLC, as reported in previous studies.25,33−35 Therefore, SphK1 is extensively used as a target for the development of anticancer therapeutics.
The earliest discovered SphK1 inhibitors were sphingosine analogs or derivatives, for example, dihydrosphingospine (DHS) and N,N-dimethylsphingosine (DMS). Both have been used for modulating S1P biosynthesis despite being weak and nonselective SphK inhibitors.36 Fingolimod (FTY720) has been clinically approved for multiple sclerosis as a functional antagonist of S1P receptors.37 FTY720 is shown to be a potent SphK1 inhibitor that induces apoptosis in prostate cancer cells.38 The crystal structures of SphK1 with sphingosine and some of the known potent inhibitors (PF-543 and SK1-II) have been useful in designing and development of selective inhibitors with improved bioavailability and less cytotoxicity.12,39,40 Some of the SphK1 inhibitors and their IC50 values are represented in Figure 1.
Figure 1.

Structures of SphK1 inhibitors.
Recently, an increasing interest in the discovery of novel antitumor agents against SphK1 from the natural resource is reported.41,42 The well-established literature validates the antioxidant and antiproliferative properties of natural products, and thus, they could be explored as an important source of new drugs for various diseases including cancer.43−46 Many scientists are trying to isolate active natural compounds that can be used for therapeutic purposes after required structural modifications.47
There are a few natural products and their derivatives that impede the growth of various cancerous cells via inhibition of the SphK1/S1P pathway.48 For instance, peretinoin inhibits the expression and activity of SphK1 in the human hepatoma cell line Huh-7.49 Likewise, 2-epi-jaspine B shows potent antitumor activity and has been used as a lead for the development of selective inhibitors of SphK1. Moreover, analog YHR17 developed from 2-epi-jaspine B possesses 125-fold selectivity over SphK2 and inhibits A375 cell line proliferation at very low concentrations (IC50 = 0.68 to 5.68 μM).50 Icaritin, a flavonoid, inhibits SphK1 activity and is cytotoxic to hepatocellular carcinoma both in vitro and in vivo.(51) Hispidulin, another flavonoid, is known to inhibit SphK1 activity and induces apoptosis in Caki-2 and A498 cell lines.52 As a result, natural products are emerging as an important source for potential leads and drug molecules for targeting cancer via SphK1 inhibition.
In the present investigation, we tried to explore the anticancer properties of harmaline (C13H15ON2), which is a major β-carboline alkaloid present in the seeds and roots of Peganum harmala.(53) The β-carboline alkaloids and their derivatives have gathered larger attention because of their prominent anticancer activity and inhibitory activity against DNA topoisomerases and cyclin-dependent kinases (CDKs).54,55 Among them, harmaline has been widely studied for its pharmacological and numerous clinical benefits including protection from radiation, reduction of inflammation, relief from psoriasis, immunosuppression, and antitumor effects.56,57 It also acts as an inhibitor of histamine N-methyltransferase58 and exhibits the ionotropic effect.59 Harmaline is responsible for reversible inhibition of monoamine oxidase.60 In another study, the harmaline and harmalol effects were evaluated on the digoxin-induced cytochrome CYP1A1 in HepG2 cells.61 The harmala alkaloids significantly inhibited CYP1A1 through transcriptional and posttranslational mechanisms.62 Harmaline exhibits potential anticancer activity in multiple cancer cell lines with a low risk of toxic side effects, as reported by previous studies.63,64 All of these studies direct that harmaline possesses the potential for using it as a novel antioxidant and antitumor agent in anticancer therapy.57
Here, we have performed isothermal titration calorimetry (ITC) and fluorescence binding studies to measure the binding affinity of harmaline with SphK1. ATPase assay was further performed to see the SphK1 inhibitory potential of harmaline. Additionally, the anticancer properties of harmaline toward human alveolar basal epithelial adenocarcinoma (A549) and lymph node-derived cancer (H1299) cells and cytotoxicity against human embryonic kidney cells (HEK293) were estimated by MTT assay. The apoptotic effect of harmaline on both NSCLC cells was investigated and was found to be linked with caspase-3 activation. To understand the binding pattern and inhibition mechanism, we identify the important amino-acid residues involved in harmaline–SphK1 binding while utilizing the molecular docking approach.
2. Results
2.1. Expression and Purification of SphK1
SphK1 gene construct was cloned and expressed in BL21 Gold (DE3) cells. The inclusion bodies were solubilized with the help of N-lauroyl sarcosine and the solubilized protein was loaded on the Ni-NTA column and subsequently purified.65 The purified SphK1 was analyzed on SDS-PAGE, which shows a single band of 45 kDa (Figure S1).
2.2. Fluorescence Binding Studies
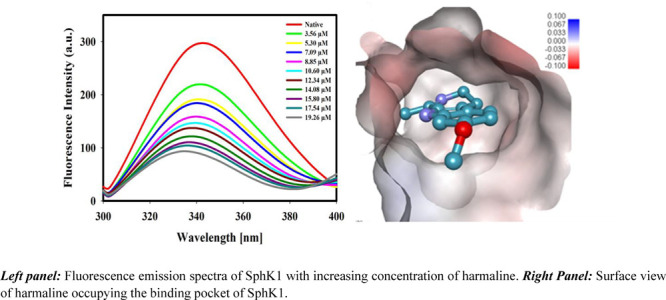
Fluorescence spectroscopy is a sensitive and simple technique for studying protein–ligand interactions and is widely used to determine the binding constants, number of binding sites, and the interaction mechanism.66 The fluorescence spectrum of SphK1 in the presence of different concentrations of harmaline is shown in Figure 2A. A decrease in fluorescence intensity with increasing concentrations of harmaline (3.56–19.26 μM) was observed. A blue shift of 3–4 nm in emission maxima (342 nm) was observed at higher concentrations of harmaline, indicating a change in polarity due to increased hydrophobicity around tryptophan and tyrosine residues of SphK1.67 A well-defined isosbestic point was observed at 396 nm for SphK1, suggesting the formation of a stable harmaline–SphK1 complex.68 The fluorescence spectra were analyzed according to eqs 1–7 and the obtained binding parameters are provided in Table S1. Figure 2B represents a modified Stern–Volmer plot, which estimated that harmaline has a single binding site with a Ka value calculated (using eq 3) to be 7.1 × 105 M–1 (Table S1). Our fluorescence result suggests that harmaline strongly binds to SphK1.
Figure 2.

Fluorescence binding studies of harmaline with SphK1. (A) Fluorescence emission spectra of SphK1 with increasing concentration of harmaline (in μM). (B) Modified Stern–Volmer plot used to estimate the binding constant (Ka) and the number of binding site (n).
2.3. Isothermal Titration Calorimetry Measurements
The actual binding affinity and the nature of the interaction of harmaline with SphK1 were studied with the help of ITC.69 During the ITC experiment, 15 μM SphK1 was titrated with increasing harmaline concentrations. The upper panel represents the experimental raw data describing the exothermic binding reaction between SphK1 and harmaline signified by negative pulses of generated heat (Figure 3). The bottom panel represents integrated heat pulses concerning time as a function of the molar ratio of [harmaline]/[SphK1] during harmaline–SphK1 complexation (Figure 3). The binding isotherm curve represents the total heat exchanged per injection during harmaline–SphK1 interaction. The heat related with the harmaline–buffer interaction was experimentally measured and deducted from the changes in the heat involved during harmaline–SphK1 titration. The thermodynamic parameters were analyzed from the pattern of the binding isotherm for harmaline–SphK1 by fitting it with the four binding site model using in-built Origin software (Table 1). The KD value falls in the micromolar range, suggesting a stronger binding affinity of harmaline toward SphK1. Following the fluorescence binding results, ITC data also suggest a strong binding affinity of harmaline to SphK1.
Figure 3.
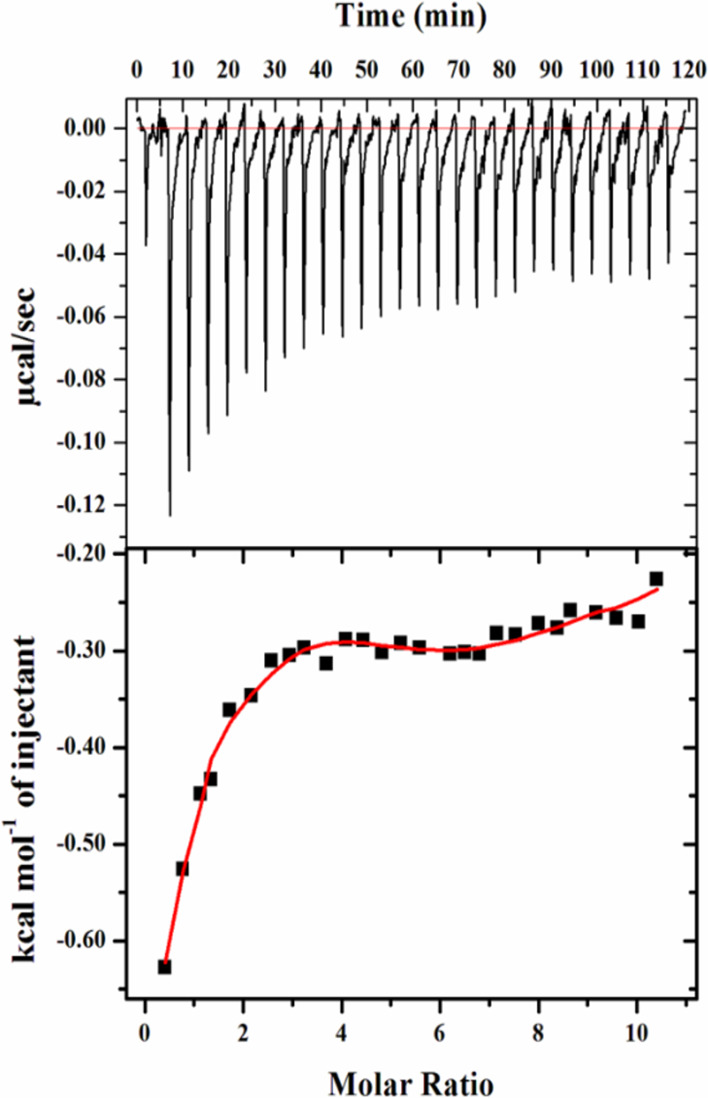
Titration of SphK1 by harmaline in the ITC experiment. The upper panel depicts the heat produced against time for the titration of SphK1 with increasing amount of harmaline. The bottom panel shows the binding isotherm formed by the integration of peak area and its normalization to yield molar enthalpy change. The red color line represents the fitted curve.
Table 1. Thermodynamic Parameters Obtained from the Calorimetric Titration of Harmaline with SphK1.
| complex | number of binding sites (N) | association constant Ka (M–1) | dissociation constant KD (μM) | enthalpy change ΔH (cal/mol) | ΔS (cal/mol/deg) | ΔG (free energy change) (kcal/mol) |
|---|---|---|---|---|---|---|
| harmaline | 4 | Ka1 = 1.89 × 104 ± 1.3 × 103 | KD1 = 52.9 | ΔH1 = −3205 ± 197 | ΔS1 = 8.82 | ΔG1 = −5.83 |
| Ka2 = 1.49 × 105 ± 8.6 × 103 | KD2 = 6.7 | ΔH2 = 1895 ± 257 | ΔS2 = 30.0 | ΔG2 = −7.04 | ||
| Ka3 = 3.50 × 104 ± 2.0 × 103 | KD3 = 28.6 | ΔH3 = 272.3 ± 190 | ΔS3 = 21.7 | ΔG3 = −5.99 | ||
| Ka4 = 4.58 × 103 ± 2.9 × 102 | KD4 = 218.3 | ΔH4 = −9574 ± 467 | ΔS4 = −15.4 | ΔG4 = −4.98 |
2.4. Enzyme Inhibition Assay
The SphK1 inhibitory potential of harmaline was determined by malachite green-based ATPase assay, which showed a significant decrease in the kinase activity on increasing harmaline concentrations (Figure 4A). The ATPase activity of SphK1 was quantified using a phosphate standard curve (Figure S5). A decrease in the activity of SphK1 was plotted as a function of percent inhibition with increasing harmaline concentrations using the formula 100 – (A/A° × 100), where A and A° are enzyme activities of SphK1 in the presence and absence of harmaline, respectively.70 The IC50 value for harmaline was calculated using SigmaPlot 10.0 based on the following equation: Sigmoidal, Hill, three-parameter, f = a × xb/(cb + xb), where f is the percent inhibition, a is the maximum inhibition, b is the hill slope (0.57), c is the IC50, and x is the harmaline concentration.71 The IC50 value was obtained as 6.13 ± 0.33 μM (Figure 4B). The enzyme inhibition result is per fluorescence and ITC data and suggested that harmaline acts as a potential SphK1 inhibitor with an admirable binding affinity.
Figure 4.

Enzyme inhibition assay of SphK1 in the presence of harmaline. (A) Decrease in the activity of SphK1 with increasing harmaline concentration (0 to 30 μM). (B) The plot represents the percent inhibition in the ATPase activity of SphK1 with increasing harmaline concentrations. The IC50 value was calculated by fitting the curve obtained from three independent experiments.
2.5. Cell Viability Studies
The expression of SphK1 is upregulated in many cancers and found to be associated with metastasis of non-small-cell lung cancer cells (NSCLC).34,72 The A549, H1299, and HEK293 cells were chosen for cell viability studies. The cell lines were incubated for about 48 h with increasing dose of harmaline (0–200 μM). MTT assay revealed that harmaline reduces the growth of H1299 and A549 cells selectively without much affecting the growth of HEK293 cells (Figure 5), although the viability of HEK293 cells was affected (up to 15%) at relatively high concentrations (>150 μM). The IC50 values of harmaline were assessed from the cell growth curve and estimated to be 48.16 ± 1.76 and 67.9 ± 2.91 μM for H1299 and A549 cells, respectively. Interestingly, these results direct that H1299 cells are more sensitive to treatment with harmaline compared to A549 cells. The results presented here suggested that harmaline obstructs the growth of cancerous cells and resonates well with earlier studies from other groups.63,64
Figure 5.
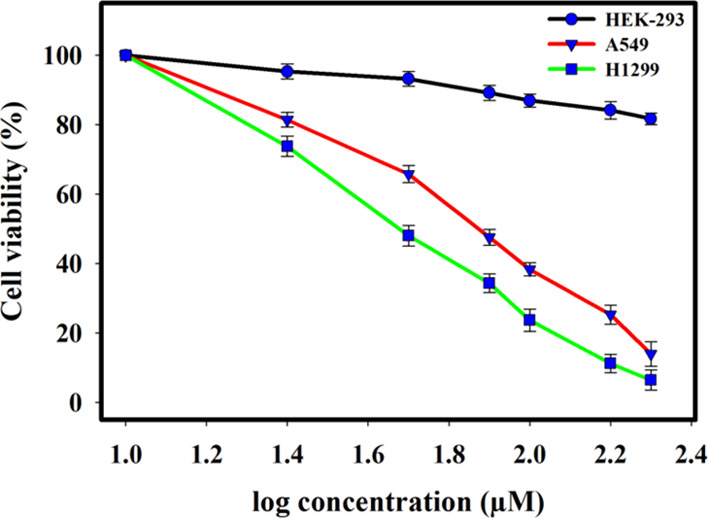
Plot of cell viability with ligand concentrations in MTT assay to assess the anticancer activity of harmaline on A549, H1299, and HEK293 cells. Each cell line was given a dose of harmaline (0–200 μM) for 48 h. The percentage of live cells was evaluated concerning the control cells without any treatment. Each data point shown is the mean ± SD from n = 3.
2.6. Determination of Caspase-3 Activity
The evasion of cell death is one of the 10 important hallmarks of cancer that guides the process of cell immortalization.73 Caspases are the biochemical markers of both the early and late stages of apoptosis.74 Caspase-3, the downstream effector caspase, cleaves and inactivates the proteins important for signal transduction, DNA repair, and cell cycle control.75,76 Caspase-3 activity through colorimetry77 was utilized to deduce the mechanism of apoptotic induction by harmaline in A549 and H1299 cells. The caspase-3 activity was evaluated by monitoring the absorbance of free pNA at wavelength 405 nm since the amount of free pNA is proportional to the amount of caspase activity present in the sample (Figure S6A). The standard pNA curve shown in Figure S6B gives absorbance values of nanomoles of free pNA at varying concentrations. pNA released in nanomole that corresponds to the caspase-3 activity was calculated from the standard curve and the final plot was obtained that clearly shows a gradual increase in the caspase 3 activity per hour (Figure 6). It can be seen that with increasing dose of harmaline, there was a gradual increase in the caspase-3 activity in both cell lines compared to untreated cells. The increase in the caspase 3 activity was more profound in H1299 cells in contrast to A549 cells on treatment with harmaline. Our results correlate well with cell viability studies and direct that the harmaline-induced apoptosis is more likely to proceed through the intrinsic pathway.
Figure 6.

Harmaline induces caspase-3-mediated cell apoptosis in non-small-cell lung cancer cell lines (A549 and H1299). Plot of caspase-3 activity (free pNA liberated per hour) in harmaline-treated A549 and H1299 cancer cells.
2.7. Interaction of Harmaline with SphK1
The molecular docking of harmaline with SphK1 was performed to see the binding pattern and identify important amino-acid residues involved in the ligand binding and subsequent inhibition. A set of 10 docking runs, each with 20 iterations, generates 200 possible docked conformations of the harmaline, which were clustered in 4 plausible binding sites on SphK1 (Figure S7). The major hydrogen bonding interactions between the SphK1-interacting residues and harmaline for all four clusters are also given in Figure S7. At cluster 1, harmaline shows the highest binding affinity (−7.9 kcal/mol) with SphK1 and preferentially occupies the substrate-binding pocket with a significantly higher number of noncovalent interactions. It is interesting to note that harmaline is binding at the same place where cocrystallized d-sphingosine ((2S,3R,4E)-2-aminooctadec-4-ene-1,3-diol) is bound to SphK1.12 Harmaline shows type 1 binding and forms a direct hydrogen bond with Asp178, which is the part of the SphK1 substrate-binding site (Figure 7A). In addition, many hydrogen bonds, alkyl, and van der Waals interactions are offered by Asp81, Gly82, Ser112, Gly113, Ala115, Leu167, Ser168, Asp178 (substrate-binding pocket), Arg191 (ATP-binding site), Phe192, Leu268, Met272, Asp341, and Gly342 to harmaline (Figure 7B). Henceforth, it is understandable that harmaline is bound to SphK1 at the catalytic site via interaction between its active site residues Asp178 and Arg191, which hampers the sphingosine and ATP availability, ultimately leading to SphK1 inhibition.
Figure 7.

Interaction of harmaline with SphK1. (A) Cartoon illustration of docked SphK1–harmaline complex and important residues participating in polar interactions. (B) Detailed interactions and their types formed between harmaline and SphK1.
3. Discussion
SphK1 is upregulated in NSCLC cells and imparts resistance to doxorubicin-mediated apoptosis via activation of the PI3K/Akt/NF-κB pathway.78 Ni et al.33 have linked the high expression of SphK1 with enhancement in the invasiveness potential of A549 cells and another group has associated the migration of the NSCLC cells via AKT pathway by regulation of E-cadherin and Snail expression.35 In another study, overexpression of LncRNA HULC was linked to upregulation of SphK1, which activated the PI3K/Akt pathway, ultimately leading to NSCLC cell proliferation.79 FTY720 is a known S1P modulator, which has been reported recently to reduce the tumor growth in the urethane-induced lung cancer murine model.80 SphK1 is an attractive drug target, but so far, none of the inhibitors have achieved the required potency to treat cancer clinically.48 There still exist challenges to develop potent SphK1 inhibitors that can decrease the S1P levels in target tissues.81 Many drug pharmaceutical companies have started to explore the potential of natural product-based compounds against cancer, inflammation, and other diseases. They have several advantages, given the low toxicity toward normal cells, metabolic stability, and fewer side effects, and the only problem is bioavailability, which can be tackled by nanotechnology-based drug delivery systems.82
Medicinal plants constitute the largest source of natural agents that are proven to have potent effects against diverse forms of diseases, including cancer.83 Several dietary agents have been reported to reduce the growth of prostate cancer cells, for example, epigallocatechin gallate, resveratrol, and polyphenols from green tea via inhibition of S1P pathway.84 Alkaloids have also gained importance in cancer treatment due to their antiproliferative and antiangiogenic effects.85−87 In the current study, we have selected a β-carboline alkaloid, harmaline, because it has well-established pharmacological importance and antioxidant and anticancerous activity. Harmaline is known to induce antitumor effects and apoptosis in HepG2, SGC-7901, HeLa, and MDA-MB-231 breast cancer cells.63,64,88
We observed that harmaline binds to SphK1 with high affinity. The binding enthalpy (ΔH) also clearly reveals that the binding of harmaline with SphK1 is a favorable enthalpy-driven process. Fluorescence binding data analysis revealed a single binding site (Figure 2B and Table S1), while ITC analysis determined four binding sites (Table 1) for harmaline. Moreover, a notable difference in the thermodynamic parameters estimated from ITC and fluorescence was also observed. It is well known that fluorescence spectroscopy measures the local changes that occur around the microenvironment of the fluorophore. Hence, the variation observed in fluorescence and ITC measurements can be because fluorescence probes the change in the microenvironment around the aromatic residues (local changes) upon ligand binding, whereas ITC measures the global change in terms of heat absorbed/released during bond formation or breaking upon protein–ligand interaction.89 In fact, such variations in thermodynamic parameters obtained from fluorescence spectroscopy and ITC have also been observed in various other protein–ligand interaction studies.90,91
Molecular docking predicted four possible binding sites for SphK1 (Figure S7) and cluster 1 (sphingosine binding site) binds with high affinity with harmaline. SphK1 activity was found to be inhibited by harmaline with IC50 calculated to be 6.1328 ± 0.33 μM (Figure 4B). Harmaline is a tricyclic aromatic compound and has a methoxyl ligand at the C-7 position and a methyl group at the C-1 position.92 The H-bond donor groups of harmaline form hydrogen bonding interactions with Asp178 (sphingosine-binding residue), Ser168, and Ser112, which are active site residues of SphK1 (Figure 7B). Additionally, harmaline forms van der Waals interaction with ATP-binding site residues including Arg191, Asp81, and Gly82. It is well known that Asp81 is a catalytically critical residue for SphK1 kinase function and plays an important role in the phosphoryl transfer step, while Gly82 is essential for ATP binding.12 Henceforth, harmaline reduces the substrate as well as ATP access to SphK1 and consequent inhibition.
Harmaline treatment proved to be cytotoxic toward NSCLCs (A549 and H1299), while a little (∼15%) loss in the cell viability of HEK293 cells was observed at concentrations >150 μM (Figure 5). P. harmala seed extracts when administered at a dose of 150 mg/kg to albino mice repeatedly for 3 weeks is reported to induce hemorrhage, degeneration, and necrosis in kidney and liver cells.93 The IC50 value of harmaline against HEK293 as studied by Storch et al.94 was found to be above 200 μmol/L after 72 h of treatment. Thus, based on the previous reports and from our studies, we can suggest that harmaline may be harmful at a relatively higher concentration (>200 μM). Interestingly, we observed a significant difference in the sensitivity of the H1299 and A549 cells toward harmaline treatment (Figure 5). Although the cytotoxic effect of harmaline toward A549 cells has been reported and we found a similar effect but with low potency,95,96 this is the first report where harmaline is found to be cytotoxic for H1299 cells. The caspase-3 activity was found to be higher in H1299 cells compared to A549 cells, thus correlating well with the MTT assay. Hence, harmaline acts as an effective inhibitor of SphK1 and induces apoptosis in NSLCs via the intrinsic pathway.
4. Conclusions
In conclusion, our findings indicate that harmaline is a promising molecule for targeting SphK1 inhibition. Harmaline can be further improved and used as a scaffold for designing selective and effective molecules for therapeutic management of lung cancer. This study would further assist in designing new anticancer drugs of semisynthetic origin to enhance their specificity with reduced cytotoxicity and improved antineoplastic abilities.
5. Materials and Methods
Harmaline, DMSO, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide), N-lauroyl sarcosine, and Tris buffer were procured from Sigma Aldrich (St. Louis, MO, USA). BIOMOL was purchased from Enzo (New York, USA). Luria broth was bought from Himedia (Mumbai, India). FBS, DMEM/F-12K medium, trypsin, and an antibiotic cocktail were procured from Gibco-Life Technologies, Thermo Fisher Scientific (USA). HEK293, A549, and H1299 cells were acquired from the NCCS, Pune-411,007, India. APOPCYTO Caspase-3 Colorimetric Assay Kit (code no. 4800) was bought from MBL Life Science (Japan). Plasmid pET28b+, DH5α, and BL21-Gold cells were bought from Qiagen. The Jasco spectrofluorometer (FP-6200, Japan), VP-ITC microcalorimeter (GE, MicroCal, USA), and ELISA reader (BioRad) were used in instrumentation. SigmaPlot 10.0, MicroCal Origin 7.0, and Graph Pad Prism were used for data analysis. All the reagents used for buffer preparation were of analytical grade.
5.1. Expression and Purification of SphK1
Plasmid pET28b+ containing SphK1 gene insert was successfully expressed in BL21-Gold cells and later purified by Ni-NTA affinity chromatography, as described in our previous communication.97 The purity of the protein was checked by SDS-PAGE and its concentration was measured by using a Jasco V-660 UV–visible spectrophotometer with a molar absorption coefficient of 48,275 M–1 cm–1 at 280 nm.
5.2. Fluorescence Measurements
The binding study of harmaline with SphK1 was performed on the Jasco spectrofluorometer (FP-6200) at 25 ± 0.1 °C. Stock solution (100 mM) of harmaline in DMSO was used for the preparation of working solution (1 mM) in 20 mM Tris and 100 mM NaCl buffer (pH 8.0). A fixed concentration of SphK1 (4 μM) was titrated with increasing harmaline concentration (3.56 to 19.26 μM). SphK1 was excited at 280 nm and the emission spectra were collected from 300–400 nm. SphK1 showed an emission peak at 342 nm. The blank titration (buffer with harmaline) was subtracted to obtain the final spectra.
Obtained data were corrected for the inner filter effect by harmaline according to the formula F = Fobs antilog [(Aex + Aem)/2], where Aex is the absorbance of harmaline at the excitation wavelength, and Aem is the absorbance of harmaline at the emission wavelength (Figure S2).98,99 The decrease in the fluorescence intensity of SphK1 with increasing harmaline concentration was plotted and the value of the binding constant (Ka) and the number of binding sites (n) for the harmaline–SphK1 interaction were calculated by analyzing the corrected fluorescence spectra by fitting into eqs 1–7. Equation 1(100) and modified Stern–Volmer eq 2 were used to analyze the quenching data and the fraction of SphK1 molecules accessible to harmaline, respectively. Equations 3–6 represent the modified Stern–Volmer equation, Lineweaver–Burk modified Stern–Volmer, Johansson equation, and Scatchard equation, respectively, as desciribed.101
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
Here, F0 denotes fluorescence intensity of SphK1 without harmaline, F is the fluorescence intensity of SphK1 at a specific concentration of harmaline at 342 nm, KSV is the Stern–Volmer quenching constant, [harmaline] denotes the concentration of harmaline titrated, K is the quenching constant, and f is the fraction of accessible SphK1 molecule to harmaline. [harmalinet] is the total quencher concentration and [SphK1t] is the total protein concentration, while Fmax denotes the concentration when the interaction is complete.
In terms of fluorescence quenching, the Langmuir isotherm102,103 can be expressed in the form of eq 7 (for one-site saturation).
| 7 |
Here, ymax is the maximum fluorescence observed and Kd is the equilibrium dissociation constant. The binding constants from all equations are given in Table S1.
5.3. Isothermal Titration Calorimetry
The interaction of harmaline binding to SphK1 was evaluated by a VP-ITC microcalorimeter from MicroCal, Inc. (GE, MicroCal, USA), at 25 °C. The protein and ligand samples were prepared in a buffer comprising 20 mM Tris and 100 mM NaCl. DMSO (0.75%, v/v) was added to the protein solution (same as in ligand solution) to minimize the signal-to-noise ratio during ITC experiments. For analysis, the heat released during the interaction of harmaline with buffer was measured to obtain the final heat of dilution during harmaline–SphK1 titrations, as described.104 The titration data were analyzed to determine the values of association constant (Ka), enthalpy change (ΔH), and entropy change (ΔS) using MicroCal Origin 7.0.
5.4. Enzyme Inhibition Assay
The SphK1 inhibitory potential of harmaline was studied with standard Malachite Green (BIOMOL GREEN reagent, Enzo Life Sciences) microtiter-plate assay, as described in our previous communications.97,105 The kinase activity of SphK1 was plotted as a function of percent inhibition using a standard phosphate curve (Figure S5) in SigmaPlot 10.0. The data points were calculated from three independent experiments in triplicates and their average was taken for data analysis.
5.5. Cell Viability Studies
The HEK293, A549, and H1299 cancer cell lines were grown in a DMEM/F12 media with 10% heat-inactivated FBS (Gibco) containing 1% penicillin and streptomycin in a 5% CO2 humidified incubator at 37 °C. Cells were cultured regularly to maintain culture homogeneity and then trypsinized after 10 passages for MTT to determine the cytotoxicity and antiproliferative activity of harmaline. The 96-well cell culture plates were used for seeding the cells at a density of 8000–10,000 cells/well and incubated overnight. The next day, cells were treated with harmaline (0–200 μM) in increasing concentration and incubated for 48 h at 37 °C in a CO2 incubator. At the end of the incubation, the culture medium was removed followed by PBS (pH 7.4) washing. Successively, MTT (0.5 mg/mL) mixed in DMEM/F12 media was added to all the wells and incubated for 4 h. Finally, the formazan crystals were mixed with DMSO (100 μL) by agitating the plate for 15–20 min on an orbital plate shaker. The ELISA reader (BioRad) was utilized for measuring the absorbance of the colored product at 570 nm. The absorbance was expressed in percentage viability for all cell lines at different concentrations of harmaline in comparison to the control cells. The graph of percent cell viability against different concentrations of the harmaline was utilized to evaluate the IC50 (50% inhibitory concentration) values.
5.6. Determination of Caspase-3 Activity
The caspase-3 assay of cell lysates was performed by using APOPCYTO caspase-3 colorimetric assay kit as per the manufacturer’s protocol. H1299 and A549 cells (approximately 1 million) were given a dose of harmaline (50 and 100 μM) and the untreated sample (media only) was used as a control. All the samples were incubated for 24 h and, later, both floating and adherent treated cells were collected in microcentrifuge tubes. After the addition of 50 μL of chilled cell lysis buffer, samples were incubated on ice for 10 min. The supernatant was collected carefully in a fresh tube after completion of a lysis reaction by centrifuging the samples for 1 min at 10,000 rpm. Fifty micrograms from all samples (treated and untreated) was added to the reaction mixture containing 10 mM DTT and 200 μM DEVD-p-NA substrate. The reaction was allowed to proceed for 3 h at 37 °C in the dark followed by the absorbance measurement at 405 nm. Three independent experiments were performed and an average of data points was taken for the calculation.
5.7. Molecular Docking
The crystal structure of human SphK1 was downloaded from the RCSB Protein Data Bank (PDB ID: 3VZB; resolution, 2.0 Å). This structure was deposited in a trimeric form with several cocrystallized water and ligand molecules including d-sphingosine, which was preprocessed considering chain “A” and deleting heteroatoms. The harmaline structure was processed in MGL tools after retrieving it from the PubChem database.106 AutoDock Vina107 was utilized for the docking experiment to produce the ligand–receptor interaction model. The docking was structurally blind with the search space having grid sizes of 50, 58, and 56 Å, centralized at 53.15, 51.79, and −1.54 for X, Y, and Z coordinates, respectively. We performed 10 docking runs, each with 20 iterations to generate 10 × 20 = 200 possible docked conformations of harmaline on SphK1. We clustered all the docked conformations of the harmaline based on their binding energy and binding mode and finally selected a docked pose having the highest affinity toward the SphK1 binding pocket. PyMOL and Discovery Studio Visualizer (Dassault Systèmes, 2016) were used for analysis to identify the structural features of the SphK1–harmaline complex to determine some important interactions.
Acknowledgments
S.R. and S.P. are thankful to the University Grants Commission and Council of Scientific and Industrial Research for the award of Senior Research Fellowship. The authors thank the Department of Science and Technology, Government of India, for the FIST support (FIST program no. SR/FST/LSI-541/2012).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02165.
SDS-PAGE profile showing a single band of SphK1 at ∼45 kDa (Figure S1); absorption spectrum of increasing concentrations harmaline (Figure S2); Stern–Volmer and modified Stern–Volmer plots for quenching of SphK1 (4 μM, pH 8.0) by harmaline at 25 °C (Figure S3); Scatchard plot and Langmuir binding fitting curve of harmaline quenching SphK1 fluorescence (Figure S4); standard phosphate hydrolysis curve showing the quantity of phosphate measured (Figure S5); absorbance at 405 nm for A549 and H1299 cells recorded after treatment with 0, 50, and 100 μM harmaline (Figure S6); plausible docking sites for harmaline on SphK1 (Figure S7); Ka and n of harmaline and SphK1 calculated using different models (Table S1) (PDF)
Author Contributions
S.R. and M.I.H. conceptualized the study. S.R., T.M., P.G., and R.D. provided the methodology. T.M. and G.M.H. provided the software. S.R., S.P., S.L., R.D., and M.I.H. conducted the validation of data. S.R., S.P., and P.G. carried out the formal analysis. T.M., G.M.H., and M.I.H. conducted the investigation. S.L. and M.I.H. provided the resources. S.R., T.M., G.M.H., and R.D. curated the data. S.R., T.M., and M.I.H. prepared and wrote the original draft of the manuscript. S.R., S. L., and M.I.H. wrote, reviewed, and edited the manuscript. T.M. contributed to the visualization of the study. S.L. and M.I.H. supervised the study. M.I.H. administered the project. S.L. and M.I.H were responsible for funding acquisition.
This work is funded by the Indian Council of Medical Research.
The authors declare no competing financial interest.
Notes
All data generated or analyzed during this study are included in this paper and supporting materials attached to this article.
Supplementary Material
References
- Torre L. A.; Siegel R. L.; Jemal A.. Lung cancer statistics. In Lung cancer and personalized medicine; Springer: 2016; 1–19. [DOI] [PubMed] [Google Scholar]
- Jemal A.; Siegel R.; Ward E.; Murray T.; Xu J.; Thun M. J. Cancer statistics, 2007. Ca-Cancer J. Clin. 2007, 57, 43–66. 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Alberg A. J.; Samet J. M. Epidemiology of lung cancer. Chest 2003, 123, 21S–49S. 10.1378/chest.123.1_suppl.21S. [DOI] [PubMed] [Google Scholar]
- Travis W. D.; Travis L. B.; Devesa S. S. Lung cancer. Cancer 1995, 75, 191–202. . [DOI] [PubMed] [Google Scholar]
- Jacques J.; Hill D.; Shier K.; Jindani A.; Miller A. Appraisal of the World Health Organization classification of lung tumours. Can. Med. Assoc. J. 1980, 122, 897. [PMC free article] [PubMed] [Google Scholar]
- Temel J. S.; Greer J. A.; Muzikansky A.; et al. Early palliative care for patients with metastatic non–small-cell lung cancer. N. Engl. J. Med. 2010, 363, 733–742. 10.1056/NEJMoa1000678. [DOI] [PubMed] [Google Scholar]
- Breindel J. L.; Haskins J. W.; Cowell E. P.; Zhao M.; Nguyen D. X.; Stern D. F. EGF Receptor Activates MET through MAPK to Enhance Non–Small Cell Lung Carcinoma Invasion and Brain Metastasis. Cancer Res. 2013, 73, 5053–5065. 10.1158/0008-5472.CAN-12-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P.; Allen M. S.; Aubry M. C.; et al. Clinical features of 5,628 primary lung cancer patients: experience at Mayo Clinic from 1997 to 2003. Chest 2005, 128, 452–462. 10.1378/chest.128.1.452. [DOI] [PubMed] [Google Scholar]
- Boolell V.; Alamgeer M.; Watkins D. N.; Ganju V. The evolution of therapies in non-small cell lung cancer. Cancers 2015, 7, 1815–1846. 10.3390/cancers7030864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raparia K.; Villa C.; DeCamp M. M.; Patel J. D.; Mehta M. P. Molecular profiling in non–small cell lung cancer: a step toward personalized medicine. Arch. Pathol. Lab. Med. 2013, 137, 481–491. 10.5858/arpa.2012-0287-RA. [DOI] [PubMed] [Google Scholar]
- Spiegel S.; Milstien S. Sphingosine 1-phosphate, a key cell signaling molecule. J. Biol. Chem. 2002, 277, 25851–25854. 10.1074/jbc.R200007200. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Min X.; Xiao S.-H.; et al. Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure 2013, 21, 798–809. 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- Hatoum D.; Haddadi N.; Lin Y.; Nassif N. T.; McGowan E. M. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: challenges for SphK as an oncotarget. Oncotarget 2017, 8, 36898. 10.18632/oncotarget.16370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K.; Yamashita T.; Olivera A.; Miller G. F.; Spiegel S.; Proia R. L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allende M. L.; Sasaki T.; Kawai H.; et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J. Biol. Chem. 2004, 279, 52487–52492. 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- Wattenberg B. W.; Pitson S. M.; Raben D. M. The sphingosine and diacylglycerol kinase superfamily of signaling kinases: localization as a key to signaling function. J. Lipid Res. 2006, 47, 1128–1139. 10.1194/jlr.R600003-JLR200. [DOI] [PubMed] [Google Scholar]
- Pitson S. M.; Moretti P. A.; Zebol J. R.; et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitson S. M.; Xia P.; Leclercq T. M.; et al. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J. Exp. Med. 2005, 201, 49–54. 10.1084/jem.20040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne N. J.; Pyne S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Spiegel S.; Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Newton J.; Lima S.; Maceyka M.; Spiegel S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195. 10.1016/j.yexcr.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.-L.; Huang W.-P.; Lee H. Roles of sphingosine 1-phosphate on tumorigenesis. World J. Biol. Chem 2011, 2, 25. 10.4331/wjbc.v2.i2.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillermet-Guibert J.; Davenne L.; Pchejetski D.; et al. Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol. Cancer Ther. 2009, 8, 809–820. 10.1158/1535-7163.MCT-08-1096. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Li W.; Ren L.; et al. The sphingosine kinase-1/sphingosine-1-phosphate axis in cancer: Potential target for anticancer therapy. Pharmacol. Ther. 2019, 195, 85–99. 10.1016/j.pharmthera.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Liu L.; Zhou X.; Zhang J.; et al. LncRNA HULC promotes non-small cell lung cancer cell proliferation and inhibits the apoptosis by up-regulating sphingosine kinase 1 (SPHK1) and its downstream PI3K/Akt pathway. Eur. Rev. Med. Pharmacol. Sci 2018, 22, 8722–8730. 10.26355/eurrev_201812_16637. [DOI] [PubMed] [Google Scholar]
- Liu S.-Q.; Huang J.-A.; Qin M.-B.; et al. Sphingosine kinase 1 enhances colon cancer cell proliferation and invasion by upregulating the production of MMP-2/9 and uPA via MAPK pathways. Int. J. Colorectal Dis. 2012, 27, 1569–1578. 10.1007/s00384-012-1510-y. [DOI] [PubMed] [Google Scholar]
- Zhu Y. J.; You H.; Tan J. X.; et al. Overexpression of sphingosine kinase 1 is predictive of poor prognosis in human breast cancer. Oncol. Lett. 2017, 14, 63–72. 10.3892/ol.2017.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava V. E.; Hobson J. P.; Murthy S.; Milstien S.; Spiegel S. Sphingosine kinase type 1 promotes estrogen-dependent tumorigenesis of breast cancer MCF-7 cells. Exp. Cell Res. 2002, 281, 115–127. 10.1006/excr.2002.5658. [DOI] [PubMed] [Google Scholar]
- Dayon A.; Brizuela L.; Martin C.; et al. Sphingosine kinase-1 is central to androgen-regulated prostate cancer growth and survival. PLoS One 2009, 4, e8048 10.1371/journal.pone.0008048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A.; Loo S. Y.; Huang B.; et al. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer. Oncotarget 2014, 5, 5920. 10.18632/oncotarget.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoure E.; Pchejetski D.; Aouali N.; et al. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting shingosine kinase-1. Leukemia 2006, 20, 95–102. 10.1038/sj.leu.2404023. [DOI] [PubMed] [Google Scholar]
- Akao Y.; Banno Y.; Nakagawa Y.; et al. High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem. Biophys. Res. Commun. 2006, 342, 1284–1290. 10.1016/j.bbrc.2006.02.070. [DOI] [PubMed] [Google Scholar]
- Ni M.; Shi X.-L.; Qu Z.-G.; Jiang H.; Chen Z.-Q.; Hu J. Epithelial mesenchymal transition of non–small–cell lung cancer cells A549 induced by SPHK1. Asian Pac. J. Trop. Med. 2015, 8, 142–146. 10.1016/S1995-7645(14)60305-9. [DOI] [PubMed] [Google Scholar]
- Song L.; Xiong H.; Li J.; et al. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-κB pathway in human non–small cell lung cancer. Clin. Cancer Res. 2011, 17, 1839–1849. 10.1158/1078-0432.CCR-10-0720. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Wang Z.; Lin Y.; et al. Sphingosine kinase 1 enhances the invasion and migration of non-small cell lung cancer cells via the AKT pathway. Oncol. Rep. 2015, 33, 1257–1263. 10.3892/or.2014.3683. [DOI] [PubMed] [Google Scholar]
- Igarashi Y.; Hakomori S.; Toyokuni T.; et al. Effect of chemically well-defined sphingosine and its N-methyl derivatives on protein kinase C and src kinase activities. Biochemistry 1989, 28, 6796–6800. 10.1021/bi00443a002. [DOI] [PubMed] [Google Scholar]
- Cohen J. A.; Barkhof F.; Comi G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- Permpongkosol S.; Wang J. D.; Takahara S.; et al. Anticarcinogenic effect of FTY720 in human prostate carcinoma DU145 cells: Modulation of mitogenic signaling, FAK, cell-cycle entry and apoptosis. Int. J. Cancer 2002, 98, 167–172. 10.1002/ijc.10178. [DOI] [PubMed] [Google Scholar]
- Wang J.; Knapp S.; Pyne N. J.; Pyne S.; Elkins J. M. Crystal structure of sphingosine kinase 1 with PF-543. ACS Med. Chem. Lett. 2014, 5, 1329–1333. 10.1021/ml5004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy K. A. O.; Obeid L. M. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway in disease: review of sphingosine kinase inhibitors. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2013, 1831, 157–166. 10.1016/j.bbalip.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P.; Mohammad T.; Dahiya R.; et al. Evaluation of binding and inhibition mechanism of dietary phytochemicals with sphingosine kinase 1: Towards targeted anticancer therapy. Sci. Rep. 2019, 9, 1–15. 10.1038/s41598-019-55199-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jairajpuri D. S.; Mohammad T.; Adhikari K.; Gupta P.; et al. Identification of Sphingosine Kinase-1 Inhibitors from Bioactive Natural Products Targeting Cancer Therapy. ACS Omega 2020, 5, 14720. 10.1021/acsomega.0c01511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan P.; Queen A.; Mohammad T.; et al. Identification of alpha-Mangostin as a Potential Inhibitor of Microtubule Affinity Regulating Kinase 4. J. Nat. Prod. 2019, 82, 2252–2261. 10.1021/acs.jnatprod.9b00372. [DOI] [PubMed] [Google Scholar]
- Khan P.; Rahman S.; Queen A.; et al. Elucidation of Dietary Polyphenolics as Potential Inhibitor of Microtubule Affinity Regulating Kinase 4: In silico and In vitro Studies. Sci. Rep. 2017, 7, 9470. 10.1038/s41598-017-09941-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz F.; Shahbaaz M.; Bisetty K.; Islam A.; Ahmad F.; Hassan M. I. Designing New Kinase Inhibitor Derivatives as Therapeutics Against Common Complex Diseases: Structural Basis of Microtubule Affinity-Regulating Kinase 4 (MARK4) Inhibition. OMICS 2015, 19, 700–711. 10.1089/omi.2015.0111. [DOI] [PubMed] [Google Scholar]
- Yousuf M.; Shamsi A.; Khan P.; Shahbaaz M.; AlAjmi M. F.; Hussain A.; Hassan G. M.; Islam A.; Haque Q. M. R.; Hassan I. Ellagic Acid Controls Cell Proliferation and Induces Apoptosis in Breast Cancer Cells via Inhibition of Cyclin-Dependent Kinase 6. Int. J. Mol. Sci. 2020, 21, 3526. 10.3390/ijms21103526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A. K.; Basu S.; Sarkar N.; Ghosh A. C. Advances in cancer therapy with plant based natural products. Curr. Med. Chem. 2001, 8, 1467–1486. 10.2174/0929867013372094. [DOI] [PubMed] [Google Scholar]
- Plano D.; Amin S.; Sharma A. K. Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors: Miniperspective. J. Med. Chem. 2014, 57, 5509–5524. 10.1021/jm4011687. [DOI] [PubMed] [Google Scholar]
- Funaki M.; Kitabayashi J.; Shimakami T.; et al. Peretinoin, an acyclic retinoid, inhibits hepatocarcinogenesis by suppressing sphingosine kinase 1 expression in vitro and in vivo. Sci. Rep. 2017, 7, 1–13. 10.1038/s41598-017-17285-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Li Y.; Chai H.; Yakura T.; Liu B.; Yao Q. Synthesis and biological evaluation of 2-epi-jaspine B analogs as selective sphingosine kinase 1 inhibitors. Bioorg. Chem. 2019, 103369. 10.1016/j.bioorg.2019.103369. [DOI] [PubMed] [Google Scholar]
- Lu P.-H.; Chen M.-B.; Liu Y.-Y.; et al. Identification of sphingosine kinase 1 (SphK1) as a primary target of icaritin in hepatocellular carcinoma cells. Oncotarget 2017, 8, 22800. 10.18632/oncotarget.15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.-Q.; Gao H.; Han M.; Liu K.-L.; Peng J.-J.; Han Y.-T. Hispidulin suppresses tumor growth and metastasis in renal cell carcinoma by modulating ceramide-sphingosine 1-phosphate rheostat. Am. J. Cancer Res. 2017, 7, 1501. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mahmoudian M.; Jalipour H.; Salehian Dardashti P. Toxicity of Peganum harmala: review and a case report. Iran. J. Pharmacol. Ther. 2002, 1, 1. [Google Scholar]
- Cao R.; Peng W.; Wang Z.; Xu A. β-Carboline alkaloids: biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. 10.2174/092986707779940998. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liang F.; Jiang W.; et al. DH334, a β-carboline anti-cancer drug, inhibits the CDK activity of budding yeast. Cancer Biol. Ther. 2007, 6, 1204–1210. 10.4161/cbt.6.8.4382. [DOI] [PubMed] [Google Scholar]
- Khan F. A.; Maalik A.; Iqbal Z.; Malik I. Recent pharmacological developments in β-carboline alkaloid “harmaline”. Eur. J. Pharmacol. 2013, 721, 391–394. 10.1016/j.ejphar.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Moloudizargari M.; Mikaili P.; Aghajanshakeri S.; Asghari M. H.; Shayegh J. Pharmacological and therapeutic effects of Peganum harmala and its main alkaloids. Pharmacogn. Rev. 2013, 7, 199. 10.4103/0973-7847.120524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming P.; Vincent S. R. Inhibition of histamine-N-methyltransferase (HNMT) by fragments of 9-amino-1, 2, 3, 4-tetrahydroacridine (tacrine) and by β-carbolines. Biochem. Pharmacol. 1992, 44, 989–992. 10.1016/0006-2952(92)90133-4. [DOI] [PubMed] [Google Scholar]
- Suleiman M.; Reeves J. Inhibition of Na+-Ca2+ exchange mechanism in cardiac sarcolemmal vesicles by harmaline. Comp. Biochem. Physiol., Part C: C, Comp. Pharmacol. Toxicol. 1987, 88, 197. 10.1016/0742-8413(87)90068-5. [DOI] [PubMed] [Google Scholar]
- Herraiz T.; González D.; Ancín-Azpilicueta C.; Arán V.; Guillén H. β-Carboline alkaloids in Peganum harmala and inhibition of human monoamine oxidase (MAO). Food Chem. Toxicol. 2010, 48, 839–845. 10.1016/j.fct.2009.12.019. [DOI] [PubMed] [Google Scholar]
- El Gendy M. A.; El-Kadi A. O. Peganum harmala L. differentially modulates cytochrome P450 gene expression in human hepatoma HepG2 cells. Drug Metab. Lett. 2009, 3, 212–216. 10.2174/187231209790218163. [DOI] [PubMed] [Google Scholar]
- El Gendy M. A.; El-Kadi A. O. Harmine and harmaline downregulate TCDD-induced Cyp1a1 in the livers and lungs of C57BL/6 mice. BioMed Res. Int. 2013, 2013, 1. 10.1155/2013/258095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Wang C.; Jiang C.; Zeng H.; He X. Novel mechanism of harmaline on inducing G2/M cell cycle arrest and apoptosis by up-regulating Fas/FasL in SGC-7901 cells. Sci. Rep. 2015, 5, 1–10. 10.1038/srep18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B.; Li M.; Yu Y.; et al. Effects of harmaline on cell growth of human liver cancer through the p53/p21 and Fas/FasL signaling pathways. Oncol. Lett. 2018, 15, 1931–1936. 10.3892/ol.2017.7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P.; Khan F. I.; Roy S.; et al. Functional implications of pH-induced conformational changes in the Sphingosine kinase 1. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 225, 117453. 10.1016/j.saa.2019.117453. [DOI] [PubMed] [Google Scholar]
- Mocz G., Ross J.A.. Fluorescence techniques in analysis of protein–ligand interactions. Protein-Ligand Interactions; Springer, 2013:169–210. [DOI] [PubMed] [Google Scholar]
- Ojha B.; Das G. Role of hydrophobic and polar interactions for BSA–amphiphile composites. Chem. Phys. Lipids 2011, 164, 144–150. 10.1016/j.chemphyslip.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Guo M.; Zou J.-W.; Yi P.-G.; Shang Z.-C.; Hu G.-X.; Yu Q.-S. Binding interaction of gatifloxacin with bovine serum albumin. Anal. Sci. 2004, 20, 465–470. 10.2116/analsci.20.465. [DOI] [PubMed] [Google Scholar]
- Freire E.; Mayorga O. L.; Straume M. Isothermal titration calorimetry. Anal. Chem. 1990, 62, 950A–959A. 10.1021/ac00217a002. [DOI] [Google Scholar]
- Buurman E. T.; Andrews B.; Gao N.; et al. In vitro validation of acetyltransferase activity of GlmU as an antibacterial target in Haemophilus influenzae. J. Biol. Chem. 2011, 286, 40734–40742. 10.1074/jbc.M111.274068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles R. H.; Poindexter C.; Evans A.; Brown M.; Cooper C. R. Nonlinear model-based estimates of IC50 for studies involving continuous therapeutic dose–response data. Contemp. Clin. Trials 2008, 29, 878–886. 10.1016/j.cct.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.; Yang Y.; Yang Y.; et al. Long noncoding RNA (lncRNA) HOTAIR affects tumorigenesis and metastasis of non-small cell lung cancer by upregulating miR-613. Oncol. Res. Featuring Preclin. Clin. Cancer Ther. 2018, 26, 725–734. 10.3727/096504017X15119467381615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington K. J. The biology of cancer. Medicine 2016, 44, 1–5. 10.1016/j.mpmed.2015.10.005. [DOI] [Google Scholar]
- Mazumder S.; Plesca D.; Almasan A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Apoptosis Cancer 2008, 13–21. 10.1007/978-1-59745-339-4_2. [DOI] [PubMed] [Google Scholar]
- Woo M.; Hakem R.; Furlonger C.; et al. Caspase-3 regulates cell cycle in B cells: a consequence of substrate specificity. Nat. Immunol. 2003, 4, 1016–1022. 10.1038/ni976. [DOI] [PubMed] [Google Scholar]
- Earnshaw W. C.; Martins L. M.; Kaufmann S. H. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Naz H.; Tarique M.; Ahamad S.; et al. Hesperidin-CAMKIV interaction and its impact on cell proliferation and apoptosis in the human hepatic carcinoma and neuroblastoma cells. J. Cell. Biochem. 2019, 120, 15119–15130. 10.1002/jcb.28774. [DOI] [PubMed] [Google Scholar]
- Payne A. W.Role of ceramide kinase in breast cancer progression; Publicly Accessible Penn Dissertations, 2014. [Google Scholar]
- Klec C.; Gutschner T.; Panzitt K.; Pichler M. Involvement of long non-coding RNA HULC (highly up-regulated in liver cancer) in pathogenesis and implications for therapeutic intervention. Expert Opin. Ther. Targets 2019, 23, 177–186. 10.1080/14728222.2019.1570499. [DOI] [PubMed] [Google Scholar]
- White C.; Alshaker H.; Cooper C.; Winkler M.; Pchejetski D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget 2016, 7, 23106. 10.18632/oncotarget.7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel G. T.; Maceyka M.; Milstien S.; Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discovery 2013, 12, 688–702. 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins R.; Wu L.; Zhang C.; Davis R. M.; Xu B. Natural product-based nanomedicine: recent advances and issues. Int. J. Nanomed. 2015, 10, 6055. 10.2147/IJN.S92162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramawat K, Dass S, Mathur M. The chemical diversity of bioactive molecules and therapeutic potential of medicinal plants. In Herbal drugs: ethnomedicine to modern medicine; Springer: 2009; 7–32, 10.1007/978-3-540-79116-4_2. [DOI] [Google Scholar]
- Brizuela L.; Dayon A.; Doumerc N.; et al. The sphingosine kinase-1 survival pathway is a molecular target for the tumor-suppressive tea and wine polyphenols in prostate cancer. FASEB J. 2010, 24, 3882–3894. 10.1096/fj.10-160838. [DOI] [PubMed] [Google Scholar]
- Kruczynski A.; Poli M.; Dossi R.; et al. Anti-angiogenic, vascular-disrupting and anti-metastatic activities of vinflunine, the latest vinca alkaloid in clinical development. Eur. J. Cancer 2006, 42, 2821–2832. 10.1016/j.ejca.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Sorensen J. B.; Østerlind K.; Hansen H. H. Vinca alkaloids in the treatment of non-small cell lung cancer. Cancer Treat. Rev. 1987, 14, 29–51. 10.1016/0305-7372(87)90049-1. [DOI] [PubMed] [Google Scholar]
- Beljanski M.; Beljanski M. Three alkaloids as selective destroyers of cancer cells in mice. Oncology 1986, 43, 198–203. 10.1159/000226363. [DOI] [PubMed] [Google Scholar]
- Shabani S. H. S.; Tehrani S. S. H.; Rabiei Z.; Enferadi S. T.; Vannozzi G. P. Peganum harmala L.’s anti-growth effect on a breast cancer cell line. Biotechnol. Rep. 2015, 8, 138–143. 10.1016/j.btre.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi N.; Ajmal M. R.; Rabbani G.; Ahmad E.; Khan R. H. A comprehensive insight into binding of hippuric acid to human serum albumin: a study to uncover its impaired elimination through hemodialysis. PLoS One 2013, 8, e71422 10.1371/journal.pone.0071422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman M. T.; Shamsi H.; Khan A. U. Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches. Mol. Pharmaceutics 2014, 11, 1785–1797. 10.1021/mp500116c. [DOI] [PubMed] [Google Scholar]
- Keswani N.; Choudhary S.; Kishore N. Interaction of weakly bound antibiotics neomycin and lincomycin with bovine and human serum albumin: biophysical approach. J. Biochem. 2010, 148, 71–84. 10.1093/jb/mvq035. [DOI] [PubMed] [Google Scholar]
- De Meester C. Genotoxic potential of β-carbolines: a review. Mutat. Res./Rev. Genet. Toxicol. 1995, 339, 139–153. 10.1016/0165-1110(95)90008-X. [DOI] [PubMed] [Google Scholar]
- Mohamed A. H. S.; AL-Jammali S. M. J.; Naki Z. J. Effect of repeated administration of Peganum harmala alcoholic extract on the liver and kidney in Albino mice: a histo-pathological study. J. Sci. Innovative Res. 2013, 2, 585–597. [Google Scholar]
- Storch A.; Hwang Y. I.; Gearhart D. A.; et al. Dopamine transporter-mediated cytotoxicity of β-carbolinium derivatives related to Parkinson’s disease: relationship to transporter-dependent uptake. J. Neurochem. 2004, 89, 685–694. 10.1111/j.1471-4159.2004.02397.x. [DOI] [PubMed] [Google Scholar]
- Bournine L.; Bensalem S.; Fatmi S.; et al. Evaluation of the cytotoxic and cytostatic activities of alkaloid extracts from different parts of Peganum harmala L.(Zygophyllaceae). Eur. J. Integr. Med. 2017, 9, 91–96. 10.1016/j.eujim.2016.10.002. [DOI] [Google Scholar]
- Ghosh T.; Sarkar S.; Bhattacharjee P.; et al. In vitro relationship between serum protein binding to beta-carboline alkaloids: a comparative cytotoxic, spectroscopic and calorimetric assays. J. Biomol. Struct. Dyn. 2020, 38, 1103–1118. 10.1080/07391102.2019.1595727. [DOI] [PubMed] [Google Scholar]
- Gupta P.; Mohammad T.; Khan P.; et al. Evaluation of ellagic acid as an inhibitor of sphingosine kinase 1: A targeted approach towards anticancer therapy. Biomed. Pharmacother. 2019, 118, 109245. 10.1016/j.biopha.2019.109245. [DOI] [PubMed] [Google Scholar]
- Raghav D.; Ashraf S. M.; Mohan L.; Rathinasamy K. Berberine induces toxicity in HeLa cells through perturbation of microtubule polymerization by binding to tubulin at a unique site. Biochemistry 2017, 56, 2594–2611. 10.1021/acs.biochem.7b00101. [DOI] [PubMed] [Google Scholar]
- Gupta K.; Panda D. Perturbation of microtubule polymerization by quercetin through tubulin binding: a novel mechanism of its antiproliferative activity. Biochemistry 2002, 41, 13029–13038. 10.1021/bi025952r. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.. Principles of fluorescence spectroscopy; Springer Science & Business Media: 2013. [Google Scholar]
- Wei X. L.; Xiao J. B.; Wang Y.; Bai Y. Which model based on fluorescence quenching is suitable to study the interaction between trans-resveratrol and BSA?. Spectrochim. Acta, Part A 2010, 75, 299–304. 10.1016/j.saa.2009.10.027. [DOI] [PubMed] [Google Scholar]
- Azevedo A. M. O.; Ribeiro D. M. G.; Pinto P. C. A. G.; Lúcio M.; Reis S.; Saraiva M. L. M. F. S. Imidazolium ionic liquids as solvents of pharmaceuticals: influence on HSA binding and partition coefficient of nimesulide. Int. J. Pharm. 2013, 443, 273–278. 10.1016/j.ijpharm.2012.12.030. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.A practical introduction to structure, mechanism, and data analysis. Enzymes, 2nd ed. John Wiley & Sons, New York, NY: 2000: 104. [Google Scholar]
- Voura M.; Khan P.; Thysiadis S.; et al. Probing the Inhibition of Microtubule Affinity Regulating Kinase 4 by N-Substituted Acridones. Sci. Rep. 2019, 9, 1676. 10.1038/s41598-018-38217-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiya R.; Mohammad T.; Gupta P.; et al. Molecular interaction studies on ellagic acid for its anticancer potential targeting pyruvate dehydrogenase kinase 3. RSC Adv. 2019, 9, 23302–23315. 10.1039/C9RA02864A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.