

Abstract
Deposition of Aβ42 aggregates in the form of amyloid plaques is a pathological hallmark of Alzheimer’s disease. A desired avenue of intervention is the inhibition of Aβ42 aggregation. Epigallocatechin gallate (EGCG), the main polyphenol in green tea, has been generally considered an inhibitor of Aβ aggregation. However, previous experiments focused on the reduction of the amount of Aβ42 aggregates, while the effect of EGCG on the rate of Aβ42 aggregation was not critically analyzed. Here we performed an experimental evaluation of Aβ42 aggregation kinetics in the absence and presence of EGCG at a wide range of concentrations. We found that EGCG reduced thioflavin T fluorescence in an EGCG concentration-dependent manner, suggesting that EGCG reduced the amount of Aβ42 fibrils. The effect of EGCG on the rate of Aβ42 aggregation appears to be bimodal. We found that higher EGCG-to-Aβ42 ratios promoted the rate of Aβ42 aggregation, while lower EGCG-to-Aβ42 ratios inhibited the aggregation rate. To confirm that the reduction of thioflavin T fluorescence is due to the lowered aggregate quantity, but not due to perturbation of thioflavin T binding to Aβ42 fibrils, we probed the effect of EGCG on Aβ42 aggregation using site-directed spin labeling. Electron paramagnetic resonance of spin-labeled Aβ42 aggregates suggests that high EGCG-to-Aβ42 ratios led to a greatly reduced amount of Aβ42 fibrils, and these aggregates adopt similar structures as the fibrils in the no-EGCG sample. Potential implications of this work in designing prevention or therapeutic strategies using EGCG are discussed.
Introduction
Alzheimer’s disease, the most common cause of dementia, has two pathological hallmarks: amyloid plaques and neurofibrillary tangles.1−3 Amyloid plaques consist of the fibrillar aggregates of Aβ protein, while tau aggregates make up the neurofibrillary tangles. Aβ proteins are produced from the proteolytic cleavage of amyloid precursor protein by β- and γ-secretases4,5 and are composed of two main isoforms: the 40-residue Aβ40 and the 42-residue Aβ42. Aβ42 is identical to Aβ40 with the exception of two extra residues at the C-terminal end. Other variants of Aβ42 with both N- and C-terminal heterogeneities also exist.6 Aβ40 is the most abundant isoform of Aβ in the brain, several folds more than Aβ42, but Aβ42 is the major Aβ isoform in the plaques.7−10 Formation of Aβ fibrils is a result of Aβ aggregation, a process that has been extensively studied.11−17 With amyloid being the central player of the amyloid cascade hypothesis, inhibiting Aβ aggregation is an obvious avenue for therapeutic intervention. Polyphenols, a group of natural compounds commonly found in fruit and vegetables,18 have attracted a lot of attention for their antiaggregation activities.19 Epigallocatechin gallate (EGCG), the main polyphenol compound in green tea, is the focus of this study.
EGCG has been associated with reduced risk for dementia or cognitive impairment. A meta-analysis of 17 studies involving 48 435 participants showed that green tea consumption was associated with a significant reduction in the risk of cognitive disorders.20 Dose–response meta-analysis showed that an increment of 100, 300, and 500 mL/day of tea consumption was associated with a 6, 19, and 29% lower risk of cognitive disorders.20 Human intervention studies also showed benefits of EGCG. In a randomized, double-blind, placebo-controlled study,21 the effect of green tea extract (720 mg, twice daily) was studied in 91 mild cognitive impairment (MCI) subjects for 16 weeks. This study showed that green tea extract improved memory and selective attention in subjects with Mini Mental State Examination-Korea scores of 21–23. Another double-blind randomized study in Japan using 2000 mg/day of green tea powder (containing 220 mg of catechins) for 12 months showed that green tea did not significantly affect cognitive function but prevented the increase of oxidative stress in the elderly population.22
EGCG has been shown to reduce the amount of amyloid plaques in transgenic animals, but the effect was largely attributed to indirect interactions. Tan and co-workers showed that EGCG reduced plaque load in mice and increased the α-secretase processing of amyloid precursor protein.23−25 Levites et al.26 reported that EGCG increased α-secretase processing of amyloid precursor protein via a protein kinase C pathway. In vitro studies suggest that EGCG has a direct effect on Aβ aggregation. Previous studies on EGCG and Aβ aggregation have mostly focused on the yield of Aβ aggregation. Bastianetto et al.,27 Sinha et al.,28 and Liu et al.29 showed that EGCG reduced thioflavin T fluorescence of Aβ42 aggregates in a concentration-dependent manner. Ehrnhoefer et al.30 studied Aβ42 aggregation in the presence of 10 and 100% EGCGs and showed the reduction of Aβ42 aggregates using both thioflavin T fluorescence and transmission electron microscopy (TEM). Sironi et al.31 showed that EGCG reduced the amount of Aβ42 fibrils using TEM. There are also studies on the effect of EGCG on preformed Aβ42 fibrils, and the general conclusion from these studies was that EGCG remodels Aβ4232 and Aβ4033 fibrils to form nonfibrillar aggregates. In two studies,28,30 kinetics experiments were performed for Aβ42 aggregation, but only limited data points were collected and no analysis on the rate of aggregation was performed. Therefore, even though EGCG has been shown to reduce the yield of Aβ42 aggregation, its effect on the aggregation rate has not been critically evaluated.
In this work, we set out to distinguish the effect of EGCG on the rate and yield of Aβ42 aggregation using kinetics experiments. In a kinetics experiment, there are two aspects of inhibiting effects: the rate and yield of aggregation. The aggregation rate refers to how fast the Aβ fibrils are formed, and the aggregation yield refers to the amount of Aβ42 fibrils upon completion of aggregation. Aβ aggregation can be described by a nucleation-dependent polymerization model.34 The kinetics curve can be separated into three phases: lag phase, growth phase, and plateau phase (Figure 1). The main event in the lag phase is the formation of fibril nuclei. For this reason, the lag phase is often called the nucleation phase. When the fibril nuclei accumulate to certain threshold concentrations, elongation of the fibril nuclei becomes the dominant event, leading to the fibril growth phase. Eventually, the Aβ monomers are largely exhausted and fibril growth reaches a plateau. There are three parameters that may represent some aspects of aggregation rate: lag time, half-time, and apparent fibril growth rate in the growth phase (Figure 1). These three parameters are also related, as shown in Figure 1. Lag time is closely related to the nucleation rate of Aβ fibrils, although secondary nucleation may complicate the exact meaning of lag time.35 The amplitude of the plateau represents the amount of Aβ fibrils at the end of the kinetics experiment.
Figure 1.

Typical sigmoidal aggregation curve with three aggregation phases and illustration of fitting parameters. The aggregation kinetics is described using the sigmoidal function shown at the top.
The thioflavin T fluorescence amplitude was used to evaluate the amount of Aβ42 fibrils.36 The kinetics data were fitted to a sigmoidal function to obtain the lag time, half-time, and apparent fibril growth rate. We found that EGCG reduces the yield of Aβ42 aggregation in a concentration-dependent manner, consistent with previous findings. The amyloid-reducing effect was confirmed using electron paramagnetic resonance (EPR) experiments. The effect on the rate of Aβ42 aggregation, however, is complicated. While low concentrations of EGCG delayed the formation of Aβ42 fibrils, high concentrations of EGCG promoted fibril formation. The implication of our findings in the context of designing human interventions is discussed.
Results and Discussion
Effect of EGCG on the Rate of Aβ42 Aggregation
Using a recombinant system to produce Aβ proteins, we have demonstrated the capability to generate aggregation kinetics data with well-resolved lag phase.37−39 This allows us to study the effect of EGCG on the rate of Aβ42 aggregation. We performed an Aβ42 aggregation at a 2 μM concentration, with EGCG concentrations at 0, 0.1, 0.2, 0.4, 1, 2, 4, 10, 20, and 40 μM, representing EGCG-to-Aβ42 ratios of 0:1, 0.05:1, 0.1:1, 0.2:1, 0.5:1, 1:1, 2:1, 5:1, 10:1, and 20:1, respectively. All of the different EGCG-to-Aβ42 ratios were performed in a single experiment, starting from a master mix containing Aβ42 at 2.2 μM and thioflavin T at 22 μM in phosphate-buffered saline (PBS). Separately, we made a series of EGCG stock solutions at 1–400 μM, using a serial dilution from 400 μM EGCG. Then, 45 μL of the master mix was mixed with 5 μL of the EGCG stock at different concentrations. With this experimental setup, the Aβ42 and thioflavin T were identical in each aggregation reaction, making any differences attributable to EGCG concentrations. Because we used a serial dilution to make different EGCG stocks from the 400 μM concentration, the relative EGCG concentration in different aggregation samples is also well controlled, even though the absolute EGCG concentration may be subject to errors such as due to EGCG purity.
The aggregation was performed in PBS buffer at 37°C without agitation. The kinetics traces are shown in Figure 2A. For clarity, these aggregation data are displayed in two figure panels. Qualitatively, the aggregation traces are shifted to the left at EGCG-to-Aβ42 ratios of 1:1 or higher, suggesting accelerated aggregation (Figure 2A, top panel). At EGCG-to-Aβ42 ratios of lower than 1:1, the aggregation traces are shifted to the right, suggesting delayed aggregation (Figure 2A, bottom panel). To quantify the lag time, half-time, and fibril growth rate (i.e., the slope in the growth phase), we fitted the data to a sigmoidal function (see Methods and Figure 1), and the fitted kinetics parameters are shown in Figure 2B–D. All of the data were fitted with the exception of the EGCG-to-Aβ42 ratio of 20:1, which shows very low amplitude in thioflavin T fluorescence and the fitting did not converge. All of the fitted traces are shown in Supporting Information Figure S1.
Figure 2.

Effect of EGCG on the rate and yield of Aβ42 aggregation. (A) Aggregation of 2 μM Aβ42 in the absence and presence of 0.1, 0.2, 0.4, 1, 2, 4, 10, 20, and 40 μM of EGCG, representing EGCG-to-Aβ42 ratios of 0.05:1, 0.1:1, 0.2:1, 0.5:1, 1:1, 2:1, 5:1, 10:1, and 20:1. The aggregation was performed in PBS buffer at 37 °C without agitation. Three replicates were performed for all samples. (B–D) Lag time, half-time, and fibril growth rate were obtained by fitting the aggregation data to a sigmoidal function (see Figure 1 and Methods). (E) Fluorescence intensity in the 27th hour of aggregation is plotted as a function of the EGCG-to-Aβ42 ratio. Columns are the mean and error bars are the standard deviation from the three aggregation repeats.
At the low end of EGCG-to-Aβ42 ratios (i.e., 0.05:1), the lag time is longer than that of Aβ42 without EGCG (Figure 2B), suggesting that small amount of EGCG delayed Aβ42 aggregation. However, the lag time decreases with increasing concentrations of EGCG from 0.05 to 2 μM, suggesting that higher EGCG-to-Aβ42 ratios promote Aβ42 aggregation. The aggregation-promoting effect of EGCG appears to have a limit because no dramatic changes in the lag time were observed when further increasing EGCG concentration from 2 to 20 μM. It is also worth noting that the lag time was not completely eliminated even when EGCG-to-Aβ42 ratios reach 10:1. The half-time data show a very similar trend (Figure 2C), which is not surprising because the lag time and the half-time are closely related (Figure 1).
The slope of the fibril growth phase represents the apparent fibril growth rate. Our data in Figure 2D shows that the fibril growth rate is higher with high EGCG concentrations (EGCG-to-Aβ42 ratios of 10:1, 5:1, and 2:1) than those with low EGCG concentrations (EGCG-to-Aβ42 ratios of 1:1 and lower). This is consistent with the accelerating effect of EGCG on the lag time and half-time at high concentrations, as shown in Figure 2B,C.
Based on the analysis of lag time, half-time, and fibril growth rate, the effect of EGCG on the Aβ42 aggregation rate is complex. High EGCG concentrations appear to accelerate Aβ42 aggregation, but the effect is limited as increasing the EGCG ratio beyond 1:1 and up to 10:1 did not completely eliminate the lag phase. Lower EGCG ratios of 0.2:1, 0.1:1, and 0.05:1 slowed down the Aβ42 aggregation by increasing the lag time.
Effect of EGCG on the Yield of Aβ42 Aggregation
In addition to the rate of aggregation, EGCG also showed a concentration-dependent effect on the amplitude of thioflavin T fluorescence (Figure 2A). To quantify this effect, we averaged the fluorescence readings in the last hour of aggregation and plotted them as a function of EGCG concentration (Figure 2E). The results show a monotonic decline in thioflavin T fluorescence with increasing concentrations of EGCG. Thioflavin T fluorescence can be affected by a number of factors. Fibril structure determines thioflavin T binding affinity and its fluorescence quantum yield. The concentration of thioflavin T and the presence of competing binders can also affect the number of thioflavin T molecules bound to the fibril. Xue et al. showed that Aβ42 fibril amount scales linearly with thioflavin T fluorescence for the same type of fibril at a fixed thioflavin T concentration.36 With the assumption that EGCG does not change the fibril structure or interfere with thioflavin T binding to a great extent, we can conclude that EGCG reduces the yield of Aβ42 aggregation in a concentration-dependent manner. This is consistent with several previous investigations27−30 in which EGCG was shown to reduce the thioflavin T fluorescence of Aβ42 aggregation, and these studies27−30 also concluded that EGCG inhibited Aβ aggregation.
The main evidence supporting that EGCG reduces Aβ42 aggregation yield came from TEM studies, which showed amorphous31 or globular aggregates29,30 in the presence of EGCG. In this work, we performed TEM studies to examine the morphology of the Aβ42 aggregates. As shown in Figure 3, fibrils are dominant in the low ratios (0.1:1 and 0.5:1) of EGCG to Aβ42, similar to those of the no-EGCG sample. In the EGCG-to-Aβ42 ratio of 5:1 sample, we observed both fibrillar and nonfibrillar aggregates (Figure 3D). Ehrnhoefer et al.30 suggested that EGCG redirects Aβ to form off-pathway oligomers. Our results are consistent with such an explanation. Unfortunately, TEM is not a quantitative technique to assess the concentration-dependent effect of EGCG on the yield of Aβ aggregation because different aggregates may have different binding and staining properties on EM grids. This motivated us to further study EGCG’s effect on Aβ aggregation yield using spin labeling and EPR spectroscopy.
Figure 3.

Transmission electron microscopy images of Aβ42 aggregates in the absence (A) and presence of EGCG at different ratios (B–D). Note that at an EGCG ratio of 5:1, both fibrils and globular oligomers were observed.
Although the decrease in thioflavin T fluorescence likely results from the reduction of aggregation yield by EGCG, it is possible that EGCG simply interfered with the binding between thioflavin T and Aβ42 fibrils without actually affecting the aggregation yield. Palhano et al.33 showed that EGCG interfered, but did not abolish, thioflavin T binding to Aβ40 fibrils. Using saturation transfer difference NMR, Roy et al.40 showed that EGCG competes with thioflavin T for the same binding sites on α-synuclein fibrils. Therefore, the explanation for the reduction in thioflavin T fluorescence may be complicated. To further investigate this, we assessed the effect of EGCG on the yield of Aβ42 aggregation using EPR spectroscopy. We used Aβ42 with a spin label named R1 introduced at residue Leu-34 to prepare fibrils in the absence and presence of EGCG with EGCG-to-Aβ42 ratios of 10:1 and 50:1 (Figure 4A). Aggregation kinetics shows that EGCG at these ratios reduced the thioflavin T fluorescence to near-background levels (Supporting Information Figure S2). The three samples have the same amount of Aβ42 at the beginning of aggregation. At the end of aggregation, all insoluble aggregates were collected by centrifugation and loaded into EPR capillaries. The parameters for EPR measurements were kept the same for the three samples to allow quantitative comparison of the insoluble aggregates. The EPR spectra of these three samples are shown in Figure 4B. There is a clear difference in the overall amplitude of the EPR spectra, suggesting that EGCG reduced the amount of Aβ42 fibrils in a concentration-dependent manner. We quantified the number of spins in the three samples by calculating the double integral of the EPR spectrum. The results show that the amount of fibrils in the 10:1 (EGCG:Aβ42) sample is 36% of the no-EGCG sample, and the fibril amount is reduced to 20% in the 50:1 (EGCG:Aβ42) sample.
Figure 4.

EPR studies of spin-labeled Aβ42 fibrils formed in the absence and presence of EGCG. (A) Schematic diagram showing the experimental procedure. (B) EPR spectra of Aβ42 fibrils spin-labeled at residue L34. R1 represents the spin label and is introduced by site-directed spin labeling. (C) EPR spectra in panel B are normalized to the same centerline amplitude to allow comparison of the EPR line shape. Note that the EPR spectra in the presence of EGCG show similar line shape as the no-EGCG sample, indicating similar fibril structure. (D) EPR spectrum of Aβ42 L34R1 globulomers, reproduced using data in Gu et al.44 (E) EPR spectrum of Aβ42 L34R1 prefibrillar oligomers, reproduced using data in Gu et al.45 The scan width is 200 G for all of the EPR spectra.
The EPR spectrum of Aβ42 L34R1 fibrils in the absence of EGCG is characteristic of the parallel in-register β-sheet structure.41−43 The EPR spectra in the presence of 10× and 50× EGCGs have a similar line shape compared to those of the no-EGCG sample (Figure 4C), suggesting that EGCG did not change the structure of Aβ42 fibrils. Previously, we have obtained the EPR spectra of Aβ42 globulomers44 (Figure 4D) and prefibrillar oligomers45 (Figure 4E) spin-labeled at the same residue position. The EPR spectra of these oligomers are reproduced here for comparison to show the distinct spectral line shape between oligomers and fibrils.
The rate and yield of aggregation normally have a positive correlation. The most straightforward example is probably the aggregation of the same protein under identical conditions but with different concentrations. At lower concentrations, the rate of aggregation is slower, and the yield of aggregation is also lower. In contrast, at higher concentrations, the rate of aggregation is faster, and there are more aggregates formed. Numerous examples for this exist in the literature, including Aβ,46 α-synuclein,47 and tau.47 This issue gets complicated when the aggregation is used to investigate the effect of mutations or aggregation-modifying compounds. For example, in the alanine scanning mutagenesis study of Aβ40, Williams et al.48 showed that Aβ40 K16A mutant had a slower rate than E22A, but a higher thioflavin T fluorescence intensity at aggregation plateau. In a spin label scanning study of Aβ42 in which the aggregation kinetics of 42 spin-labeled Aβ42 was analyzed, Hsu et al.37 found that there was no correlation between the half-time of aggregation and thioflavin T fluorescence amplitude. Although inhibiting aggregation can mean either slowing down aggregation rate or reducing aggregation yield, it is important to make the distinction because the rate and yield may not change at the same direction.
Our findings that EGCG promotes the rate of Aβ42 aggregation at high EGCG-to-Aβ ratios while at the same time reduces the amount of Aβ42 fibrils have important implications for using EGCG as a potential treatment or prevention for Alzheimer’s disease. If the goal is to reduce the amyloid load, it is clear that higher concentrations of EGCG have a more pronounced effect. Therefore, it is desirable to use the highest dosage that is safely achievable. At the time of diagnosis, patients with Alzheimer’s disease already have a large amount of amyloid deposits in the brain. At this stage, one main therapeutic goal is to reduce amyloid load, so high concentrations of EGCG are desired. If the goal is to delay the appearance of amyloid plaques, however, caution should be taken to investigate the effect of EGCG on Aβ42 aggregation at physiologically relevant concentrations. Our results show that low concentrations of EGCG delayed Aβ42 aggregation, while higher concentrations of EGCG promoted Aβ42 aggregation (Figure 2). It is important to uncover this EGCG concentration-dependent effect at physiologically relevant Aβ concentrations. There are reports that EGCG reduces the amount of amyloids in transgenic mice, but the authors attribute the effect to the α-secretase pathway.23,26 The effect of EGCG on the rate of amyloid formation in transgenic animals has not yet been investigated. In vitro, concentrations in the picomolar and nanomolar ranges can be studied using highly sensitive fluorescence methods,49 which may provide further insights into the interactions between Aβ and EGCG.
Our findings also have implications on the mechanism by which EGCG affects Aβ aggregation (Figure 5). Previously, it has been observed that EGCG redirects Aβ42 aggregation toward off-pathway oligomerization.30 This effect can, in essence, reduce the effective Aβ42 concentration and thus leads to a reduced aggregation rate. Furthermore, EGCG can also remodel preformed Aβ42 fibrils.32 This effect can reduce the rate of secondary nucleation, which is catalyzed by fibrillar aggregates.50 Perhaps, more surprising is the increased aggregation rate at high EGCG-to-Aβ ratios (Figure 2). We speculate that this is due to the reversibility of the EGCG’s remodeling effect on Aβ fibrils (shown as a red arrow in the schematic illustration in Figure 5). In other words, we think that the EGCG-induced oligomers can also form fibrils, but the EGCG-induced oligomers are much more stable than fibrils, thus leading to a net reduction of the amount of fibril. The rate of the fibril formation, however, may be accelerated by EGCG when EGCG-induced oligomers accumulate to high concentrations.
Figure 5.
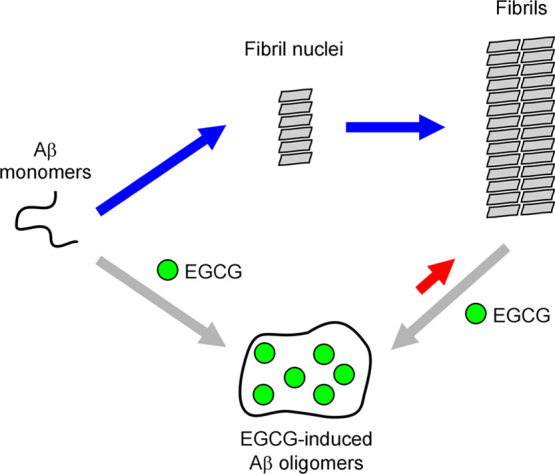
Schematic illustration of Aβ aggregation showing a potential mechanism of action by EGCG. A simplified nucleation-dependent polymerization mechanism for Aβ aggregation is depicted. EGCG can induce the formation of oligomers from Aβ monomers, leading to a reduction of the effective Aβ monomer concentration. EGCG can also remodel Aβ fibrils to form oligomers, thus decreasing the fibril-catalyzed secondary nucleation. We speculate that EGCG-induced oligomers can also form fibrils, as represented by the red arrow. In the cases of high EGCG-to-Aβ ratios, this aggregation pathway may significantly contribute to the overall aggregation rate. If the aggregation rate from EGCG-induced oligomers (red arrow) is faster than the spontaneous Aβ aggregation rate (blue arrows), EGCG would show a net acceleration effect for the rate of Aβ aggregation.
In recent years, sophisticated kinetic analysis has greatly improved our understanding of the Aβ aggregation process. Notably, the concept of secondary nucleation50 has led to breakthrough investigations into the microscopic steps of the Aβ aggregation process.51−53 Kinetic data have also been used to estimate the size of the nuclei in both primary and secondary nucleation.54,55 Knowles and colleagues developed an online platform called AmyloFit that allows the testing of various aggregation models by nonexpert users.56 A range of aggregation mechanisms including secondary nucleation, fragmentation, and saturation can be tested using AmyloFit. Furthermore, global fitting of the kinetic parameters can be used to investigate the specific microscopic steps of the aggregation pathway. For aggregation inhibitors such as EGCG, AmyloFit can be used to provide mechanistic insights into the action of EGCG on Aβ aggregation. It has been previously shown that Aβ42 aggregates via a secondary nucleation mechanism.50 As a preliminary analysis, we investigated whether the effect of EGCG in our studies can be explained by the action on either primary or secondary nucleation processes. In AmyloFit, we either fit secondary nucleation globally and primary nucleation individually (Figure S4A) or fit primary nucleation globally and secondary nucleation individually (Figure S4B). We found that neither approach led to good fits for all of the aggregation data across the full range of EGCG concentration (Figure S4). As pointed out by Meisl et al.,56 this means that the action of EGCG on Aβ aggregation is complex and may involve different microscopic processes at different EGCG concentrations. EGCG at high concentrations may mainly affect primary nucleation, while EGCG at low concentrations may primarily affect secondary nucleation, and this is consistent with our proposed model in Figure 5. Aβ aggregation is a complicated system, which may include primary nucleation, secondary nucleation, fibril fragmentation, and nucleated conformational conversion. In addition to the more common mechanism of monomer addition, oligomer addition was also proposed as a mechanism of fibril growth.57,58 To conclusively investigate these microscopic steps using aggregation kinetics, future studies of Aβ aggregation at a broad range of Aβ and EGCG concentrations will be needed to delineate the detailed mechanism of interactions between EGCG and various Aβ-aggregated species.
Methods
Preparation of Aβ42 Proteins
Aβ protein was expressed in Escherichia coli as a fusion protein, GroES-ubiquitin-Aβ, as described previously.36,38 The fusion protein partners were then cleaved with a deubiquitylating enzyme to obtain full-length Aβ without any extra residues.59 Afterward, Aβ was buffer-exchanged to 30 mM ammonium acetate (pH 10.0) and lyophilized in small-volume aliquots. The lyophilized Aβ powder was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at a 100 μM concentration with shaking (1000 rpm) at room temperature for 24 h. The HFIP was dried overnight in a chemical hood and the HFIP-treated Aβ film was stored at −80°C.
Aggregation Kinetics of Aβ42 in the Absence and Presence of EGCG
One tube of HFIP-treated Aβ42 film was dissolved in 1 mL of a high-pH denaturing buffer called CG (20 mM CAPS, 7 M guanidine hydrochloride, pH 11) and bath-sonicated for 5 min. Then, the sample was buffer-exchanged to PBS (50 mM phosphate, 140 mM NaCl, pH 7.4) using a 5 mL HiTrap desalting column (GE Healthcare). Afterward, the sample was filtered through a 100 kD ultrafiltration filter (Sartorius Vivaspin 500) and was referred to as 100 kD filtrate. The concentration of Aβ42 was determined to be 2.7 μM using the fluorescamine method (see below). A fresh thioflavin T (Sigma-Aldrich, catalog #T3516) solution was made in PBS, and the concentration was determined using the extinction coefficient of 36 mM–1·cm–1 at 412 nm.36 Then, we made a master mix containing 2.2 μM Aβ42 and 22 μM thioflavin T in PBS. The purpose of the master mix is to ensure that Aβ42 and thioflavin T are identical in all of the aggregation reactions and any differences can be attributed to EGCG concentrations.
A stock solution of EGCG (Tocris Bioscience, catalog #4524) was made at 400 μM in water using the weight of EGCG. The EGCG stock was then serially diluted to concentrations of 200, 100, 40, 20, 10, 4, 2, and 1 μM. To set up aggregation, 45 μL of master mix was mixed with 5 μL of EGCG stock solutions at different concentrations or 5 μL of water for the no-EGCG sample. There are three replicates for each aggregation condition. Finally, all 50 μL of the aggregation mix was transferred to a black 384-well nonbinding surface microplate with a clear bottom (Corning, catalog #3655) and sealed with a polyester-based sealing film (Corning, catalog #PCR-SP). All of the mixing steps were performed on ice. The fibrillization was started by incubating the plate in a Victor 3V plate reader (Perkin Elmer) at 37°C without agitation. The thioflavin T fluorescence was measured from the bottom of the plate with an excitation filter of 450 nm and an emission filter of 490 nm. We reported the aggregation data as fold change in fluorescence intensity by dividing the average of thioflavin T-alone sample at each time of measurements. We would like to point out that the fold change in thioflavin T fluorescence as a result of Aβ aggregation may vary greatly when different fluorescence readers are used. This is likely due to the type of light source, measurement settings, detector sensitivity, and other instrument-specific factors of the plate reader, which gives various levels of background readings.
Measurement of Aβ Concentration Using the Fluorescamine Method
Because the Aβ42 concentration is low and could not be reliably determined using absorbance at 280 nm, we have developed a fluorescamine method that allows the accurate measurement of Aβ concentration with sub-micromolar sensitivity.60 The standard assay mix contains 40 μL of PBS, 5 μL of sample, and 5 μL of 5 mM fluorescamine (Acros Organics, pure grade, catalog #AC19167) dissolved in acetonitrile. Upon mixing, the sample is transferred to a black 384-well microplate (Corning, catalog #3655). The fluorescence measurement was taken using a Gemini EM plate reader (Molecular Devices) with excitation at 390 nm and emission at 478 nm. A standard curve was prepared using hen egg white lysozyme (Fisher Bioreagents, crystalline powder, catalog #BP535) with a concentration range of 0.1–1 μM, corresponding to amine concentrations of 0.7–7 μM. Aβ was diluted if needed so that the fluorescence reading falls in the middle range of the standard curve.
Analysis of Aggregation Data
The aggregation data were fitted to the equation below, which was based on the work of Nielsen et al.61
Here S is the fluorescence intensity, t is the aggregation time, t50 (half-time) is the time to reach 50% of the maximum fluorescence, and τ is the time it takes for the fluorescence signal to change from 26.9 to 50%, or from 50 to 73.1% of the maximum fluorescence. The change in fluorescence in the lag phase is described by SI + kI·t, and the fluorescence in the plateau phase is described by SF + kF·t. The lag time is given by t50 – 2τ, and the maximum fibril growth rate at t50 is given by 1/4τ. All of the fits are shown in Supporting Information Figure S1.
Fitting of the aggregation data using AmyloFit was performed as previously described by Meisl et al.56 using the unseeded secondary nucleation-dominated model. The fitting results are shown in Supporting Information Figure S3. The secondary nucleation model and associated fitting parameters have been described in detail in Cohen et al.50 and Meisl et al.62
Transmission Electron Microscopy
For transmission electron microscopy, 5 μL of the aggregation sample described above after approximately 48 h of incubation was applied to glow-discharged copper grids (400 mesh formvar/carbon film, Ted Pella, catalog #01754-F) and stained with 2% uranyl acetate. The grids were then examined under an FEI T12 electron microscope with an accelerating voltage of 120 kV.
Site-Directed Spin Labeling
First, the residue L34 was mutated to a cysteine using site-directed mutagenesis. Then, Aβ42 L34C was purified and fusion partners were cleaved as described above. To attach the spin label R1, the Aβ42 L34C protein was incubated with the spin labeling reagent MTSSL (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl methanethiosulfonate, AdipoGen Life Sciences) at 5-fold molar excess at room temperature for 1 h. A buffer exchange was then performed to remove the free spin label and change the buffer to 30 mM ammonium acetate (pH 10) for lyophilization. Lyophilized Aβ42 L34R1 was treated with HFIP as described above and stored at −80°C.
Preparation of Spin-Labeled Aβ42 Fibrils in the Absence and Presence of EGCG
One tube of HFIP-treated Aβ42 L34R1 protein was dissolved in CG buffer to a final concentration of 400 μM, which was determined using absorbance at 280 nm. Then, 25 μL of the Aβ42 sample in CG buffer was mixed with 475 μL of PBS to obtain the no-EGCG sample. Two other Aβ42 samples at 25 μL volume were mixed with 475 μL of PBS containing 200 μM and 1 mM EGCG to obtain the EGCG-to-Aβ42 ratio of 10:1 and 50:1, respectively. These three samples were incubated at 37 °C for 3 days without agitation. At the end of incubation, the three samples were centrifuged at 20 000g for 20 min to pellet the Aβ42 aggregates. The pellet was surface-washed twice with PBS to remove loosely associated Aβ42 proteins.
EPR Spectroscopy
The pellets of Aβ42 L34R1 after centrifugation were resuspended in 15 μL of PBS and then loaded in glass capillary tubes (VitroCom) sealed at one end. EPR spectra were collected at X-band using a Bruker EMXnano spectrometer at room temperature using a microwave power of 15 mW. A modulation frequency of 100 kHz was used, and the modulation amplitude was set at 4 G based on the line width of the EPR spectrum. The scan width was 200 G. The same parameters for data acquisition, including the number of scans, were used for all three Aβ42 samples prepared with or without EGCG. To quantify the amount of spin-labeled aggregates from the EPR spectra, double integrals were calculated using an in-house program written by Dr. Christian Altenbach at the University of California, Los Angeles. Errors were estimated by adjusting the parameters for baseline correction to obtain the lower and upper ranges of the calculated double integrals. The double integrals were then normalized as a percentage of the no-EGCG sample, which is set at 100%.
Acknowledgments
The authors thank the members of the Guo laboratory for their helpful discussions and technical assistance in protein expression and purification. This work was supported by the National Institute of Health (Grant R01AG050687).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02063.
Sigmoidal fits for fibrillization kinetics of Aβ42 samples in the absence and presence of EGCG (Figure S1); aggregation of Aβ42 spin-labeled at position 34 in the absence and presence of EGCG (Figure S2); and fitting of aggregation data using AmyloFit (Figure S3) (PDF)
Author Contributions
G.P., C.X., and H.W. designed and carried out experiments, analyzed data, and drafted the manuscript. Z.G. conceived and supervised the study, designed the experiments, and drafted the manuscript. All authors gave final approval for publication.
The authors declare no competing financial interest.
Supplementary Material
References
- Selkoe D. J. Alzheimer’s Disease. Cold Spring Harbor Perspect. Biol. 2011, 3, a004457 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon E. E.; Sigurdsson E. M. Tau-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 399–415. 10.1038/s41582-018-0013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I.; Karran E.; Strooper B. D. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Zhou R.; Yang G.; Guo X.; Zhou Q.; Lei J.; Shi Y. Recognition of the Amyloid Precursor Protein by Human γ-Secretase. Science 2019, 363, aaw0930 10.1126/science.aaw0930. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Osswald H. L. BACE1 (β-Secretase) Inhibitors for the Treatment of Alzheimer’s Disease. Chem. Soc. Rev. 2014, 43, 6765–6813. 10.1039/C3CS60460H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildburger N. C.; Esparza T. J.; LeDuc R. D.; Fellers R. T.; Thomas P. M.; Cairns N. J.; Kelleher N. L.; Bateman R. J.; Brody D. L. Diversity of Amyloid-Beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 9520 10.1038/s41598-017-10422-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina S. A.; Ho L.; Eckman C. B.; Long K. E.; Otvos L.; Younkin L. H.; Suzuki N.; Younkin S. G. Amyloid β Protein (Aβ) in Alzheimer’s Disease Brain. Biochemical and Immunocytochemical Analysis with Antibodies Specific for Forms Ending at Aβ40 or Aβ42(43). J. Biol. Chem. 1995, 270, 7013–7016. 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T.; Odaka A.; Suzuki N.; Mizusawa H.; Nukina N.; Ihara Y. Visualization of Aβ42(43) and Aβ40 in Senile Plaques with End-Specific Aβ Monoclonals: Evidence That an Initially Deposited Species Is Aβ42(43). Neuron 1994, 13, 45–53. 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Mak K.; Yang F.; Vinters H. V.; Frautschy S. A.; Cole G. M. Polyclonals to β-Amyloid(1–42) Identify Most Plaque and Vascular Deposits in Alzheimer Cortex, but Not Striatum. Brain Res. 1994, 667, 138–142. 10.1016/0006-8993(94)91725-6. [DOI] [PubMed] [Google Scholar]
- Miller D. L.; Papayannopoulos I. A.; Styles J.; Bobin S. A.; Lin Y. Y.; Biemann K.; Iqbal K. Peptide Compositions of the Cerebrovascular and Senile Plaque Core Amyloid Deposits of Alzheimer′s Disease. Arch. Biochem. Biophys. 1993, 301, 41–52. 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- Vaquer-Alicea J.; Diamond M. I. Propagation of Protein Aggregation in Neurodegenerative Diseases. Annu. Rev. Biochem. 2019, 88, 785–810. 10.1146/annurev-biochem-061516-045049. [DOI] [PubMed] [Google Scholar]
- Iadanza M. G.; Jackson M. P.; Hewitt E. W.; Ranson N. A.; Radford S. E. A New Era for Understanding Amyloid Structures and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. 10.1038/s41580-018-0060-8. [DOI] [PubMed] [Google Scholar]
- Panza F.; Lozupone M.; Logroscino G.; Imbimbo B. P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88. 10.1038/s41582-018-0116-6. [DOI] [PubMed] [Google Scholar]
- Galzitskaya O. V. Oligomers Are Promising Targets for Drug Development in the Treatment of Proteinopathies. Front. Mol. Neurosci. 2019, 12, 319. 10.3389/fnmol.2019.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren O.; Ben Zichri S.; Taube R.; Jelinek R.; Papo N. An Aβ42 Double Mutant Inhibits Aβ42-Induced Plasma and Mitochondrial Membrane Disruption in Artificial Membranes, Isolated Organs and Intact Cells. ACS Chem. Neurosci. 2020, 1027 10.1021/acschemneuro.9b00638. [DOI] [PubMed] [Google Scholar]
- Xi W.; Vanderford E. K.; Hansmann U. H. E. Out-of-Register Aβ42 Assemblies as Models for Neurotoxic Oligomers and Fibrils. J. Chem. Theory Comput. 2018, 14, 1099–1110. 10.1021/acs.jctc.7b01106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K.-C.; Chen Y.-R.; Liang C.-T.; Huang S.-J.; Tzeng C.-Y.; Chang C.-F.; Huang S.-J.; Huang H.-B.; Lin T.-H. Conformational Characterization of Native and L17A/F19A-Substituted Dutch-Type β-Amyloid Peptides. Int. J. Mol. Sci. 2020, 21, 2571 10.3390/ijms21072571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieślik E.; Gręda A.; Adamus W. Contents of Polyphenols in Fruit and Vegetables. Food Chemistry 2006, 94, 135–142. 10.1016/j.foodchem.2004.11.015. [DOI] [Google Scholar]
- Lakey-Beitia J.; Berrocal R.; Rao K. S.; Durant A. A. Polyphenols as Therapeutic Molecules in Alzheimer’s Disease through Modulating Amyloid Pathways. Mol. Neurobiol. 2014, 51, 466–479. 10.1007/s12035-014-8722-9. [DOI] [PubMed] [Google Scholar]
- Liu X.; Du X.; Han G.; Gao W. Association between Tea Consumption and Risk of Cognitive Disorders: A Dose-Response Meta-Analysis of Observational Studies. Oncotarget 2017, 8, 43306–43321. 10.18632/oncotarget.17429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.-K.; Jung I.-C.; Lee W. K.; Lee Y. S.; Park H. K.; Go H. J.; Kim K.; Lim N. K.; Hong J. T.; Ly S. Y.; Rho S. S. A Combination of Green Tea Extract and L-Theanine Improves Memory and Attention in Subjects with Mild Cognitive Impairment: A Double-Blind Placebo-Controlled Study. J. Med. Food 2011, 14, 334–343. 10.1089/jmf.2009.1374. [DOI] [PubMed] [Google Scholar]
- Ide K.; Yamada H.; Takuma N.; Kawasaki Y.; Harada S.; Nakase J.; Ukawa Y.; Sagesaka Y. M. Effects of Green Tea Consumption on Cognitive Dysfunction in an Elderly Population: A Randomized Placebo-Controlled Study. Nutr. J. 2016, 15, 49. 10.1186/s12937-016-0168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh K.; Shytle D.; Sun N.; Mori T.; Hou H.; Jeanniton D.; Ehrhart J.; Townsend K.; Zeng J.; Morgan D.; Hardy J.; Town T.; Tan J. Green Tea Epigallocatechin-3-Gallate (EGCG) Modulates Amyloid Precursor Protein Cleavage and Reduces Cerebral Amyloidosis in Alzheimer Transgenic Mice. J. Neurosci. 2005, 25, 8807–8814. 10.1523/JNEUROSCI.1521-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh K.; Arendash G. W.; Hou H.; Fernandez F.; Jensen M.; Runfeldt M.; Shytle R. D.; Tan J. Green Tea Epigallocatechin-3-Gallate (EGCG) Reduces Beta-Amyloid Mediated Cognitive Impairment and Modulates Tau Pathology in Alzheimer Transgenic Mice. Brain Res. 2008, 1214, 177–187. 10.1016/j.brainres.2008.02.107. [DOI] [PubMed] [Google Scholar]
- Giunta B.; Hou H.; Zhu Y.; Salemi J.; Ruscin A.; Shytle R. D.; Tan J. Fish Oil Enhances Anti-Amyloidogenic Properties of Green Tea EGCG in Tg2576 Mice. Neurosci. Lett. 2010, 471, 134–138. 10.1016/j.neulet.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y.; Amit T.; Mandel S.; Youdim M. B. H. Neuroprotection and Neurorescue against Abeta Toxicity and PKC-Dependent Release of Nonamyloidogenic Soluble Precursor Protein by Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate. FASEB J. 2003, 17, 952–954. 10.1096/fj.02-0881fje. [DOI] [PubMed] [Google Scholar]
- Bastianetto S.; Yao Z.-X.; Papadopoulos V.; Quirion R. Neuroprotective Effects of Green and Black Teas and Their Catechin Gallate Esters against Beta-Amyloid-Induced Toxicity. Eur. J. Neurosci. 2006, 23, 55–64. 10.1111/j.1460-9568.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Du Z.; Maiti P.; Klärner F.-G.; Schrader T.; Wang C.; Bitan G. Comparison of Three Amyloid Assembly Inhibitors: The Sugar Scyllo-Inositol, the Polyphenol Epigallocatechin Gallate, and the Molecular Tweezer CLR01. ACS Chem. Neurosci. 2012, 3, 451–458. 10.1021/cn200133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Yu L.; Dong X.; Sun Y. Synergistic Effects of Negatively Charged Hydrophobic Nanoparticles and (-)-Epigallocatechin-3-Gallate on Inhibiting Amyloid β-Protein Aggregation. J. Colloid Interface Sci. 2016, 491, 305–312. 10.1016/j.jcis.2016.12.038. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D. E.; Bieschke J.; Boeddrich A.; Herbst M.; Masino L.; Lurz R.; Engemann S.; Pastore A.; Wanker E. E. EGCG Redirects Amyloidogenic Polypeptides into Unstructured, off-Pathway Oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- Sironi E.; Colombo L.; Lompo A.; Messa M.; Bonanomi M.; Regonesi M. E.; Salmona M.; Airoldi C. Natural Compounds against Neurodegenerative Diseases: Molecular Characterization of the Interaction of Catechins from Green Tea with Aβ1-42, PrP106-126, and Ataxin-3 Oligomers. Chemistry 2014, 20, 13793–13800. 10.1002/chem.201403188. [DOI] [PubMed] [Google Scholar]
- Bieschke J.; Russ J.; Friedrich R. P.; Ehrnhoefer D. E.; Wobst H.; Neugebauer K.; Wanker E. E. EGCG Remodels Mature Alpha-Synuclein and Amyloid-Beta Fibrils and Reduces Cellular Toxicity. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 7710–7715. 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palhano F. L.; Lee J.; Grimster N. P.; Kelly J. W. Toward the Molecular Mechanism(s) by Which EGCG Treatment Remodels Mature Amyloid Fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. 10.1021/ja3115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R.; Moreno-Gonzalez I.; Soto C. Cross-Seeding of Misfolded Proteins: Implications for Etiology and Pathogenesis of Protein Misfolding Diseases. PLoS Pathog. 2013, 9, e1003537 10.1371/journal.ppat.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P.; Knowles T. P. J.; Linse S. On the Lag Phase in Amyloid Fibril Formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. 10.1039/C4CP05563B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C.; Lin T. Y.; Chang D.; Guo Z. Thioflavin T as an Amyloid Dye: Fibril Quantification, Optimal Concentration and Effect on Aggregation. R. Soc. Open Sci. 2017, 4, 160696 10.1098/rsos.160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F.; Park G.; Guo Z. Key Residues for the Formation of Aβ42 Amyloid Fibrils. ACS Omega 2018, 3, 8401–8407. 10.1021/acsomega.8b00887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C.; Tran J.; Wang H.; Park G.; Hsu F.; Guo Z. Aβ42 Fibril Formation from Predominantly Oligomeric Samples Suggests a Link between Oligomer Heterogeneity and Fibril Polymorphism. R. Soc. Open Sci. 2019, 6, 190179 10.1098/rsos.190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran J.; Chang D.; Hsu F.; Wang H.; Guo Z. Cross-Seeding between Aβ40 and Aβ42 in Alzheimer’s Disease. FEBS Lett. 2017, 591, 177–185. 10.1002/1873-3468.12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D.; Bhattacharyya D.; Bhunia A. Do Catechins (ECG and EGCG) Bind to the Same Site as Thioflavin T (ThT) in Amyloid Fibril? Answer From Saturation Transfer Difference NMR. Nat. Prod. Commun. 2019, 14, 1934578X19849791 10.1177/1934578X19849791. [DOI] [Google Scholar]
- Gu L.; Tran J.; Jiang L.; Guo Z. A New Structural Model of Alzheimer’s Aβ42 Fibrils Based on Electron Paramagnetic Resonance Data and Rosetta Modeling. J. Struct. Biol. 2016, 194, 61–67. 10.1016/j.jsb.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Lee Y. K.; Xue C.; Guo Z. Site-Specific Structural Order in Alzheimer’s Aβ42 Fibrils. R. Soc. Open Sci. 2018, 5, 180166 10.1098/rsos.180166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Duo L.; Hsu F.; Xue C.; Lee Y. K.; Guo Z. Polymorphic Aβ42 Fibrils Adopt Similar Secondary Structure but Differ in Cross-Strand Side Chain Stacking Interactions within the Same β-Sheet. Sci. Rep. 2020, 10, 5720 10.1038/s41598-020-62181-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L.; Liu C.; Guo Z. Structural Insights into Aβ42 Oligomers Using Site-Directed Spin Labeling. J. Biol. Chem. 2013, 288, 18673–18683. 10.1074/jbc.M113.457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L.; Liu C.; Stroud J. C.; Ngo S.; Jiang L.; Guo Z. Antiparallel Triple-Strand Architecture for Prefibrillar Aβ42 Oligomers. J. Biol. Chem. 2014, 289, 27300–27313. 10.1074/jbc.M114.569004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrand E.; Boland B.; Walsh D. M.; Linse S. Amyloid β-Protein Aggregation Produces Highly Reproducible Kinetic Data and Occurs by a Two-Phase Process. ACS Chem. Neurosci. 2010, 1, 13–18. 10.1021/cn900015v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. W. P.; Meisl G.; Knowles T. P. J.; Buell A. K.; Dobson C. M.; Galvagnion C. Kinetic Barriers to α-Synuclein Protofilament Formation and Conversion into Mature Fibrils. Chem. Commun. 2018, 54, 7854–7857. 10.1039/C8CC03002B. [DOI] [PubMed] [Google Scholar]
- Williams A. D.; Shivaprasad S.; Wetzel R. Alanine Scanning Mutagenesis of Aβ(1-40) Amyloid Fibril Stability. J. Mol. Biol. 2006, 357, 1283–1294. 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Iljina M.; Garcia G. A.; Dear A. J.; Flint J.; Narayan P.; Michaels T. C. T.; Dobson C. M.; Frenkel D.; Knowles T. P. J.; Klenerman D. Quantitative Analysis of Co-Oligomer Formation by Amyloid-Beta Peptide Isoforms. Sci. Rep. 2016, 6, 28658 10.1038/srep28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I. A.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. Proliferation of Amyloid-B42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 9758–9763. 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törnquist M.; Michaels T. C. T.; Sanagavarapu K.; Yang X.; Meisl G.; Cohen S. I. A.; Knowles T. P. J.; Linse S. Secondary Nucleation in Amyloid Formation. Chem. Commun. 2018, 54, 8667 10.1039/C8CC02204F. [DOI] [PubMed] [Google Scholar]
- Michaels T. C. T.; Šarić A.; Habchi J.; Chia S.; Meisl G.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J. Chemical Kinetics for Bridging Molecular Mechanisms and Macroscopic Measurements of Amyloid Fibril Formation. Annu. Rev. Phys. Chem. 2018, 69, 273–298. 10.1146/annurev-physchem-050317-021322. [DOI] [PubMed] [Google Scholar]
- Scheidt T.; Łapińska U.; Kumita J. R.; Whiten D. R.; Klenerman D.; Wilson M. R.; Cohen S. I. A.; Linse S.; Vendruscolo M.; Dobson C. M.; Knowles T. P. J.; Arosio P. Secondary Nucleation and Elongation Occur at Different Sites on Alzheimer’s Amyloid-β Aggregates. Sci. Adv. 2019, 5, eaau3112 10.1126/sciadv.aau3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovidchenko N. V.; Finkelstein A. V.; Galzitskaya O. V. How to Determine the Size of Folding Nuclei of Protofibrils from the Concentration Dependence of the Rate and Lag-Time of Aggregation. I. Modeling the Amyloid Protofibril Formation. J. Phys. Chem. B 2014, 118, 1189–1197. 10.1021/jp4083294. [DOI] [PubMed] [Google Scholar]
- Dovidchenko N. V.; Glyakina A. V.; Selivanova O. M.; Grigorashvili E. I.; Suvorina M. Y.; Dzhus U. F.; Mikhailina A. O.; Shiliaev N. G.; Marchenkov V. V.; Surin A. K.; Galzitskaya O. V. One of the Possible Mechanisms of Amyloid Fibrils Formation Based on the Sizes of Primary and Secondary Folding Nuclei of Aβ40 and Aβ42. J. Struct. Biol. 2016, 194, 404–414. 10.1016/j.jsb.2016.03.020. [DOI] [PubMed] [Google Scholar]
- Meisl G.; Kirkegaard J. B.; Arosio P.; Michaels T. C. T.; Vendruscolo M.; Dobson C. M.; Linse S.; Knowles T. P. J. Molecular Mechanisms of Protein Aggregation from Global Fitting of Kinetic Models. Nat. Protoc. 2016, 11, 252–272. 10.1038/nprot.2016.010. [DOI] [PubMed] [Google Scholar]
- Galzitskaya O. V.; Surin A. K.; Glyakina A. V.; Rogachevsky V. V.; Selivanova O. M. Should the Treatment of Amyloidosis Be Personified? Molecular Mechanism of Amyloid Formation by Aβ Peptide and Its Fragments. J. Alzheimer’s Dis. Rep. 2018, 2, 181–199. 10.3233/ADR-180063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selivanova O. M.; Surin A. K.; Marchenkov V. V.; Dzhus U. F.; Grigorashvili E. I.; Suvorina M. Y.; Glyakina A. V.; Dovidchenko N. V.; Galzitskaya O. V. The Mechanism Underlying Amyloid Polymorphism Is Opened for Alzheimer’s Disease Amyloid-β Peptide. J. Alzheimer’s Dis. 2016, 54, 821–830. 10.3233/JAD-160405. [DOI] [PubMed] [Google Scholar]
- Shahnawaz M.; Thapa A.; Park I.-S. Stable Activity of a Deubiquitylating Enzyme (Usp2-Cc) in the Presence of High Concentrations of Urea and Its Application to Purify Aggregation-Prone Peptides. Biochem. Biophys. Res. Commun. 2007, 359, 801–805. 10.1016/j.bbrc.2007.05.186. [DOI] [PubMed] [Google Scholar]
- Xue C.; Lee Y. K.; Tran J.; Chang D.; Guo Z. A Mix-and-Click Method to Measure Amyloid-β Concentration with Sub-Micromolar Sensitivity. R. Soc. Open Sci. 2017, 4, 170325 10.1098/rsos.170325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen L.; Khurana R.; Coats A.; Frokjaer S.; Brange J.; Vyas S.; Uversky V. N.; Fink A. L. Effect of Environmental Factors on the Kinetics of Insulin Fibril Formation: Elucidation of the Molecular Mechanism. Biochemistry 2001, 40, 6036–6046. 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- Meisl G.; Yang X.; Hellstrand E.; Frohm B.; Kirkegaard J. B.; Cohen S. I. A.; Dobson C. M.; Linse S.; Knowles T. P. J. Differences in Nucleation Behavior Underlie the Contrasting Aggregation Kinetics of the Aβ40 and Aβ42 Peptides. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 9384–9389. 10.1073/pnas.1401564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.