

Abstract
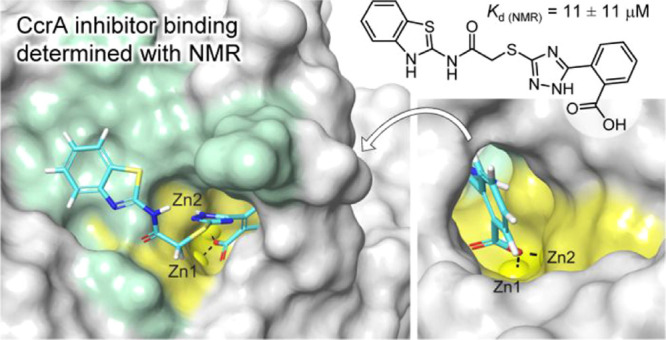
Metallo-β-lactamase (MBL)-producing bacteria resistant to β-lactam antibiotics are a serious threat to human health. Despite great efforts and important progress in the discovery of MBL inhibitors (MBLIs), there is none in clinical use. Herein, inhibitor complexes of the MBL CcrA were investigated by NMR spectroscopy to provide perspectives on the further development of 2-(triazolylthio)acetamide-type MBLIs. By using the NMR-based chemical shift perturbation (CSP) and direction of CSP methodologies together with molecular docking, the spatial orientation of three compounds in the CcrA active site was investigated (4–6). Inhibitor 6 showed the best binding affinity (Kd ≈ 2.3 ± 0.3 μM), followed by 4 (Kd = 11 ± 11 μM) and 5 (Kd = 34 ± 43 μM), as determined from the experimental NMR data. Based on the acquired knowledge, analogues of other MBLIs (1–3) were designed and evaluated in silico with the purpose of examining a strategy for promoting their interactions with the catalytic zinc ions.
Introduction
The evolution and spread of a variety of resistance mechanisms in bacteria to withstand the effects of antibiotics constitutes one of the largest human health issues of today. Thus, in 2015, a global action plan on antimicrobial resistance with the aim of ensuring the “prevention and treatment of infectious diseases with safe and effective medicines” was commenced by WHO.1 One strategy to preserve the clinical use of current classes of antibiotics is to use them in combination with compounds capable of restoring their antibacterial activity by targeting the resistance mechanisms.2 Such compounds are referred to as antibiotic resistance breakers. Among Gram-negative bacteria, the production of β-lactamase enzymes is the most significant mechanism of resistance to β-lactam antibiotics, which remains the main class of drugs to treat bacterial infections. The development of metallo-β-lactamase (MBL) inhibitors for coadministration with β-lactam antibiotics plays a pivotal role in reaching the goal of WHO’s global action plan.
β-Lactamase enzymes deactivate β-lactam antibiotics by hydrolyzing the crucial β-lactam ring in an active site containing either a seryl hydroxy group or a zinc-activated water molecule (hydroxide ion).3 Accordingly, the enzymes are termed serine-β-lactamases (SBLs) and MBLs, respectively. They are commonly divided into four classes based on amino acid sequence similarity.4,5 The A, C, and D classes consist of SBLs, and class B are MBLs. Class B is further divided into subclasses B1–B4. Generally, the B1, B3, and B4 MBLs require two Zn(II) ions in their active sites, while subgroup B2 requires only one Zn(II) ion for catalytic activity. Today, there are six SBL inhibitors in clinical use but still no clinical MBL inhibitors (MBLIs). The development of broad spectrum inhibitors with therapeutic potential is challenging because of the required selectivity over other zinc metalloenzymes as well as the flexibility and structural differences among the MBLs.3,6 Among the candidates in late clinical development, there is only one B1 type MBLI, VNRX-5133. The compound is effective against the widespread lactamases NDM-1 and VIM-2. Because MBLs, and the B1 class in particular, are capable of disabling almost all types of β-lactam antibiotics, including last resort carbapenems and third generation cephalosporins, MBLI discovery and development is currently an area of intense research effort.
The Bacteroides fragilis group of Gram-negative anaerobic bacteria is a part of the gastrointestinal flora and is also one of the more important pathogenic anaerobes.7,8 It is frequently associated with both anaerobic and mixed infections, which can occur upon displacement into the bloodstream or the tissue surrounding the gastrointestinal tract. Furthermore, it is the most virulent and antimicrobial resistant group of anaerobic bacteria. Carbapenem resistance in B. fragilis is often mediated by the ccrA gene, also named cifA, which encodes the B1-type MBL CcrA (or CfiA).9,10 Recent surveillance studies across the world show a low (i.e., < 5%) but emerging prevalence of the gene in clinical strains.11−13
For many MBLs, including CcrA, the identification of potent inhibitors is sparsely reported in the literature (see the Supporting Information Section S3.2 for details). Because emerging and new MBLs are continuously being identified in clinically significant pathogenic bacteria, initiatives to broaden the scope of existing inhibitors are important. Herein, complexes of CcrA with diaryl-substituted 2-(triazolylthio)acetamide (2TTA)-type MBLIs 1–6(14,15) (Figure 1) were investigated by NMR spectroscopy and molecular docking. NMR chemical shift mapping and direction of chemical shift perturbation (CSP) analysis were used to identify the binding site and spatial orientation of 4–6. The benzoic acid moiety in 4–6 allowed for these compounds to be dissolved in the same buffer as the protein without using dimethyl sulfoxide (DMSO) as cosolvent. This is advantageous because the use of DMSO requires knowledge of its effects on ligand binding.16,17 Four prospective analogues of the most potent 2TTA-type CcrA inhibitors 1–3, in which the aryl substituent of the triazole moiety is varied to promote zinc interactions, was developed based on the results of the aforementioned NMR and docking studies.
Figure 1.

Diaryl-substituted 2TTA-type MBLIs 1–6, with terminal aryl groups connected to the amide and triazole moieties.
Among the previously reported series of diaryl-substituted 2TTAs,14,15,20−221–3 were selected for further studies as they showed the highest CcrA inhibitory activity and had a high inhibitory activity against a broad spectrum of MBLs (Table 1).14 Compounds 4–6 showed selectivity for ImiS over CcrA, NDM-1, and L1.15 However, 4 and 6 induced a 32-fold reduction in the minimal inhibitory concentration of cefalexin for Escherichia coli expressing CcrA (Table 2).15 This is the largest enhancement of antibacterial activity observed among the 2TTA type CcrA inhibitors, thus making 4–6 interesting for further studies.
Table 1. Inhibition Constants of 2TTA-Type MBLIs 1–3.
|
Ki ± SD (μM) |
||||
|---|---|---|---|---|
| class
B1 |
class B2 | class B3 | ||
| no | CcrA | NDM-1 | ImiS | L1 |
| 1 | 6.3 ± 0.1 | 0.35 ± 0.01 | >300 | 0.25 ± 0.03 |
| 2 | 0.3 ± 0.1 | 0.43 ± 0.03 | 2.20 ± 0.2 | 0.07 ± 0.01 |
| 3 | 8.5 ± 0.2 | 5.60 ± 0.1 | 0.75 ± 0.02 | 0.34 ± 0.05 |
Table 2. Antibacterial Activities of β-Lactam Antibiotics in the Presence of 2TTA-Type MBLIs 4–6.
| MIC (μg mL–1) |
||||
|---|---|---|---|---|
| antibiotic-resistant bacteria |
||||
| no | E. coli-CcrAa | E. coli-NDM-1a | E. coli-ImiSb | E. coli-L1a |
| inhibitor blank | 4 | 4 | 1 | 8 |
| 4 | <0.125 | 4 | 1 | 8 |
| 5 | 4 | 4 | 1 | 8 |
| 6 | <0.125 | 4 | 0.5 | 8 |
Antibiotic: cefalexin.
Antibiotic: imipenem.
Results and Discussion
To identify the binding sites of 4–6, NMR chemical shift mapping was applied (Supporting Information, Section S2).23 In this method, the chemical shift changes of the backbone amide protons and nitrogens are monitored by acquiring 1H,15N HSQC spectra upon ligand titration. Both the amide protons and amide nitrogens in proteins are significantly influenced by “through space” as well as “through bond” interactions, which makes the 1H,15N HSQC experiment appropriate for determining the location of ligand binding sites. The data analysis requires the NMR resonance assignment of the protein backbone. Although the observed CSPs can result from both ligand binding and ligand-induced conformational changes, the areas with the highest density of residues showing a significant CSP (SCSP) are likely to outline the ligand binding site.23 A weighted average CSP (1H, 15N Δδ) larger than the population mean (μ) plus one standard deviation (1σ) is considered as significant (see the Supporting Information Section S2.2 for details).23
The affinity of zinc-chelating MBL-inhibitors capable of removing zinc ions from the active site is stronger to zinc than the zinc affinity of the enzyme. To be effective, such compounds should have a 1 nM to 1 pM zinc binding affinity (Kd).24 For CcrA, the affinities of both zinc sites have an upper limit of ≤10 μM, but there is evidence for a tight binding, and for an actual affinity much lower than 10 μM.25 Although the zinc affinities of the 2TTA-type MBLIs have not yet been determined experimentally, these are presumed to interact with, and not deprive the zinc ions from the active site of CcrA.14,26 Being zinc coordination compounds, they are suitable for chemical shift mapping studies.
Similar to the first NMR characterization of CcrA in the presence of an inhibitor,27 the majority of the residues displaying a SCSP upon addition of 4–6 were located in the vicinity of the catalytic zinc site (Figures 2 and 3). For 4 and 5, the residues in the active site added up to 64 and 62% of the significantly shifted residues, respectively, whereas for 6, 45% of the significantly shifted residues were located in the active site. A second site with a significant density was not identified. The remaining residues of CcrA that underwent a SCSP upon binding of the ligands were situated adjacent to the active site, or separately either at the surface or buried inside the protein. Looking at the density of CSPs in the active site falling within the thresholds of μ + 1σ, μ + 2σ and μ + 3σ, and above μ + 3σ, respectively, 54, 67, and 100% were identified for 4; 50, 78, and 67% for 5, and 45, 60, and 33% for 6 (Figures 2 and 3).
Figure 2.

Weighted average amide CSPs of 15N-labeled CcrA upon titration with 4 (A), 5 (B), and 6 (C), plotted as a function of the CcrA residue numbers. Active site residues are highlighted in black. Text labels are shown for the significantly shifted active site residues (Δδ > μ + 1σ). The solid and dashed lines represent the population mean (μ) plus one, two, and three standard deviations (σ), respectively.
Figure 3.
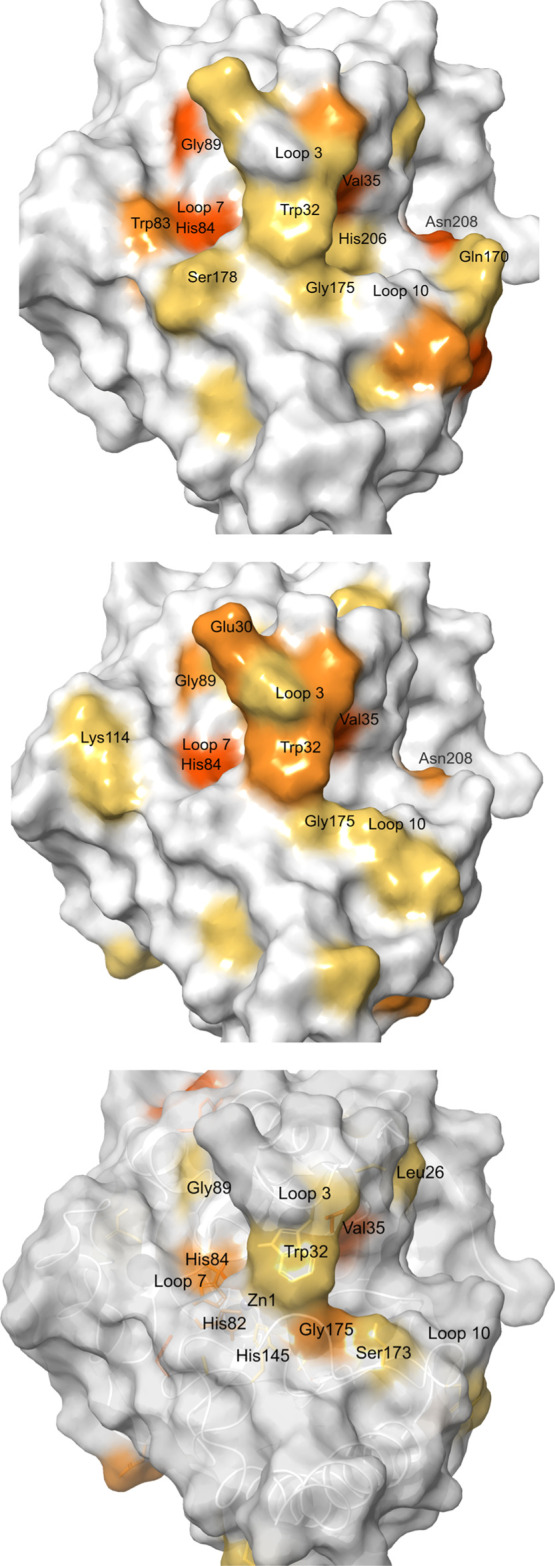
Residues of CcrA that underwent a SCSP upon binding of 4 (top) 5 (middle) and 6 (bottom) are mapped onto the reported crystal structure PDB ID 1A8T.18,19 The larger the CSP, the darker orange color (≥μ + 1σ, ≥μ + 2σ and ≥μ + 3σ). Trp32, Val35, His84(Zn1), Gly89 and Gly175 underwent a SCSP in titration with all three compounds. The active site is located in the cavity extending from loop 10, underneath loop 3, and past loop 7.
The mobile loop that is folding over the catalytic site (loop 3, Figure 3) is a common feature among the B1 MBLs. It is known to change position upon and to play crucial roles in ligand binding and in the catalytic activity of the enzymes.27,28 Several residues in loop 3 of CcrA were influenced upon ligand binding (29–37; which are 61–69 according to the BBL numbering scheme29). Significant changes were also found for residues in the other loops forming the active site, including loops 5 (55), 7 (82–89), 9 (145), 10 (164–178), and 12 (206–208). Some of the residues that were identified are known to be involved in ligand binding within the B1 class MBLs.27,28 For example, hydrophobic interactions and π-interactions are often formed with the residues in positions 29, 32, 35, and 55 (61, 64, 67, and 87, BBL numbering29). In CcrA, these positions are occupied by Ile, Trp, Val, and Ile residues, respectively.
Because the MBLs have a shallow active-site groove, flanked by two flexible loops (L3 and L10), the CSPs observed for CcrA upon ligand binding should be a result of both protein–ligand interactions and ligand-induced conformational changes. Thus, based on the chemical shift mapping, we concluded the active site to be the most likely binding site of 4–6. A more exact location could not be determined judged by the distribution pattern and the magnitude of the CSPs in the different parts of the active site.
To get a further insight into the location and orientation of the studied compounds in the binding site, modeling via molecular docking and MM-GBSA rescoring30 of docking poses were performed (Supporting Information, Sections S3.1–3.3).
A crystal structure of CcrA obtained in the presence of a biphenyl tetrazole inhibitor of similar size as the 2TTA inhibitors was used for the studies (PDB ID 1A8T).18 Structural interaction fingerprints31 were used for analysis and clustering of the top-ranked ligand-binding poses. Three major clusters of docking poses, referred to as L3-L5-L7, L3-L10, and L10-L12, were identified for 4–6 and are shown with 4 in Figure 4A. The labeling of the clusters is linked to the loops (abbreviated as L#) of CcrA forming the binding site where the benzothiazolyl, phenyl, and 4-clorophenyl groups of 4, 5, and 6 (respectively) are located.
Figure 4.

Top-ranked poses of 4 (MM-GBSA ΔGbind) in the L3-L5-L7 [turquoise, in (A,C)], L3-L10 [apricot, in (A,B)], and L10-L12 [green, in (A,D)] clusters of binding poses predicted by molecular docking. The labeling of the binding poses is based on the loops (abbreviated as L#) of CcrA forming the binding sites of the benzothiazole moiety. The CcrA residues displaying different direction of chemical shift perturbations (DCSPs) are colored in orange [no residues in (B); Glu30, Ile55, Trp83, Ile88, and Gly89 in C; Asp168 and Gln170 in (D)], whereas the residues showing the same DCSP in the presence of 4–6 are colored in yellow [Gly31, Trp32, and Ser178 in (B); Ile29 and Gly85 in (C); Lys167, Thr171, Ser173, and Asn208 in (D)]. The fraction of residues displaying different DCSPs is the largest in the L3-L5-L7 binding site, which is one of three sites where the benzothiazole, phenyl, and 4-chlorophenyl aryl groups in 4, 5, and 6 (respectively) are likely to bind. Hence, the L3-L5-L7 type binding poses shows the best fit to the experimental NMR data.
Compounds 4, 5, and 6 only vary at their amide substituents, which make them suitable for DCSP analysis for the determination of their binding location and orientation.32,33 In DCSP analysis, the direction and the magnitude of the CSPs are compared for structurally related ligands. The binding regions of a moiety are identified by examining the CSP effects caused by variation of the moiety.32,33 If the chemical environment of the amide protons and nitrogen upon ligand binding is different, the direction of the CSPs is expected to diverge.34 Conversely, if the environment is identical or highly similar, the direction and the magnitude of the CSPs are expected to be similar. On the basis of the molecular docking results, the DCSPs for the residues surrounding the benzothiazolyl, phenyl, and 4-clorophenyl groups of 4, 5, and 6 (respectively) were compared (Table S7 and Figures S12–S14). The residues are shown in Figure 4. Diverging DSCPs were found in five of seven residues in the L3-L5-L7 binding site, zero of three in the L3-L10 binding site, and two of seven residues in the L10-L12 binding site (Figure 4, Table S7). The percentage of residues showing diverging DCSP was largest in the L3-L5-L7 binding site, which suggests the benzothiazole, phenyl, and 4-chlorophenyl groups of 4–6 to bind here. The same result was obtained upon comparison of all, as well as of all three pairs of compounds. The large CSPs and conforming DCSPs observed for Gly31, Trp32, Gly33, and Gly175 support the binding of the identical parts of 4–6 in and close to the catalytic site (Figures 3 and 4C, and Table S7).
A similar binding arrangement was observed by Christopeit et al. in the crystal structure of 4 in complex with VIM-2 (PDB ID 5LSC,26Figure 5). The dynamic aryl-substituted triazole moiety of the 2TTA-type compounds makes them adaptable to the structural differences of the binding sites of different MBLs. Hence, their resonance, tautomerization and ionization states, and the characteristics of their individual aryl groups may differ (Table S11), which all influence the binding poses, for instance, their ability to coordinate the zinc ions trough one or both aryl moieties. In the VIM-2 crystal structure 5LSC(26) compound 4 coordinates the zinc ions through both its triazole and its carboxylate functionalities (Z1 and Z2, and Z2 only, respectively). Here, the carboxylate group hydrogen bonds to Asn176 (BBL number 233) and to two water molecules (omitted in Figure 5) as well. Among the top scoring docking poses of 4 with CcrA within the L3-L5-L7 cluster, 4 is slightly shifted as compared to its VIM binding mode, and it coordinates the zinc ions via its carboxylate group only (Figures 5, S19–S20). The different L3-L5-L7-type binding pose of 4–6 with CcrA and VIM-2 is dominantly due to the shorter distance between the zinc ions in CcrA (2.84 Å in 1A8T) as compared to VIM-2 (3.88 Å in 5LSC).35 This shorter Zn–Zn distance does not allow simultaneous coordination of the inhibitors to both zinc ions without putting the triazole in a different angle (Figure S23). It should be underlined that the docking of 1–6 into the active site of VIM-2, using the crystal structure 5LSC, reproduces the native binding mode observed by X-ray crystallography, in which the triazole coordinates the zinc ions (Figure S24). This demonstrates that the docking protocol is appropriate and can be applied for prediction. Docking of the same ligands into the active site of CcrA (PDB 1A8T), the above type of binding pose with coordination to both zinc ions was only seen in four poses of 2 (Figures S22 and S23), independent of the triazole protonation state. This is not surprising, as the active site of CcrA and VIM-2 are different, and accordingly provide different interactions at different spatial arrangements. Hence, in addition to the Zn–Zn distance, the residues Ile 29, Trp 32, Val35, Ile55, and Trp83 in CcrA correspond to Phe61, Ala64, Tyr67, Trp87, and Phe117 in VIM-2. Moreover, Gly89, which is localized to the helix after loop 7, corresponds to a larger and more polar Asp119 in VIM-2, influencing both the possible interactions with the ligand and the size of the binding pocket (Figure 5). With the reservation that the docking poses are candidate binding modes that might not represent the native binding modes,36 we wish to emphasize that 4 shares the same location and orientation in the active site of CcrA and VIM-2 even if the specific interactions with the ligand in the active site may differ. These differences in the binding of the biaryl moiety do not change the overall conclusions.
Figure 5.
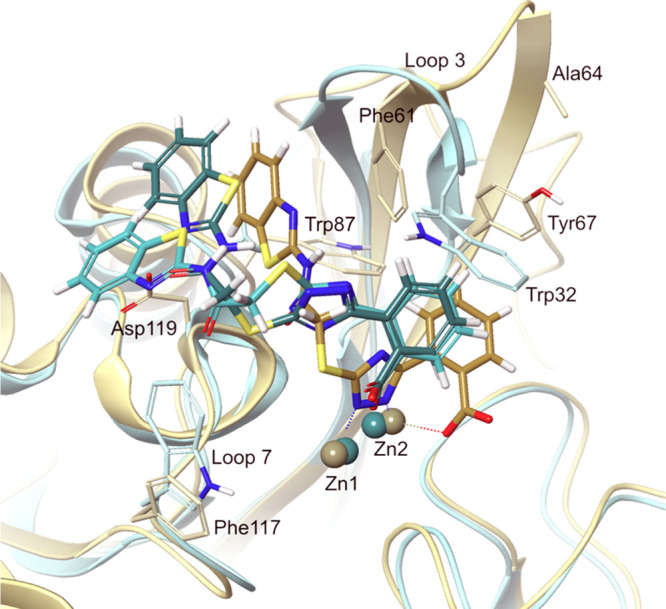
Top- (turquoise, triazole in neutral protonated form) and the fifteenth (green, triazole in deprotonated form)-ranked poses (MM-GBSA ΔGbind) from the docking of 4 into the active site of CcrA (PDB 1A8T(18)); superimposed with the crystal structure of 4 (light brown) in complex with VIM-2 (PDB 5LSC(26)). Residues Ile29, Trp32, Val35, Ile55, Trp83, and Gly89 in CcrA, are corresponding to Phe61, Ala64, Tyr67, Trp87, Phe117, and Asp119 in VIM-2. Some of these side-chains, as well as the side-chains of the residues involved in coordination with Zn1 and Zn2, were omitted for clarity.
In investigations of protein–ligand interactions by 1H,15N HSQC NMR titration experiments, the rate of the exchange process of the protein-bound and the free ligand is reflected in the chemical shift data.23,37,38 For exchange processes that are slow on the chemical shift timescale, the NMR signals representing the free and bound forms are simultaneously present in the spectra and vary in intensity over the course of the titration. The fraction of each peak is proportional to its concentration if the relaxation rates of the two states are identical. In these cases, it is possible to use the peak integrals to determine the dissociation constant (Kd). Ligands in slow exchange are often found to be strong binders. Before the protein is saturated, the small Kd will result in low free ligand concentrations. As a consequence, the exchange rate is much lower for ligands with strong interactions where low concentrations of the free ligand are needed to populate the bound state.23,37,38 Intermediate exchange processes are characterized by signal broadening, whereas the observed chemical shift for processes with a fast exchange rate is a population-weighted average of the chemical shifts of the free and the bound species. For processes in fast exchange, the Kd can be determined by least square fitting of the measured protein 1H and 15N chemical shifts as a function of the concentration of the ligands.
Fast exchange was detected for the majority of CcrA residues upon titration with 4 and 5, and slow exchange upon titration with 6 (see the Supporting Information Section S2.2 for details). To get the most representative dissociation constants for the compounds, only the data corresponding to the significantly shifted residues in the active site representing the L3-L5-L7 poses were used in the Kd calculations.23,37,38 Counterintuitively to the results of a previous study, in which 4 showed a superior reduction in the MIC of cephalexin V as compared with 5 (32-fold vs no change), the binding affinity of 5 (Kd = 34 ± 43 μM, Table S5) was only slightly weaker than that of 4 (Kd = 11 ± 11 μM, Table S4). By comparing the relative changes in the peak intensities during the titration of 6 its Kd was estimated to 2.3 ± 0.3 μM. Although 6 showed a stronger binding to CcrA as compared to 4, their antibiotic activity is analogous (MIC assay, Table 2). The higher binding affinity of 6 might partly be explained by the formation of a favorable hydrogen or halogen bond, or a salt bridge to CcrA via the 4-chlorophenyl group.
Next, the results from the investigation of 4–6 were used as a basis for the further development of the best-known 2TTA-type CcrA inhibitors 1–3 (see Table 1). In these compounds, the triazole ring is substituted with a 2-hydroxyphenyl or 4-pyridine moiety instead of a 2-carboxyphenyl moiety.14 For an initial comparison between the series, the compounds were evaluated by molecular docking (Supporting Information, Section S3.5). Both the phenol and phenolate forms of 1 and 2 shared the three main clusters of binding poses identified for 4–6 (i.e., L3-L5-L7, L3-L10, and L10-L12). A fourth pose was identified for 2, in which the phenyl group is positioned close to Trp83 in loop 7 (Trp83 is shown in Figure 4C). The phenolic hydroxy group or the triazole moiety of 1 and 2 are involved in interactions with the zinc ions. For 3, the L7(Trp83) and L10-L12 poses were the main ones identified, and zinc–ligand interactions were found solely for the triazole moiety (i.e., in two L10-L12 poses).
The relationship observed in in vitro CcrA inhibition constants for 1 and 2 (i.e., 6.3 and 0.3 μM) is in agreement with the in silico binding energies for the top L3-L5-L7 poses (i.e., −65.2 and −81.7 kcal mol–1). For 3, the pose ranked as number eleven (−55.2 kcal mol–1) is of L3-L5-L7 type, but the arrangement of the molecule is flipped (i.e., the benzothiazole moiety is located in the catalytic zinc site, and the pyridine in the L3-L5-L7 binding site). Thus, the in silico binding energy for 3 (8.5 μM and −55.2 kcal mol–1) is not directly comparable with those of 1 and 2.
All-known MBLs possess two conserved metal binding sites and require zinc ions as enzymatic cofactors; two zinc ions for class B1, B3, and B4 MBLs, and one zinc ion for class B2 MBLs. As long as a compound is able to prevent the targeted metallo-beta-lactamase enzymes from deactivating beta-lactam antibiotics, zinc coordination is not a prerequisite. But an efficient coordination with one or two zinc ions, as well as specific interactions with conserved active site residues and less specific interactions with nonconserved residues, would be beneficial for achieving potent broad-spectrum inhibitors with delayed resistance development.39 It is not surprising that the majority of reported MBLIs are forming coordination or chelation complexes with the zinc ions.3,6 To further explore and promote the zinc coordination ability of the biaryl moiety in 1–3, alternatives to the 2-hydroxyphenyl group holding a permanent negative charge were assessed by molecular docking (7–10, Figure 6). Heterocyclic N-oxide motifs, such as 2-pyridine-N-oxide in 7 and 8, have been successfully used as carbonyl bioisosteres, as well as ligands in coordination chemistry.40 While the potential of this group is relatively unexplored, the analogous pyrithione moiety in 9 and 10, is a well-known zinc(II) coordinating motif with a wide range of medical applications.41
Figure 6.

Compounds 7–10 are prospective analogues of the broad spectrum MBLIs 1 and 2, containing two different triazole substituents.
The predicted pKa’s (Table S11) of the triazole nitrogen of 7–10 are 6.9 and 8.8 for tautomer A and B (Figure 7), respectively, as compared to 7.5 and 10.5 in 1 and 2. For the pyridine-N-oxide and pyrithione, the pKa’s are 0.2 and 0.7 for tautomer A and B, respectively, whereas 8.9 and 9.4 for the corresponding 2-hydroxyphenyl group of 1 and 2. Thus, the degree of ionization is larger for both parts of the biaryl moiety. While the triazole is ionized to as much as 56%, the aryl group is fully ionized at pH 7.
Figure 7.

General structures of the 1H-tautomers A and B of the diaryl-substituted 2TTA-type MBLIs 1–10, in which Ar and Ar′ represents the aryl substituents.
The molecular docking study showed that L3-L5-L7-type binding poses were favored by the suggested structural modifications (Tables S13 and S14). The MM-GBSA binding free energies of the top L3-L5-L7 poses were −71.3, −56.4, −80.4, and −77.2 kcal mol–1 for 7–10, respectively. In comparison to the binding free energies of the top L3-L5-L7 poses of the reference compounds 1 and 2 in the deprotonated form (ΔGbind = −65.2 and −81.7 kcal mol–1), compounds 7 and 9 showed an improvement to 1; whereas 8 showed a reduction and 10 was comparable to 2. Interactions with Zn1 and Zn2 were mainly observed for the oxygen atoms of the pyridine-N-oxide and pyrithione, but also in combination with the triazole. The latter poses are attractive and interesting as the binding of the biaryl moiety resembles the binding mode of 4 in complex with VIM-2 (Figures 5, S25–S27) in the crystal structure (PDB 5LSC(26)). These type of poses were generally not observed for 1–6.42 In addition, the sulfur atom in 9 and 10 is close enough to form a hydrogen bond with Lys167 (BBL number 224). In previously reported L3-L5-L7-type docking poses of 2TTA MBLIs, including 2, the triazole has been identified as the main zinc coordinating group; and the hydroxyl group (or corresponding carboxyl group as in 4–6) to interact with the Lys167 residue.14,15 Based on the previous and current observations, the biaryl moiety is likely to adapt to the structural differences among the MBLs, and coordinate or chelate one or both zinc ions, resulting in a slight offset in the position of the thioacetamide moiety, thereby enabling a broad spectrum inhibitory activity of the 2TTA type MBLIs.
We observed a correlation between the binding pose and the main 1H-1,2,4-triazole tautomeric form among the top scoring poses. For all compounds except 9, the L3-L5-L7 pose was predicted to favor 1H-tautomer B (Figure 7 and Table S15). Tautomer A was the favored 1H-tautameric form of 9. Further studies are suggested to identify the opportunities that are contained in the 1H-1,2,4-triazole tautomerization.
Conclusions
NMR chemical shift mapping and DCSP analysis of the structurally closely related compounds 4–6 suggest them to bind to the active site of CcrA in one of three possible binding poses. In this arrangement, the benzothiazolyl, phenyl, and 4-clorophenyl groups of 4–6, respectively, are located in the binding site that is formed by loops 3, 5, and 7 (L3-L5-L7). The biaryl moiety, that is, the 2-carboxyphenyl substituted triazole, is located in the catalytic zinc site. Because the predicted free energy of binding of the top scoring poses are comparable, the spatial arrangement could possibly switch upon structural modifications, or upon interaction with other MBLs.20 Such a feature offers great possibilities in the development of broad spectrum MBLIs.
Inspired by the knowledge obtained for 4–6, possible directions for the further development of the closely related broad spectrum MBLIs 1–3 were investigated using molecular docking. The focus was to promote their zinc binding properties, as well as binding poses, in line with the experimental NMR data of 4–6 and with the previous literature. By replacing the 2-hydroxyphenyl group of the biaryl moiety in 1 and 2 by the significantly more acidic pyridine-N-oxide or pyrithione moiety, poses displaying similar in silico binding affinities and a stronger preference for the desired spatial arrangement were acquired. It remains to be seen whether the trends in vitro conform to the trends seen in silico; however, both the pyridine-N-oxide and the pyrithione moieties are suggested to be promising alternatives to the 2-hydroxyphenyl moiety.
The MBL activity spectrum needed for a compound to be clinically relevant is constantly changing because of the emergence and spread of MBL-producing bacterial pathogens. Filling existing and emerging knowledge gaps is important for maintaining and increasing the therapeutic potential of MBLIs.
Materials and Methods
Compounds 4–6, used in the NMR study, were prepared as described previously.15 Uniformly 13C- and 15N-labeled CcrA, as well as 15N-labeled CcrA, was supplied by Lund Protein Production Platform (LP3) based at Lund University. For NMR backbone resonance assignment, a sample with 0.6 mM uniformly 13C,15N-labeled CcrA in 10 mM phosphate buffer (pH 7) with 1.0 mM ZnSO4 was prepared and added into a 3 mm Shigemi tube. NMR spectra were acquired at 35 °C on a 800 MHz spectrometer equipped with a 3 mm TCI cryogenic probe. The data was acquired with sweep ranges without folding, and with nonuniform sampling using a 25% sampling density. The spectra were reconstructed via the compressed sensing algorithm within the MddNMR software and processed using NMRPipe. CCPN software (V2) was used for peak assignment and peak picking. The 26.5 kDa CcrA protein likely forms a dimer, and accordingly some 3D experiments gave a low signal-to-noise ratio because of relaxation. For assignment, we predominantly used HNCA and HNcoCA that gave good s/n, and the HNCACB experiment, which gave reasonable s/n. The assignment was performed by conventional peak picking, and walking along the backbone connecting each amino acid residue with the 1–n amino acid residue through the Cα atom, which is identified both via the intra- (HNCA and HNCACB) and interresidue correlation (HNcoCA) experiments. Assignment of at least one of the H–N and H–C peaks was achieved for 98% of the amino acid residues. The 13C chemical shift data is presented in Table S2, and the 1H and 15N chemical shift data is presented in Tables S4–S6 in the Supporting Information. Further experimental details are given in the Supporting Information.
Acknowledgments
The Swedish Research Council is acknowledged for financial support (grant 2013-8804). The Swedish NMR Centre at the University of Gothenburg is acknowledged for instrument access and support (Cecilia Persson). Anna Rasmussen of Lund Protein Production Platform (LP3) at Lund University, managed by Wolfgang Knecht, is acknowledged for supplying stable isotope-labeled CcrA. Vasanthanathan Poongavanam is acknowledged for sharing his expertise in molecular docking. We thank Alan Vanderkooy for the helpful reading of and instructive comments to this text.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02187.
Detailed information about CcrA, NMR experiments, and molecular docking (PDF)
Author Present Address
∥ Karolinska Experimental Research and Imaging Center, Karolinska University Hospital Solna, SE-171 76 Stockholm, Sweden.
Author Contributions
The compounds were synthesized by S.-K.Y. in the lab of K.-W.Y. The NMR experiments and NMR data processing was performed by P.J. in the lab of M.E.. The NMR data analysis was performed by H.A. and P.J. in the lab of M.E. The molecular docking study and design of inhibitor analogues were performed by H.A. in the lab of M.E.. The manuscript was written by H.A. and M.E., with contributions to the Supporting Information from P.J.. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Antibacterial Agents in clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline, Including Tuberculosis; World Health Organization: Geneva, 2017.
- Laws M.; Shaaban A.; Rahman K. M. Antibiotic resistance breakers: current approaches and future directions. FEMS Microbiol. Rev. 2019, 43, 490–516. 10.1093/femsre/fuz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooke C. L.; Hinchliffe P.; Bragginton E. C.; Colenso C. K.; Hirvonen V. H. A.; Takebayashi Y.; Spencer J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. 10.1016/j.jmb.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambler R. P. The structure of beta-lactamases. Philos. Trans. R. Soc. London, Ser. B 1980, 289, 321–331. 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- Bush K.; Jacoby G. A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. 10.1128/aac.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docquier J.-D.; Mangani S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updates 2018, 36, 13–29. 10.1016/j.drup.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Wexler H. M. Bacteroides: the Good, the Bad, and the Nitty-Gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. 10.1128/cmr.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pence M. A. Antimicrobial Resistance in Clinically Important Anaerobes. Clin. Microbiol. Newsl. 2019, 41, 1–7. 10.1016/j.clinmicnews.2018.12.003. [DOI] [Google Scholar]
- Rasmussen B. A.; Gluzman Y.; Tally F. P. Cloning and sequencing of the class B beta-lactamase gene (ccrA) from Bacteroides fragilis TAL3636. Antimicrob. Agents Chemother. 1990, 34, 1590–1592. 10.1128/aac.34.8.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. S.; Malamy M. H. Sequencing the gene for an imipenem-cefoxitin-hydrolyzing enzyme (CfiA) from Bacteroides fragilis TAL2480 reveals strong similarity between CfiA and Bacillus cereus beta-lactamase II. J. Bacteriol. 1990, 172, 2584–2593. 10.1128/jb.172.5.2584-2593.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho P.-L.; Yau C.-Y.; Ho L.-Y.; Chen J. H. K.; Lai E. L. Y.; Lo S. W. U.; Tse C. W. S.; Chow K.-H. Rapid detection of cfiA metallo-β-lactamase-producing Bacteroides fragilis by the combination of MALDI-TOF MS and CarbaNP. J. Clin. Pathol. 2017, 70, 868–873. 10.1136/jclinpath-2017-204335. [DOI] [PubMed] [Google Scholar]
- Gao Q.; Wu S.; Xu T.; Zhao X.; Huang H.; Hu F. Emergence of carbapenem resistance in Bacteroides fragilis in China. Int. J. Antimicrob. Agents 2019, 53, 859–863. 10.1016/j.ijantimicag.2019.02.017. [DOI] [PubMed] [Google Scholar]
- Jeverica S.; Sóki J.; Premru M. M.; Nagy E.; Papst L. High prevalence of division II (cfiA positive) isolates among blood stream Bacteroides fragilis in Slovenia as determined by MALDI-TOF MS. Anaerobe 2019, 58, 30–34. 10.1016/j.anaerobe.2019.01.011. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-L.; Yang K.-W.; Zhou Y.-J.; LaCuran A. E.; Oelschlaeger P.; Crowder M. W. Diaryl-Substituted Azolylthioacetamides: Inhibitor Discovery of New Delhi Metallo-β-Lactamase-1 (NDM-1). ChemMedChem 2014, 9, 2445–2448. 10.1002/cmdc.201402249. [DOI] [PubMed] [Google Scholar]
- Yang S.-K.; Kang J. S.; Oelschlaeger P.; Yang K.-W. Azolylthioacetamide: A Highly Promising Scaffold for the Development of Metallo-β-lactamase Inhibitors. ACS Med. Chem. Lett. 2015, 6, 455–460. 10.1021/ml500534c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senac C.; Desgranges S.; Contino-Pépin C.; Urbach W.; Fuchs P. F. J.; Taulier N. Effect of Dimethyl Sulfoxide on the Binding of 1-Adamantane Carboxylic Acid to β- and γ-Cyclodextrins. ACS Omega 2018, 3, 1014–1021. 10.1021/acsomega.7b01212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallerstein J.; Akke M. Minute Additions of DMSO Affect Protein Dynamics Measurements by NMR Relaxation Experiments through Significant Changes in Solvent Viscosity. ChemPhysChem 2019, 20, 326–332. 10.1002/cphc.201800626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toney J. H.; Cleary K. A.; Hammond G. G.; Yuan X.; May W. J.; Hutchins S. M.; Ashton W. T.; Vanderwall D. E. Structure-activity relationships of biphenyl tetrazoles as metallo-β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 1999, 9, 2741–2746. 10.1016/s0960-894x(99)00458-8. [DOI] [PubMed] [Google Scholar]
- Toney et al. reported that since there was no clear electron density at the position of the zinc-bridging nucleophilic water molecule (hydroxide ion) its presence could not be confirmed. Thus, it is not shown in the reported crystal structure (PDB ID 1A8T).
- Zhai L.; Zhang Y.-L.; Kang J. S.; Oelschlaeger P.; Xiao L.; Nie S.-S.; Yang K.-W. Triazolylthioacetamide: A Valid Scaffold for the Development of New Delhi Metallo-β-Lactmase-1 (NDM-1) Inhibitors. ACS Med. Chem. Lett. 2016, 7, 413–417. 10.1021/acsmedchemlett.5b00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Chang Y.-N.; Ge Y.; Kang J. S.; Zhang Y.-L.; Liu X.-L.; Oelschlaeger P.; Yang K.-W. Azolylthioacetamides as a potent scaffold for the development of metallo-β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5225–5229. 10.1016/j.bmcl.2017.10.038. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yan Y.; Liang L.; Feng J.; Wang X.; Li L.; Yang K. Halogen-Substituted Triazolethioacetamides as a Potent Skeleton for the Development of Metallo-β-Lactamase Inhibitors. Molecules 2019, 24, 1174. 10.3390/molecules24061174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Schnaars C.; Kildahl-Andersen G.; Prandina A.; Popal R.; Radix S.; Le Borgne M.; Gjøen T.; Andresen A. M. S.; Heikal A.; Økstad O. A.; Fröhlich C.; Samuelsen Ø.; Lauksund S.; Jordheim L. P.; Rongved P.; Åstrand O. A. H. Synthesis and Preclinical Evaluation of TPA-Based Zinc Chelators as Metallo-β-lactamase Inhibitors. ACS Infect. Dis. 2018, 4, 1407–1422. 10.1021/acsinfecdis.8b00137. [DOI] [PubMed] [Google Scholar]
- Crowder M. W.; Wang Z.; Franklin S. L.; Zovinka E. P.; Benkovic S. J. Characterization of the Metal-Binding Sites of the β-Lactamase from Bacteroides fragilis. Biochemistry 1996, 35, 12126–12132. 10.1021/bi960976h. [DOI] [PubMed] [Google Scholar]
- Christopeit T.; Yang K.-W.; Yang S.-K.; Leiros H.-K. S. The structure of the metallo-β-lactamase VIM-2 in complex with a triazolylthioacetamide inhibitor. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 813–819. 10.1107/s2053230x16016113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrofani S. D. B.; Chung J.; Huntley J. J. A.; Benkovic S. J.; Wright P. E.; Dyson H. J. NMR Characterization of the Metallo-β-lactamase from Bacteroides fragilis and Its Interaction with a Tight-Binding Inhibitor: Role of an Active-Site Loop. Biochemistry 1999, 38, 14507–14514. 10.1021/bi990986t. [DOI] [PubMed] [Google Scholar]
- Palzkill T. Metallo-β-lactamase structure and function. Ann. N.Y. Acad. Sci. 2013, 1277, 91–104. 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galleni M.; Lamotte-Brasseur J.; Rossolini G. M.; Spencer J.; Dideberg O.; Frère J.-M. Standard Numbering Scheme for Class B β-Lactamases. Antimicrob. Agents Chemother. 2001, 45, 660–663. 10.1128/aac.45.3.660-663.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães C. R. W.; Cardozo M. MM-GB/SA Rescoring of Docking Poses in Structure-Based Lead Optimization. J. Chem. Inf. Model. 2008, 48, 958–970. 10.1021/ci800004w. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chuaqui C.; Singh J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein–Ligand Binding Interactions. J. Med. Chem. 2004, 47, 337–344. 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- Abulwerdi F. A.; Liao C.; Mady A. S.; Gavin J.; Shen C.; Cierpicki T.; Stuckey J. A.; Showalter H. D. H.; Nikolovska-Coleska Z. 3-Substituted-N-(4-Hydroxynaphthalen-1-yl)arylsulfonamides as a Novel Class of Selective Mcl-1 Inhibitors: Structure-Based Design, Synthesis, SAR, and Biological Evaluation. J. Med. Chem. 2014, 57, 4111–4133. 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedinger C.; Endicott J. A.; Kemp S. J.; Smyth L. A.; Watson A.; Valeur E.; Golding B. T.; Griffin R. J.; Hardcastle I. R.; Noble M. E.; McDonnell J. M. Analysis of Chemical Shift Changes Reveals the Binding Modes of Isoindolinone Inhibitors of the MDM2-p53 Interaction. J. Am. Chem. Soc. 2008, 130, 16038–16044. 10.1021/ja8062088. [DOI] [PubMed] [Google Scholar]
- The DCSP diverge if the signs of (Δ1H,Δ15N) are not all = (+,+), (+,−), (−,+) or (−,−); and the DSCP does not diverge if the signs of (Δ1H,Δ15N) are all = (+,+), (+,−), (−,+) or (−,−).
- Rivière G.; Oueslati S.; Gayral M.; Créchet J.-B.; Nhiri N.; Jacquet E.; Cintrat J.-C.; Giraud F.; van Heijenoort C.; Lescop E.; Pethe S.; Iorga B. I.; Naas T.; Guittet E.; Morellet N. NMR Characterization of the Influence of Zinc(II) Ions on the Structural and Dynamic Behavior of the New Delhi Metallo-β-Lactamase-1 and on the Binding with Flavonols as Inhibitors. ACS Omega 2020, 5, 10466–10480. 10.1021/acsomega.0c00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X.; Zhu T.; Zhang J. Z. H.; He X. Automated Fragmentation QM/MM Calculation of NMR Chemical Shifts for Protein-Ligand Complexes. Front. Chem. 2018, 6, 150. 10.3389/fchem.2018.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding L. NMR Methods for the Determination of Protein- Ligand Dissociation Constants. Curr. Top. Med. Chem. 2003, 3, 39–53. 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- Teilum K.; Kunze M. B. A.; Erlendsson S.; Kragelund B. B. (S)Pinning down protein interactions by NMR. Protein Sci. 2017, 26, 436–451. 10.1002/pro.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez-Campoy A.; Freire E. Incorporating target heterogeneity in drug design. J. Cell. Biochem. 2001, 84, 82–88. 10.1002/jcb.10068. [DOI] [PubMed] [Google Scholar]
- Mfuh A. M.; Larionov O. V. Heterocyclic N-Oxides - An Emerging Class of Therapeutic Agents. Curr. Med. Chem. 2015, 22, 2819–2857. 10.2174/0929867322666150619104007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandiford L.; Holmes A. M.; Mangion S. E.; Mohammed Y. H.; Zvyagin A. V.; Roberts M. S. Optical Characterization of Zinc Pyrithione. Photochem. Photobiol. 2019, 95, 1142–1150. 10.1111/php.13100. [DOI] [PubMed] [Google Scholar]
- Control experiments, in which the water molecule was removed and Lys167(224) was protonated, were performed, as well as experiments using a different crystal structure (PDB 1A7T). The result of these controls was comparable to those presented in the article and are therefore not shown.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.