

ABSTRACT
Host-directed therapies are gaining considerable impetus because of the emergence of drug-resistant strains of pathogens due to antibiotic therapy. Therefore, there is an urgent need to exploit alternative and novel strategies directed at host molecules to successfully restrict infections. The C-type lectin receptor CLEC4E and Toll-like receptor TLR4 expressed by host cells are among the first line of defense in encountering pathogens. Therefore, we exploited signaling of macrophages through CLEC4E in association with TLR4 agonists (C4.T4) to control the growth of Mycobacterium tuberculosis (Mtb). We observed significant improvement in host immunity and reduced bacterial load in the lungs of Mtb-infected mice and guinea pigs treated with C4.T4 agonists. Further, intracellular killing of Mtb was achieved with a 10-fold lower dose of isoniazid or rifampicin in conjunction with C4.T4 than the drugs alone. C4.T4 activated MYD88, PtdIns3K, STAT1 and RELA/NFKB, increased lysosome biogenesis, decreased Il10 and Il4 gene expression and enhanced macroautophagy/autophagy. Macrophages from autophagy-deficient (atg5 knockout or Becn1 knockdown) mice showed elevated survival of Mtb. The present findings also unveiled the novel role of CLEC4E in inducing autophagy through MYD88, which is required for control of Mtb growth. This study suggests a unique immunotherapeutic approach involving CLEC4E in conjunction with TLR4 to restrict the survival of Mtb through autophagy.
Abbreviations
3MA: 3 methyladenine; AO: acridine orange; Atg5: autophagy related 5; AVOs: acidic vesicular organelles; BECN1: beclin 1, autophagy related; BMDMs: bone marrow derived macrophages; bw: body weight; C4.T4: agonists of CLEC4E (C4/TDB) and TLR4 (T4/ultra-pure-LPS); CFU: colony forming unit; CLEC4E/Mincle: C-type lectin domain family 4, member e; CLR: c-type lectin receptor; INH: isoniazid; LAMP1: lysosomal-associated membrane protein 1; MφC4.T4: Mtb-infected C4.T4 stimulated macrophages; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MDC: monodansylcadaverine; MTOR: mechanistic target of rapamycin kinase; MYD88: myeloid differentiation primary response 88; NFKB: nuclear factor of kappa light polypeptide gene enhance in B cells; NLR: NOD (nucleotide-binding oligomerization domain)-like receptors; PFA: paraformaldehyde; PPD: purified protein derivative; PtdIns3K: class III phosphatidylinositol 3-kinase; RELA: v-rel reticuloendotheliosis viral oncogene homolog A (avian); RIF: rifampicin; RLR: retinoic acid-inducible gene-I-like receptors; TDB: trehalose-6,6´-dibehenate; TLR4: toll-like receptor 4; Ultra-pure-LPS: ultra-pure lipopolysaccharide-EK; V-ATPase: vacuolar-type H+ ATPase.
KEYWORDS: Anti-TB drugs, atg5 knockout, autophagy, Becn1 knockdown, CLEC4E/Mincle, host-directed immunotherapy, macrophages, Mycobacterium tuberculosis, TLR4, tuberculosis
Introduction
Tuberculosis (TB) is one of the top ten leading causes of death and responsible for the mortality of 1.6 million people in 2017 [1,2]. Nearly one third of the global populace is infected with Mycobacterium tuberculosis (Mtb), and 5–10% of infected individuals develop active TB [1,2]. Even though potent anti-TB drugs are available to treat the disease, their use is restricted owing to the emergence of drug resistant strains [3–5]. In addition, the lengthy treatment is associated with serious side-effects. Consequently, there is an urgent need to bring new approaches to the current regime and/or identify alternative strategies to effectively manage TB. Innovative therapies involving immunomodulators, humanized antibodies, adoptive cell therapies, host-directed remedies, etc., are being currently employed for treating cancer, autoimmunity, and cardiac diseases [6–8]. Highlighting the importance of certain immunomodulators, WHO recommends their use as adjunct therapy, along with the current TB regimen [9].
Macrophages express an array of costimulatory molecules, and different pattern recognition receptors (PRRs) [10]. During infection, recognition of pathogen-associated molecular patterns (PAMPs) through these molecules can activate macrophages [10]. CLEC4E (c-type lectin domain family 4, member e) is a PRR that contributes to the recognition of Mtb and enhances adaptive immune responses [11]. Mice deficient in clec4e−/- show impaired cytokine release by Mtb-infected macrophages [11]. The first CLEC4E ligand identified was a nucleo-protein, SAP130, which is involved in responses to stroke and traumatic brain injury [12–14]. Furthermore, macrophages expressing CLEC4E can recognize Candida albicans [15–17], Malassezia spp [18,19], and Fonsecaea monophora [20]. Consequently, CLEC4E contributes to the release of pro-inflammatory cytokines and protective immune responses.
The mycobacterial cord factor trehalose-6,6´-dimycolate (TDM) is an important virulence factor of Mtb. In addition, it has adjuvant properties and causes inflammatory response [21,22]. The synthetic analogue of TDM is trehalose-6,6´-dibehenate (TDB), which is the active constituent of TB vaccine CAF01 that has entered into phase I clinical trials [23,24]. TDB is non-toxic, safe and induces strong cell mediated and humoral immunity [25–27], and is an agonist for CLEC4E.
Further, toll-like receptors (TLRs) play a crucial function in macrophage maturation and activation [28,29]. Even though innate immune responses against Mtb can signal via TLR2, 4 and 9, their role appears to be limited and requires extensive investigation to be fully elucidated [30]. Recently, the Food and Drug Administration (FDA) approved a TLR4 agonist (mono phosphoryl lipid A/LPS) for human therapy [31]. The optimum dose of LPS does not induce adverse effects [32,33]. C-type lectin receptors (CLRs) in conjunction with TLRs further bolster the immune response [34,35].
The role of autophagy in protection against Mtb is well-known [36–39]. Autophagy targets antigens for lysosomal degradation and transports microbial peptides for MHC presentation [38,39]. At the same time, it increases cell survival by inhibiting inflammatory responses [37]. TLR4 is a strong activator of autophagy [40,41]. In the current study, we are reporting an interesting and novel role for CLEC4E in inducing autophagy against Mtb infection. We activated Mtb infected macrophages by stimulating the cells with CLEC4E and TLR4 (denoted as C4.T4) agonists TDB and ultra-pure-LPS, respectively, and noted significant augmentation in the bactericidal activity of macrophages. Of key importance, macrophages from Becn1/Beclin1 knockdown and atg5 knockout (atg5fl/fl-Lyz2/LysM-Cre) mice exhibited increased survival of Mtb even when treated with C4.T4. The mechanism uncovered in C4.T4-induced killing of Mtb by macrophages was through MYD88-mediated activation of the class III phosphatidylinositol 3-kinase (PtdIns3K) pathway, which led to the induction of autophagy. In the future, immunotherapy involving C4.T4 agonists may serve as an alternative strategy to treat TB patients.
Results
Signaling through CLEC4E and TLR4 enhanced macrophage activation
Macrophage activation is crucial in combating pathogens [42] and can be assessed by morphology, expression of costimulatory molecules, cytokine release and antigen processing and presentation. We monitored whether stimulation of macrophages using CLEC4E and TLR4 agonists (C4.T4) could enhance their ability to activate T cells. Interestingly, we observed that bone marrow derived macrophages (BMDMs) triggered by C4.T4, exhibited an activation phenotype, as evidenced by increased size and granularity, as depicted by scanning electron microscopy (SEM), and significantly (p < 0.001) improved secretion of nitric oxide (NO), as compared to unstimulated control or controls stimulated with either C4 or T4 (Figure 1A,B). Next, we were curious about the specificity of the signals provided through CLEC4E and TLR4 in the activation of macrophages. CLEC4E signaling regulates through SYK (spleen tyrosine kinase) [43,44]. Therefore, the activities of CLEC4E and TLR4 were blocked using their inhibitors piceatannol and CLI-095, respectively. A significant (p < 0.001) reduction in the release of NO and IL6 was observed in cell cultures with the inhibitors. Therefore, these data confirmed the specificity of CLEC4E and TLR4 signaling (Figure 1B,C). Next, titration was performed to identify the optimum dose of C4.T4 agonists. CLEC4E and TLR4 agonists led to the optimum release of IL6 at 24 µg/ml and 10 ng/ml, respectively. Further, these concentrations showed no effect on the viability of macrophages (Figures S1 and S2). These concentrations were chosen for all subsequent experiments. We observed homogeneous expression of CLEC4E and TLR4 on the macrophages by fluorescence microscopy and flow cytometry (Figure S3A–C). We noticed that the expression of CLEC4E and TLR4 was upregulated on Mtb-infected macrophages (Figure S3D,E).
Figure 1.
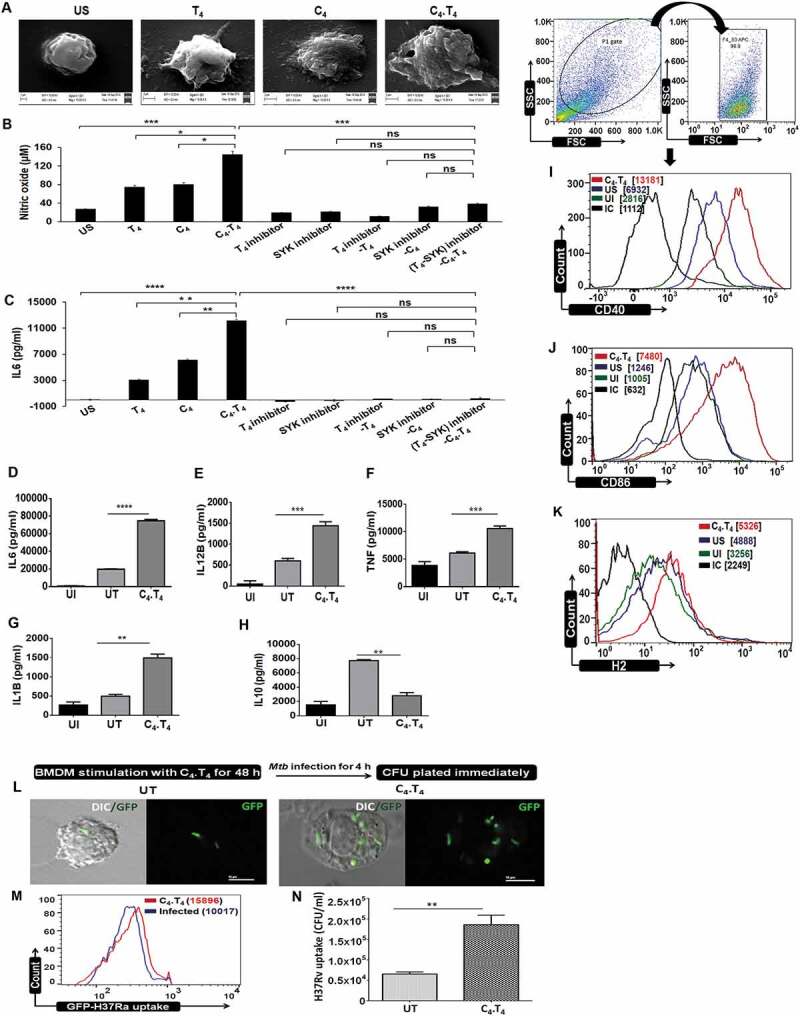
CLEC4E and TLR4 signaling induced the activation and maturation of macrophages. BMDMs were stimulated with the agonists of CLEC4E (C4 [24 µg/ml]) and TLR4 (T4 [10 ng/ml]) individually or in combination (C4.T4) for 48 h. (A) The cells were placed on glass coverslips and fixed with modified Karnoyses fixative and processed for scanning electron microscopy (SEM). Scale bar: 2 µm; magnification: 15000X. Data are representative of 2 independent experiments. (B,C) BMDMs were treated with CLEC4E (SYK-piceatannol) and TLR4 (CLI-095) signaling inhibitors for 1 h prior to C4.T4 stimulation, and then SNs were collected. The specificity of signaling was established through NO and IL6 release. (D–K) BMDMs were infected with Mtb for 4 h, treated with C4.T4 for 48 h and SNs were collected for the estimation of (D) IL6; (E) IL12B; (F) TNF, (G) IL1B, and (H) IL10 by ELISA. Data represent the mean ± SEM of 4 wells and are from 3 independent experiments. Further, the cells were evaluated for the expression of (I) CD40; (J) CD86; and (K) H2/MHC-II on ADGRE1/F4/80 gated cell populations by flow cytometry. All pooled MFI values from all replicates have been added to Figure S4D-F. Data represent the mean ± SEM of 2 wells and are from 2–3 independent experiments. (L–N) BMDMs were stimulated with the ligand of CLEC4E (C4 [24 µg/ml]) and TLR4 (T4 [10 ng/ml]) for 48 h. The cells were infected with GFP-H37Ra and phagocytosis was analyzed by (L) confocal microscopy (DIC and GFP); (M) flow cytometry. Scale bar: 10 µm. Data in each histogram overlay depicted the MFI, which was shown as bar diagram in Figure S4. (N) BMDMs were stimulated with C4.T4 for 48 h, followed by infection with H37Rv and phagocytosis was validated by CFU after 4 h. Data represents the mean ± SEM of 4 wells and are from 2–3 independent experiments. (A–N) IC, isotype control; US, unstimulated; UI, uninfected; UT, untreated and infected; C4, CLEC4E agonist (TDB); T4, TLR4 agonist (ultra-pure LPS); ns, non-significant. Data were analyzed by one-way ANOVA repeated measure *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
The release of pro-inflammatory cytokines contributes to protection against Mtb [45]. We observed a significant increase in the release of IL6 (p < 0.0001), IL12B (p < 0.001), TNF (p < 0.001), and IL1B (p < 0.01) upon stimulation of Mtb-infected macrophages with C4.T4 combinatorial therapy (MφC4.T4) (Figure 1D–G). In contrast, a decrease in the level of IL10 (p < 0.01) was noted (Figure 1H). The increased levels of Il6 (p < 0.001), Il12b (p < 0.05) and Il1b (p < 0.01) was further verified by RT-qPCR (Figure S4A–C).
For optimum T cell activation, two signals are essential; occupancy of TCRs by peptide-MHC complexes and subsequent activation of costimulatory molecules. Without the activation of costimulatory molecules, T cells undergo anergy [45]. Here, we observed up-regulated expression of costimulatory molecules CD40, CD86, and H2/MHC-II (histocompatibility 2) on MφC4.T4. These result suggested that MφC4.T4 attain augmented capacity to activate T cells (Figures 1I–K and S4D–F).
Triggering with C4.T4 enhanced antigen uptake potency of macrophages
Macrophages are highly potent phagocytic cells [46–49]. MφC4.T4 increased uptake of GFP-H37Ra, as demonstrated by confocal microscopy and flow cytometry (Figures 1L,M and S5). Further, these results were corroborated with CFU assays (p < 0.01) with H37Rv (Figure 1N). These experiments indicated that combinatorial signaling upon stimulation with C4.T4 agonists boosted the macrophage potential to phagocytose Mtb.
CLEC4E and TLR4 agonists enhanced macrophage bactericidal activity
Based on the increased phagocytic activity, we subsequently analyzed the bactericidal activity of the MφC4.T4. Mφ were first infected with Mtb, followed by stimulation through C4.T4 for 48 h. Further, to establish the specificity of the CLEC4E and TLR4 signaling, we used their inhibitors (piceatannol, SYK inhibitor) and CLI-095, respectively, and enumerated Mtb killing by CFU. As compared to unstimulated macrophages, MφC4.T4 showed enhanced bactericidal activity, as evidenced by significant reduction (p < 0.0001) in CFUs (Figure 2). Intriguingly, we noticed a significant (p < 0.01) increase in the survival of Mtb in the MφC4.T4 when signaling through CLEC4E and TLR4 was blocked (Figure 2A). These experiments confirmed the specificity of C4.T4 in signaling through CLEC4E and TLR4 to regulate macrophage function. Further, we validated our results with the attenuated Mtb-H37Ra strain (p < 0.01) and M. smegmatis (p < 0.001) (Figure S6A,B).
Figure 2.
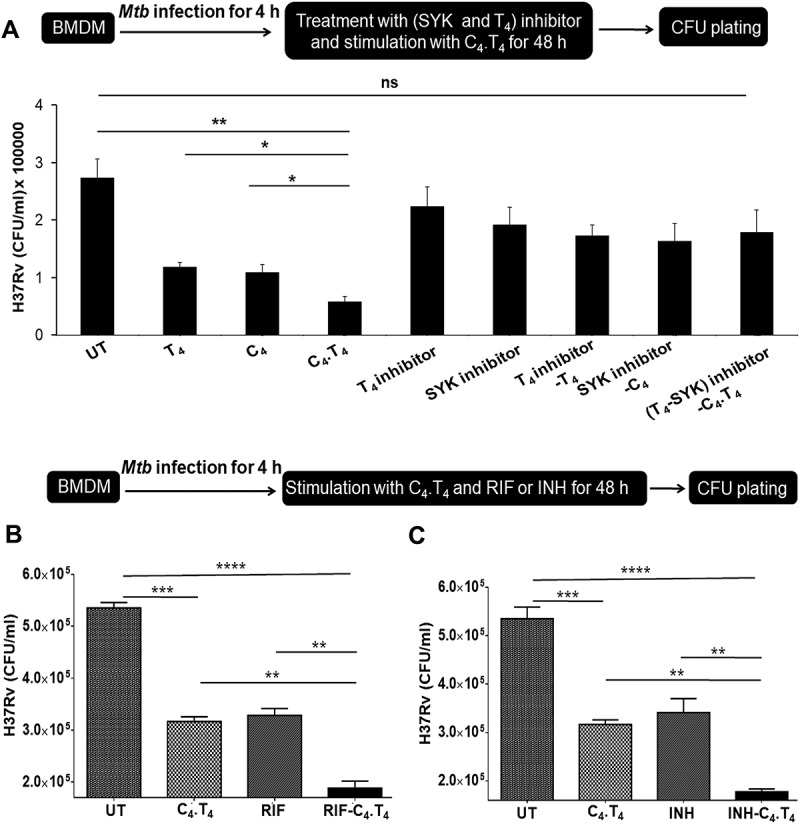
Adjunct therapy with C4.T4 in combination with anti-TB drugs restricted the survival of Mtb. (A) BMDMs were infected with H37Rv for 4 h and treated with CLEC4E (SYK-piceatannol) and TLR4 (CLI-095) signaling inhibitors for 1 h, prior to C4.T4 stimulation for 48 h, then (B,C) infected cells were treated with (B) rifampicin (0.5 µg/ml) or (C) isoniazid (2.5 µg/ml) along with C4.T4 for 48 h. The infected macrophages were lysed and CFUs were enumerated after 21 d. UT, untreated and Mtb infected; C4, CLEC4E agonist (TDB); T4, TLR4 agonist (ultra-pure LPS); ns, non-significant; RIF, rifampicin; INH, isoniazid. Data present as the mean ± SEM of 4 wells and are from 3 independent experiments. Data were analyzed by one-way ANOVA repeated measure *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Next, we investigated whether activating macrophages with C4.T4 could potentiate the efficacy of rifampicin (RIF) and isoniazid (INH) to kill Mtb. We noted a significant enhancement in the efficacy of RIF (p < 0.01) and INH (p < 0.01) to kill Mtb in MφC4.T4, as compared to the drugs alone (Figure 2B,C). These results suggested that adjunct therapy involving the agonists C4.T4 along with RIF and INH would improve the efficacy of anti-TB drugs, at a substantially lower drug dose than that of the currently used regimen.
Treatment with the agonists C4.T4 restricted the growth of Mtb and improved the potency of drugs in the murine and guinea pig models of TB
We next assessed in vivo efficacy of the agonists C4.T4. Administration of C4.T4 to Mtb-challenged mice protected them, as indicated by a statistically significant decline in bacterial burden in the lungs (p < 0.0001), liver (p < 0.0001) and spleen (p < 0.0001), as compared to the PBS (placebo) group (Figure 3A). Further, C4.T4 treatment substantially decreased the number (p < 0.001) of granulomas in the lung, as evidenced by lung morphology and granulomatous lesions, and perivascular and peribronchiolar cuffing in histopathological samples. We noted that granulomas were less consolidated with more normal alveolar structures in C4.T4-immunized lungs. More infiltration and accumulation of lymphocytes were observed in the non-immunized placebo control mice (Figure 3B,C). Next, we investigated whether treatment with C4.T4 agonists potentiate the efficacy of anti-TB drugs even at a lower dose than the recommended concentration [50]. Interestingly, we observed that treatment of mice with C4.T4 in combination with RIF considerably (p < 0.001) boosted the killing efficacy of the drug, when compared to the drug alone (Figure 3D). It is important to note that 1 mg/kg body weight (bw) of RIF in combination with C4.T4 decreased Mtb CFUs more than 10 mg/kg bw of RIF alone. Furthermore, improved killing efficacy of Mtb was achieved with only 2 oral doses (1 mg/kg bw) in combination with C4.T4, as compared to the recommended daily drug regime (Figure 3D).
Figure 3.
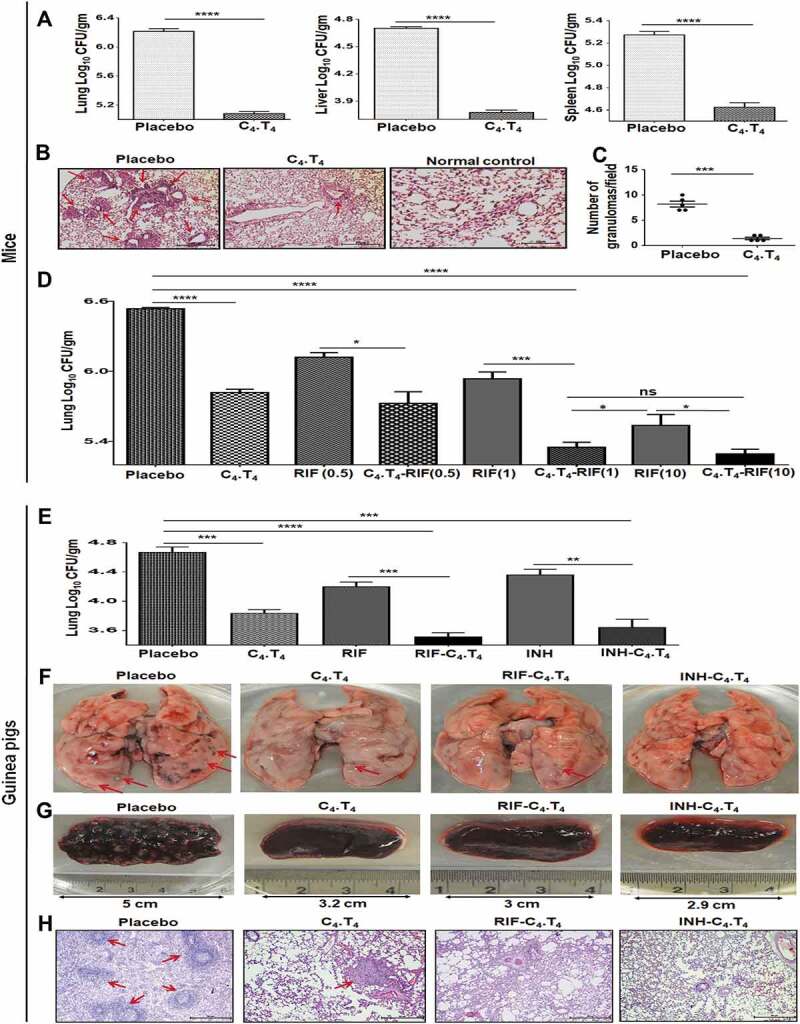
C4.T4 improved the efficacy and reduced the dose of anti-TB drugs in mice and guinea pigs. (A–D) Mtb-infected mice were treated with C4.T4. After 45 d, mycobacterial load in the (A) lungs, liver and spleen were enumerated by CFU plating. (B) Photomicrographs (10X) of H & E stained lung sections; (C) the number of granulomas/field. Scale bar: 50 µm. (D) Mtb-infected mice were administered C4.T4 in combination with anti-TB drugs (RIF: 0.5 mg, 1 mg and 10 mg/kg bw). After 45 d, mycobacterial load in the lungs was enumerated by CFU plating. Data represent CFU/gm wt of lungs. Control group was injected with only PBS (placebo). Data are mean ± SEM from 3 independent experiments (n = 4 mice/group). Data were analyzed by one-way ANOVA repeated measure *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001. (E–H) Guinea pigs infected with Mtb were treated with the C4.T4 along with anti-TB drugs (RIF: 6 mg/kg bw; INH: 5 mg/kg bw). After 45 d, (E) Mtb load in the lungs was enumerated by CFU plating. Data represent CFU/gm wt of lungs. Gross pathology photomicrograph showed granulomas in the (F) lungs and (G) spleen (n = 4 animals/group). Size of the spleen is indicated by the scale bar. (H) Photomicrographs (10X) of H & E stained lung sections. Scale bar: 50 µm. Control group of guinea pigs was administered only PBS (placebo). Red arrows indicated the granulomas. Normal, animals not exposed with Mtb; Placebo, animals exposed to Mtb; C4.T4, Mtb-exposed animals treated with C4.T4; RIF, rifampicin; INH, isoniazid. Data (mean ± SEM) are from 2 independent experiments (n = 3 guinea pigs/group unless otherwise indicated). Data were analyzed by one-way ANOVA repeated measure **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
The guinea pig model of TB is a gold standard for validating the efficacy of murine results. We observed that treatment with C4.T4 significantly (p < 0.001) decreased Mtb burden in guinea pig lungs (Figure 3E), and substantially increased the killing efficacy of RIF (p < 0.001) and INH (p < 0.01), as illustrated by decreased lung CFUs. Gross pathology of the lungs and spleen suggested nearly normal morphology of animals injected with C4.T4, as compared to placebo control. Improved lung and spleen architecture were also observed (Figure 3F,G). The lungs were less consolidated with more normal alveolar structures in C4.T4-immunized animals, which was further improved in the groups that received C4.T4 with RIF or INH (Figure 3H). These results indicated the in vivo efficacy of C4.T4 in confining the growth of Mtb in both murine and guinea pig models.
Treatment of mice with C4.T4 expanded the pool of Th1 cells and Th17 cells against Mtb
Next, we administered C4.T4 to the Mtb-challenged mice. The animals were sacrificed, and splenocytes were cultured in vitro with PPD to assess Mtb-specific T cell responses. In splenocytes from C4.T4 treated mice, Mtb-reactive CD4 T cells (p < 0.0001) and CD8A T cells (p < 0.01) proliferated significantly more than those isolated from the placebo (PBS) control (Figure 4A–D). Likewise, significantly (p < 0.001) greater proliferation of T cells was noticed in the case of splenocytes isolated from C4.T4-treated guinea pigs (Figure 4E,F). These results demonstrated that signaling of macrophages through C4.T4 improved the activation of Mtb-specific T cells.
Figure 4.
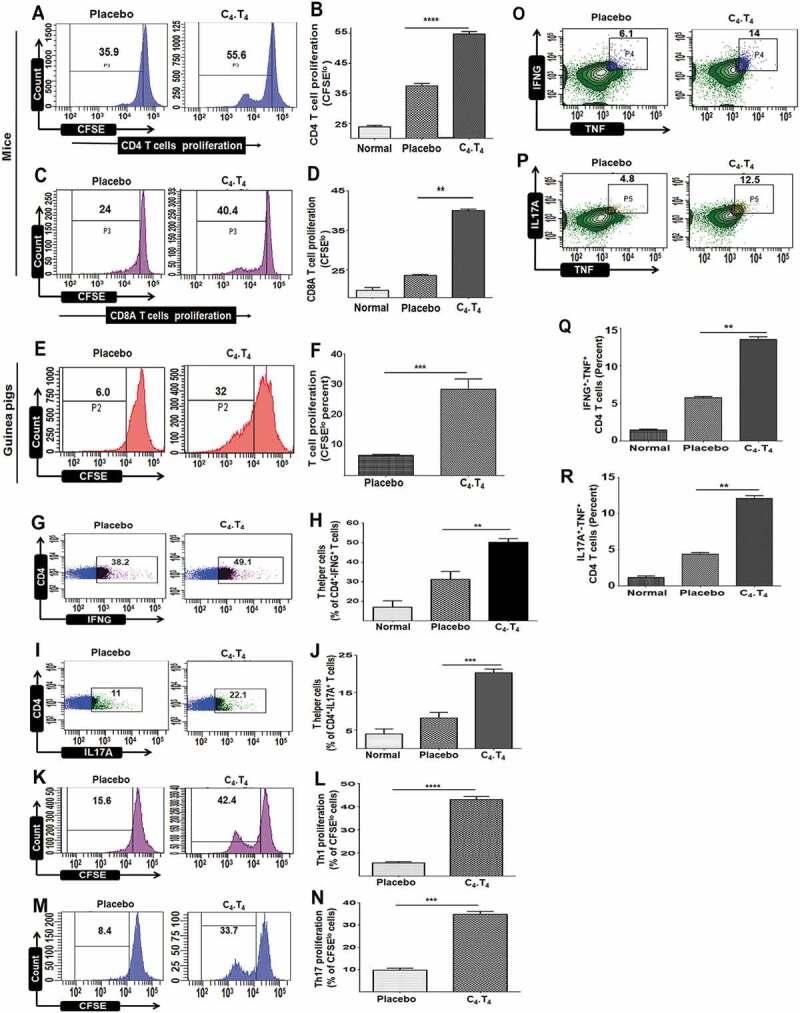
Treatment with C4.T4 augmented the proliferation of T cells and evoked Th1 and Th17 immune responses in Mtb-infected mice and guinea pigs. Mtb-challenged animals were treated with C4.T4. After 45 d, splenocytes were isolated and CFSE labeled. Proliferation of (A,B) CD4 T cells and (C,D) CD8A T cells was examined. (E,F) T cell proliferation of cells isolated from the spleen of guinea pigs. Data in the inset of flow cytometer histograms depicted percentage of CFSElo cells and were correspondingly shown as bar diagrams (B,D,F). Intracellular staining of (G,H) IFNG+ and (I,J) IL17A+ was measured by flow cytometry in CD4 T cells isolated from the lung. The CD4 gated T cells showed intracellular expression of (K,L) IFNG and (M,N) IL17A were examined for proliferation through CFSE-dye dilution assay. The number in the inset of the flow cytometry plots depicted percentage of cells (G,I,K,M). Bar diagrams show pooled data from 2–3 independent experiments (H,J,L,N). (O–R) The cells isolated from lungs were stimulated with C4.T4 for 72 h and intracellular staining was done for (O,Q) IFNG-TNF, (P,R) and IL17A-TNF in CD4 gated polyfunctional T cells. Numbers in the inset of the flow cytometry plots depicted the percentage of cells. The flow cytometry data are also shown as bar diagrams and are pooled data from 2–3 independent experiments (Q,R). Normal, animals not exposed to Mtb; Placebo, animals exposed to Mtb; C4.T4, Mtb-exposed animals treated with C4.T4. Data are mean ± SEM from 2–3 independent experiments (n = 4 mice/group), each performed in triplicate. Data were analyzed by one-way ANOVA repeated measure **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Besides Th1 cells, Th17 cells are also crucial for protection against Mtb [51,52]. We noted Th1 cells and Th17 cells expansion in the animals that were given C4.T4, as indicated by significantly higher production of IFNG (p < 0.01) and IL17A (p < 0.001) by CD4-positive T cells that had been isolated from the lungs and in vitro stimulated with PPD (Figures 4G–J and S7). Similarly, significantly increased expression of IFNG (p < 0.05) and IL17A (p < 0.001) was observed in CD8A-positive T cells (Figure S8A–D). Furthermore, we observed notable proliferation of T cells gated on either CD4-IFNG (p < 0.001) or CD4-IL17A (p < 0.001) (Figure 4K–N).
Polyfunctional T cells produce multiple cytokines and are thought to function better at protection against Mtb than their counterparts that secrete only single cytokines [53–55]. The cells isolated from the mice injected with C4.T4 showed a higher frequency of IFNG+-TNF+ (p < 0.01) and IL17A+-TNF+ (p < 0.01) polyfunctional CD4 T cells, as compared to placebo control (Figure 4O–R). Secretion of IFNG in combination with TNF by T cells has direct correlation with protection against Mtb by promoting the induction of autophagy [56]. Further, IL17A imparts protection by chemokine-mediated recruitment of Th1 cells at the site of infection [57].
We also demonstrated the role of MφC4.T4 in stimulating T cells in vitro. CD4-positive T cells were isolated from Mtb-challenged mice and the macrophages were derived from BMDMs. CD4-positive T cells were carboxyfluorescein diacetate N-succinimidyl ester (CFSE)-labeled and co-cultured with Mtb-infected MφC4.T4. We observed increased T cell proliferation with MφC4.T4, compared to control Mφ (Figure S9A). Our data suggested the immunomodulatory potential of C4.T4 bolstered both in vivo and in vitro immunity against Mtb.
Treatment of mice with C4.T4 generated memory CD4 T cells
Mice were challenged with Mtb and then administered C4.T4. The animals were sacrificed, and lung cells were isolated to monitor memory CD4-positive T cells. As expected, Mtb infection increased the percentage of central memory (CD44hi-SELL/CD62Lhi) (p < 0.0001) and effector memory (CD44hi-SELL/CD62Llo) (p < 0.01) CD4 T cells, and these populations were further increased in the group treated with C4.T4 (Figures 5A–C and S10). Further, we observed a significant (p < 0.001) increase in the frequency of memory cells (CD4+-IL7R/CD127hi); thereby confirming the above data (Figure 5D). We further confirmed our results by observing an enhanced level of memory CD4 T cells (CD4+-CD44hi-SELL/CD62Lhi) in vitro by co-culturing CD4-positive T cells isolated from Mtb-challenged mice with Mtb-infected MφC4.T4 (Figure S9B). Activated APCs capture antigen from the infection site and then migrate towards the draining lymph nodes to activate naive T cells. CCR7 is responsible for the migration of APCs [58,59]. We observed a significantly larger (p < 0.0001) population of CD44+-CCR7hi expressing T cells in Mtb-infected animals that received C4.T4 (Figure 5E). Overall, these data suggest that C4.T4 therapy enhanced the immunological memory and migratory potential of CD4 T cells.
Figure 5.
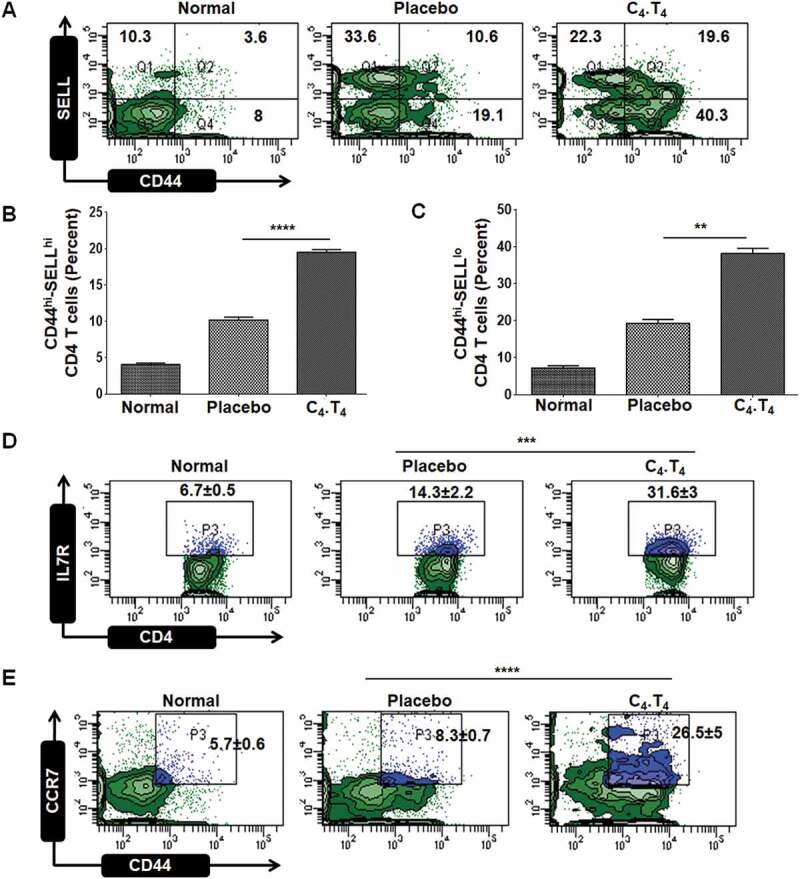
Signaling through CLEC4E and TLR4 generated sustained memory CD4 T cells. Mice were challenged with Mtb and treated with C4.T4. The control group was administered PBS (placebo). After 45 d, cells were isolated from the lungs, in vitro cultured with PPD for 48 h and analyzed for the expression of (A,B) central memory (CD4+-CD44hi-SELL/CD62Lhi); (A,C) effector memory (CD4+-CD44hi-SELL/CD62Llo); (D) CD4+-IL7R/CD127hi; (E) CD4+-CD44+-CCR7hi markers by flow cytometry. The number in the inset of the flow cytometry dot plots depicted the percentage of cells. Normal, animals not exposed to Mtb; Placebo, animals exposed to Mtb; C4.T4, Mtb-exposed animals treated with C4.T4. Data are mean ± SEM and are from 3 independent experiments (n = 4 mice/group). Data were analyzed by one-way ANOVA repeated measure **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Treatment of mice with C4.T4 enhanced autophagy gene expression
Autophagy has been suggested to play an important role in protection against Mtb [47,60,61]. Consequently, we next wanted to monitor autophagy in the Mtb-challenged mice that were treated with C4.T4. Interestingly, the lung cells of these animals showed a substantial reduction in Mtb burden (Figure 3D) and a significant increase in the expression of the autophagy genes Atg5 (p < 0.0001), Atg7 (p < 0.0001), Lc3 (p < 0.01), Becn1 (p < 0.0001), Atg12 (p < 0.001), Lamp1 (p < 0.0001), and Eea1 (p < 0.01) (Figure 6A–G). Further, reduced Il4 (p < 0.0001) and Il10 (p < 0.01) gene expression was noted (Figure 6H–I). Il4 and Il10 genes are known to inhibit autophagy [56,62,63]. These results indicated a correlation between increased autophagy gene expression and reduced Mtb growth after C4.T4 treatment of mice.
Figure 6.
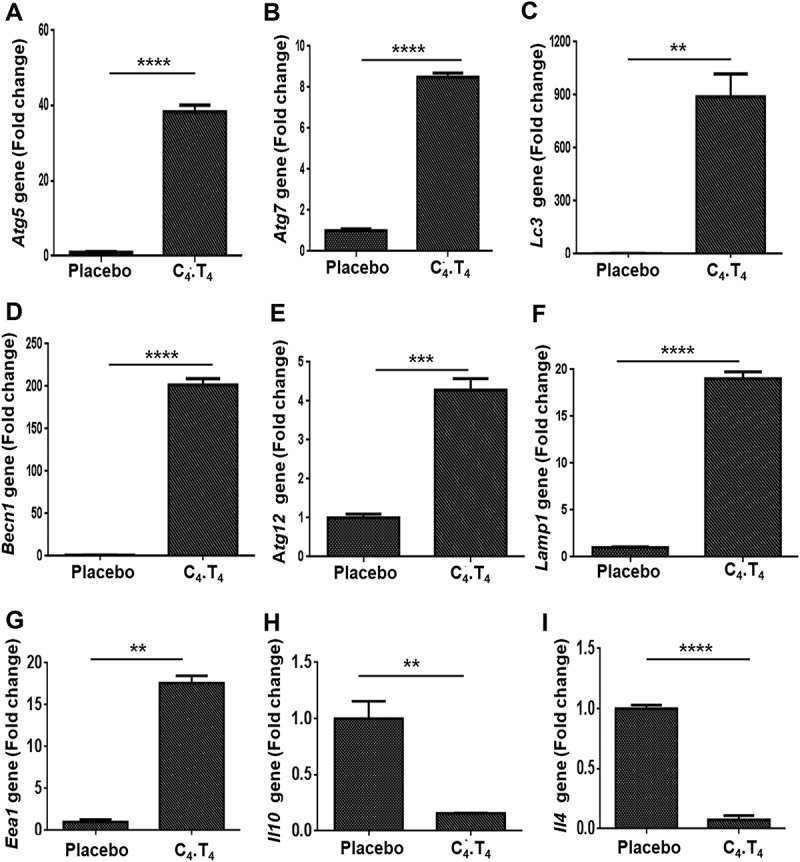
Signaling through C4.T4 agonists induced autophagy in vivo. Mtb-challenged animals were treated with C4.T4 and RNA was isolated from lung cells. Quantitative RT-PCR was performed to monitor the expression of (A) Atg5; (B) Atg7; (C) Lc3; (D) Becn1; (E) Atg12; (F) Lamp1; (G) Eea1; (H) Il10; (I) Il4. Placebo, animals exposed to Mtb; C4.T4, Mtb-exposed animals treated with C4.T4. Data are the mean ± SEM from 3 independent experiments, performed in triplicate wells. Data were analyzed by unpaired Student’s “t” test ** p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
C4.T4 enhanced autophagy flux through MYD88-mediated, MTOR-independent activation of PtdIns3K
We next determined if the in vivo findings of C4.T4 induced autophagy gene expression correlates with autophagy activation in macrophages. The conversion of LC3-I to LC3-II is one of the fundamental indicators of autophagy [47,61,64,65]. We observed conversion of LC3-I to LC3-II in C4.T4-stimulated, Mtb-infected macrophages (Figure 7A). This finding demonstrated the novel role for CLEC4E in inducing autophagy. We measured autophagic flux by probing for SQSTM1/p62 degradation and inhibited this with bafilomycin A1. Bafilomycin A1 prevents activation and maturation of autophagic vacuoles by impeding vacuolar-type H+ ATPase (V-ATPase), thus avoiding degradation of autophagosomes [66,67]. Interestingly, we observed a substantial decrease in SQSTM1 expression in C4.T4-stimulated macrophages (Figure 7B) and expression of SQSTM1 was induced by bafilomycin A1 treatment in Mtb-infected macrophages. This observation suggested that the augmented conversion to LC3-II by C4.T4 was due to increased autophagic flux. In addition, C4.T4 induced autophagy in macrophages infected with either virulent (H37Rv) or avirulent (H37Ra) strains of Mtb (Figure S11A,B). We also observed increased LC3 puncta formation, as visualized using confocal microscopy (Figure 7C). As expected, C4.T4 induced autophagy was inhibited by PtdIns3K and autophagy inhibitors (Ly294002 and 3MA, respectively) (Figure 7D). Rapamycin was used as a positive control (Figure 7D). We also determined that CLEC4E and TLR4 pathways were MTOR-independent (Figure 7E). Taken together, these results provided evidence that the induction of autophagy in macrophages through C4.T4 signaling was linked with the MTOR-independent PtdIns3K pathway.
Figure 7.
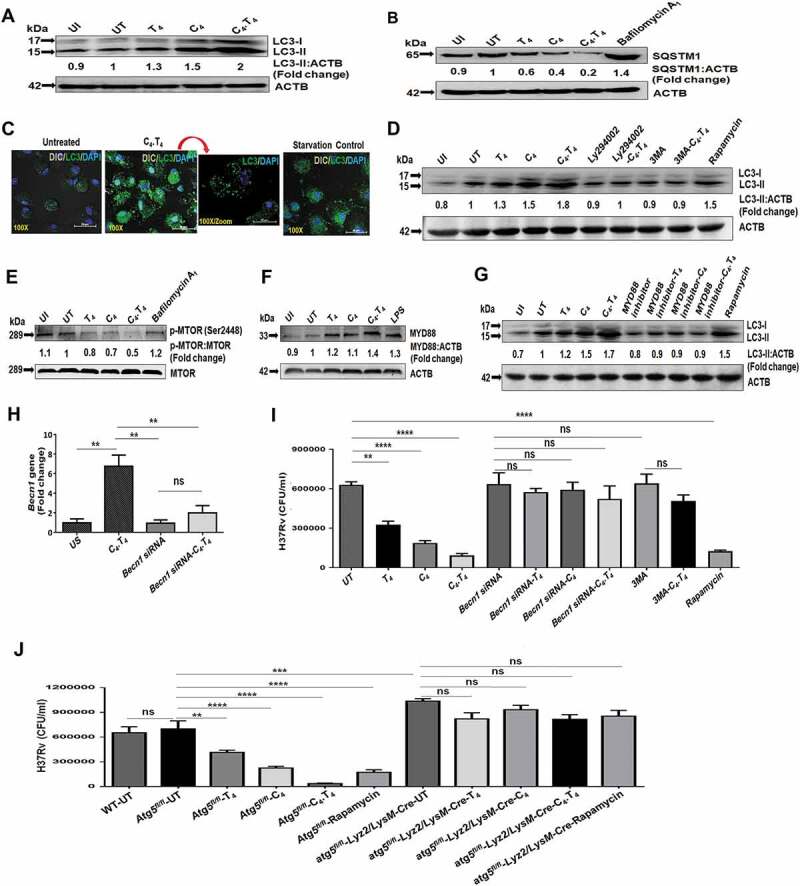
Signaling of Mtb-infected macrophages through C4.T4 induced autophagy flux through the PtdIns3K pathway. BMDMs were infected with H37Rv for 4 h, then stimulated with CLEC4E (C4 [24 µg/ml]) and TLR4 (T4 [10 ng/ml]) individually or in combination (C4.T4) for 4 h and checked for the (A) conversion of LC3-I to LC3-II and (B) expression of SQSTM1/P62 by western blot. Bafilomycin A1 was used as a lysosomal enzymatic activity inhibitor, which prevents the degradation of autophagosomes. (C) Uninfected BMDMs stimulated with C4.T4 for 4 h were stained for the expression of LC3 puncta formation with anti-LC3 Abs (green). The nucleus was stained with DAPI (blue). Scale bar: 20 µm. (D) The specificity of the induction of autophagy was determined by blocking PtdIns3K to inhibit autophagy (Ly294002 and 3MA). Rapamycin was used as a positive control for autophagy and ACTB was a loading control. (E) Phosphorylation of MTOR was measured by western blot. Fold change was calculated relative to UT controls. (F,G) Infected BMDMs were treated with a MYD88 inhibitor (pepinh-MYD) for 1 h and then stimulated with C4.T4 for 4 h. (F) Expression of MYD88 and (G) conversion of LC3-I to LC3-II was observed by western blot. Data are representative of 3 independent experiments. (H,I) BMDMs were treated with Becn1 specific siRNA for 72 h, and then cells were infected with Mtb for 4 h. Becn1 expression was determined by (H) RT-qPCR. (I) Becn1 specific siRNA treated BMDMs were infected with H37Rv for 4 h and treated with the autophagy inhibitor 3MA for 1 h. Then the cells were stimulated with C4.T4 for 48 h. Infected macrophages were lysed and CFUs were enumerated after 21 d. CFU assay showed the blocking of autophagy by siRNA or its inhibitor 3MA, increased the survival of Mtb. Data are presented as the mean ± SEM and are from 3 independent experiments. (J) BMDMs were collected from wild type (WT), atg5 knockout (atg5fl/fl-Lyz2/LysM-Cre), control flox (Atg5fl/fl) mice and infected with H37Rv. The cells were then stimulated with C4.T4 for 48 h. Infected macrophages were lysed and CFUs were enumerated after 21 d. Data are presented as the mean ± SEM and are from 2 independent experiments. ns, non-significant; UI, uninfected; UT, untreated and Mtb-infected; C4, CLEC4E agonist (TDB); T4, TLR4 agonist (ultra-pure LPS). Data were analyzed by one-way ANOVA repeated measure ** p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Previous reports suggested that a MYD88-dependent pathway is involved in both CLEC4E and TLR4 signaling [68–70]. We found that MYD88 expression was induced by both CLEC4E and TLR4 signaling (Figure 7F) and that MYD88 signaling was important for the induction of autophagy. Western blot results depicted abrogation of the conversion of LC3-I to LC3-II when MφC4.T4 were cultured with a MYD88 inhibitor (Figure 7G). These results indicated that C4.T4 activated macrophages through MYD88-mediated, MTOR-independent activation of the PtdIns3K pathway, which led to the induction of autophagy and inhibition of Mtb growth.
Abrogation of autophagy negated C4.T4-mediated restriction of Mtb growth in macrophages
BECN1 is a master regulator of autophagy [64]. To confirm that C4.T4 mediated killing of Mtb through autophagy, we knocked down Becn1 expression in macrophages (Figure 7H), or inhibited autophagy with 3MA. C4.T4-induced clearance (p < 0.0001) of Mtb was abolished upon Becn1 knockdown (Figure 7I). Similar results were obtained if macrophages were treated with 3MA (Figure 7I) or if macrophages from atg5 knockout (atg5fl/fl-Lyz2/LysM-Cre) mice were used, confirming that C4.T4-mediated elimination of Mtb was through autophagy (Figure 7J).
Importantly, we found significantly (p < 0.0001) reduced Mtb burden in C4.T4-stimulated human THP-1 macrophages as well (Figure S11C). C4.T4-mediated Mtb growth was inhibited after treatment with an autophagy inhibitor. These data confirmed that C4.T4 induced autophagy decreased Mtb burden in both murine and human macrophages (Figure 7I,J and S11C).
Stimulation of macrophages with C4.T4 augmented the clearance of Mtb in macrophages through the autophagic flux
We next studied the induction of autophagy in Mtb-infected macrophages by microscopy. We noted increased co-localization of GFP-Mtb with LC3 (GFP/green; LC3/red fluorescence) and increased LC3 puncta in the C4.T4 treated macrophages (Figure 8A). There was also an increased number of lysosomes and Mtb co-localization with lysosomes by immunostaining of LAMP1 (lysosomal-associated membrane protein 1) (Figure 8B). Furthermore, to confirm the presence of Mtb in lysosomes, we performed LysoTracker Red staining and observed co-localization of GFP-Mtb with acidic vacuoles in MφC4.T4 (Figure 8C). These data provided further evidence that the improved clearance of Mtb in C4.T4-treated macrophages was through a xenophagy process (the autophagy-mediated clearance of pathogens).
Figure 8.
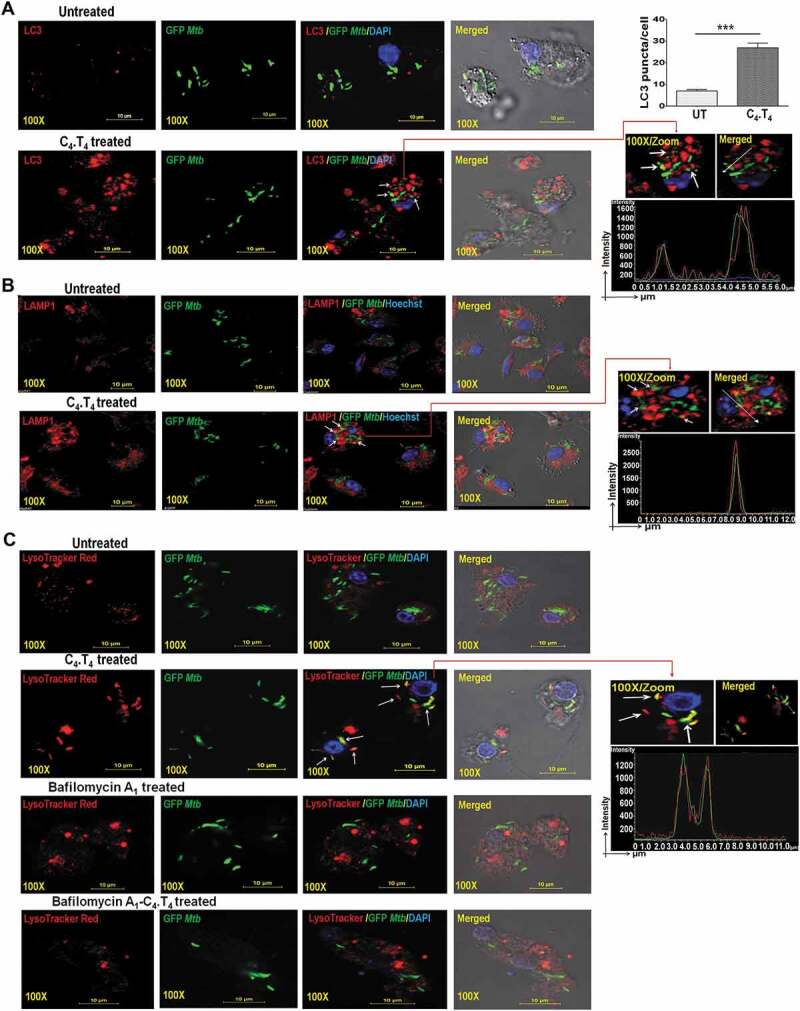
Stimulation of Mtb-infected macrophages by C4.T4 induced autophagy flux and enhanced the number of autolysosomes and transported Mtb to the lysosomes of macrophages. BMDMs were infected with GFP-Mtb for 4 h then stimulated with C4.T4 for 4 h and 10 h. The cells were immunostained with anti-LC3 and anti-LAMP1 Abs along with DAPI or Hoechst to visualize nucleus (blue) and examined for the formation of (A) autophagosomes (red) and (B) lysosomes/autolysosomes (red). (C) BMDMs were infected with GFP-Mtbfor 4 h and stimulated with C4.T4 for 10 h. LysoTracker Red staining was performed to monitor acidic vacuoles and LysoTracker co-localization with GFP-Mtb by confocal microscopy. The histogram data (green and red) showed the co-localization of GFP-Mtb (green) with LC3 (red) or LAMP1 (red) or LysoTracker (red) in C4.T4-treated macrophages indicated by white arrows. Scale bar: 10 µm and 100X magnification. (A) The bar diagram showed the number of LC3 puncta/cell. The LC3-II puncta in each cell were counted in 10 different fields. Quantification of the LC3 puncta per cell are presented as the mean ± SD. UT, untreated and GFP-Mtb-infected; C4.T4, C4.T4 treated and GFP-Mtb-infected. The data are representative of 4 independent experiments. Data were analyzed by unpaired Student’s “t” test *** p ≤ 0.001.
One hallmark of autophagy is an increase in acidic vesicular organelles (AVOs) in the cytosol [71]. Our data show an increased number of AVOs in Mtb-infected MφC4.T4 that is specific to autophagy since there is a decrease in the frequency of AVOs with 3MA inhibitor treatment (Figure 9A,B). The data were substantiated using monodansylcadaverine (MDC) staining, which is a specific dye for locating acidic compartments (Figure 9C). Therefore, these data suggested that CLEC4E and TLR4 signaling in macrophages enhanced acidic vacuole formation. We also performed intracellular staining of LAMP1 in the macrophages treated with C4.T4 by flow cytometry. A significantly (p < 0.01) higher accumulation of LAMP1 in MφC4.T4 was seen (Figures 9D,E and S12), which was further validated by staining MφC4.T4 with LysoTracker Red dye (Figure 9F).
Figure 9.
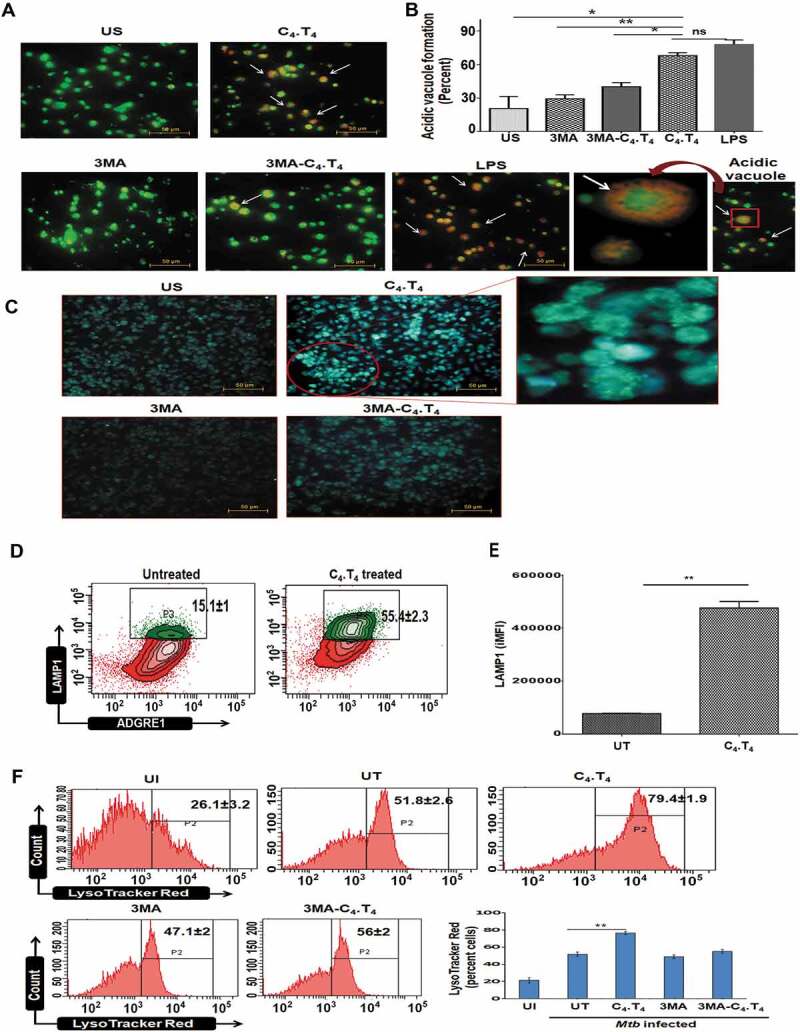
Activation of macrophages through C4.T4 induced autophagosome and lysosome formation. BMDMs were treated with 3MA (10 mM) for 1 h. Next, they were stimulated with C4.T4 for 4 h and the cells were stained with (A,B) acridine orange or (C) MDC to monitor the acidic compartments by fluorescence microscopy. Scale bar: 50 µm and 20X magnification. (B) The number of acidic vacuoles in each cell was counted in 4 individual fields for each group. White arrows indicated lysosomal acidification (orange). LPS was used as a positive control for acidic vacuoles formation and 3MA was a negative control for autophagosomes formation. Data were analyzed by one-way ANOVA repeated measure *p ≤ 0.05, ** p ≤ 0.01. Late endosomal-lysosomal acidification was assessed through (D,E) LAMP1 staining by flow cytometry; data were expressed as a bar diagram (E). Numbers in the inset of contour plots depicted the percentage of cells. The integrated MFI (iMFI) is calculated by multiplying the relative frequency (% positive population) of cells expressing LAMP1 with the mean fluorescence intensity (MFI) of that population. (F) BMDMs were infected with GFP-Mtbfor 4 h. Next, the cells were treated with 3MA (10 mM) for 1 h and then stimulated with C4.T4 for 10–12 h. Control cultures were kept without 3MA. The cells were stained with LysoTracker Red dye and monitored by flow cytometry. US, unstimulated; UI, uninfected cells; UT, untreated and Mtb-infected cells. Scale bar: 10 μm. 100X magnification. Data are expressed as mean ± SEM and are from 3 independent experiments. Data were analyzed by unpaired Student’s “t” test ** p ≤ 0.01.
CLEC4E and TLR4 signaling induced phosphorylation of PtdIns3K and STAT1 and nuclear translocation of RELA/NFKB
Activating macrophages with C4.T4 augmented NOS2 (nitric oxide synthase 2, inducible), IL6, TNF, and IL1B production. These molecules play important roles in the activation of different transcription factors like RELA/NFKB. We observed increased STAT1 and PtdIns3K phosphorylation in Mtb-infected MφC4.T4 (Figure 10A,B). The PtdIns3K inhibitor LY294002 was used as a control (Figure 10B). Interestingly, we observed decreased STAT6 phosphorylation in MφC4.T4 (Figure S13A). CLEC4E and TLR4 may enhance the survival of macrophages. We noticed increased levels of the anti-apoptotic molecule BCL2L1/Bcl-xL in MφC4.T4 (Figure 10C). Furthermore, there was downregulation in the expression of negative regulators of immunity like FAS, HAVCR2/TIM-3, and CD274/PD-L1 (Figure S13B–D). This phenotype denoted a possibility of better survival of MφC4.T4.
Figure 10.
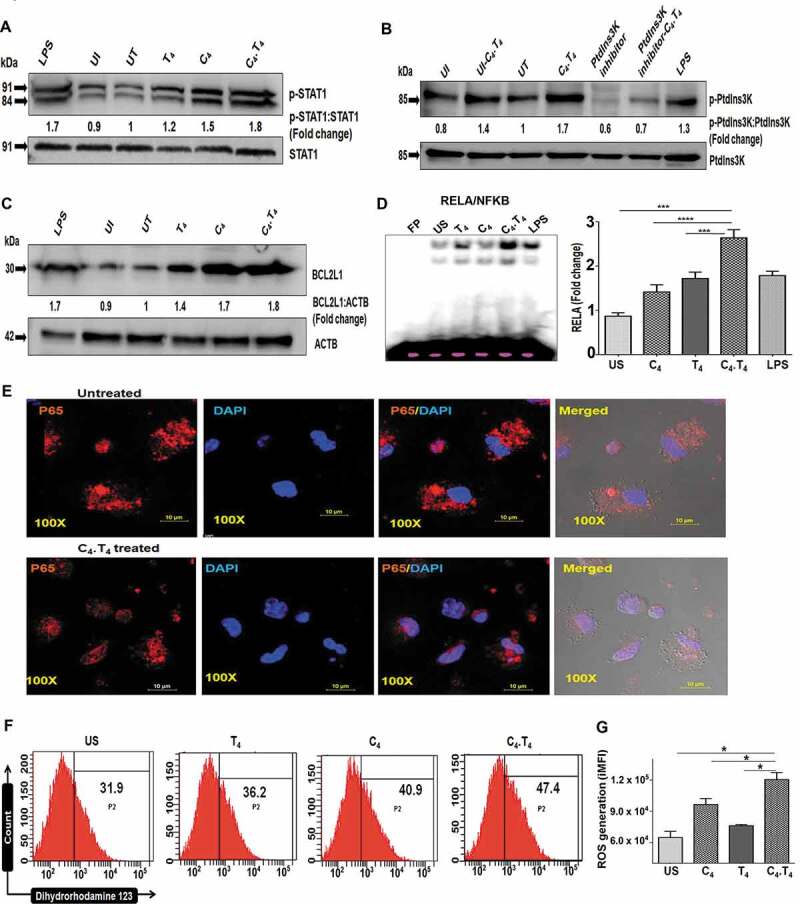
C4.T4 induced phosphorylation of STAT1, PtdIns3K and enhanced nuclear translocation of RELA/NFKB. Macrophages were infected with H37Rv for 4 h and cultured with C4.T4 for 15–30 min and 24 h. The cell lysate was prepared, and western blot was performed to monitor the expression of (A) p-STAT1; (B) p-PtdIns3K; (C) BCL2L1. The densitometry data represent fold change. The ratio for untreated cells was considered to be 1. LPS was used as a positive control. UI, uninfected; UT, untreated and Mtb-infected; C4, CLEC4E agonist (TDB); T4, TLR4 agonist (ultra-pure LPS). The data are representative of 2–3 independent experiments. (D–G) BMDMs were stimulated with C4 (24 µg/ml) and T4 (10 ng/ml) individually or in combination (C4.T4) for 30 min. (D) Nuclear translocation of RELA/NFKB was monitored by EMSA. The data were graphed as fold change in the form of a bar diagram. Unstimulated cells were considered as 1. FP, free probe; US, unstimulated. The data are shown as the mean ± SEM and are from 2 independent experiments. Data were analyzed by one-way ANOVA repeated measure. ***p ≤ 0.001, ****p ≤ 0.0001. (E) Translocation of RELA/NFKB p65 into the nucleus was further monitored by confocal microscopy and (F,G) the generation of ROS was observed by flow cytometry after labeling with dihydrorhodamine 123 dye. (F) The histogram depicted ROS generation (percentage). (G) The bar diagram represented integrated MFI of ROS generation. The data represented in each histogram or contour plot depicted the percentage of cells from the ADGRE1/F4/80 gated population. US, unstimulated; UT, untreated and Mtb-infected; C4, CLEC4E agonist (TDB); T4, TLR4 agonist (ultra-pure LPS). The integrated MFI (iMFI) is calculated by multiplying the relative frequency (% positive population) of cells generating ROS with the mean fluorescence intensity (MFI) of that population. Data are shown as the mean ± SD and are representative of 2–3 independent experiments. Data were analyzed by one-way ANOVA repeated measure. *p ≤ 0.05.
Signaling through CLEC4E via SYK induces reactive oxygen species (ROS) generation [72], which can directly activate RELA/NFKB [72–74]. RELA/NFKB is responsible for cellular growth and inflammatory responses. Considering all these facts, we monitored RELA/NFKB activation in MφC4.T4. Interestingly, we observed increased (p < 0.0001) RELA/NFKB activation and translocation into the nucleus, by EMSA (Figure 10D). We confirmed these results by confocal microscopy (Figure 10E). In addition, we noted increased (p < 0.05) ROS generation by flow cytometry (Figure 10F,G). Therefore, it can be concluded that C4.T4 signaling involved ROS and RELA/NFKB pathway, which may be responsible for the activation of macrophages. We also monitored NOS2 expression and NO release by MφC4.T4, since NO mediates killing of intracellular Mtb in murine macrophages [75,76]. Our western blot results indicated that C4.T4 enhanced the expression of NOS2, as compared to controls (Figure S14A). We confirmed that this change correlated with increased NO release and validated the specificity of the results by blocking the production of NO by its inhibitor N-mono-methyl L-arginine (NM). Decreased (p < 0.001) C4.T4-stimulated NO release was observed with NM treatment (Figure S14B). Notably, we observed significantly (p < 0.05) enhanced NO secretion from the cells isolated from the lungs of C4.T4 injected Mtb-challenged mice (Figure S14C). RT-qPCR performed on the same lung tissues revealed a significant (p < 0.01) augmentation in Nos2 gene expression in the mice administered C4.T4 (Figure S14D). Therefore, the results indicated that C4.T4-mediated killing of Mtb could occur through autophagy, as well as ROS and NO production.
Overall, the study provided evidence that combinatorial signaling through CLEC4E and TLR4 activated macrophages via the MYD88-PtdIns3K-STAT1-RELA/NFKB pathway that ultimately leads to the induction of autophagy to control the growth of Mtb (Figure 11A,B).
Figure 11.
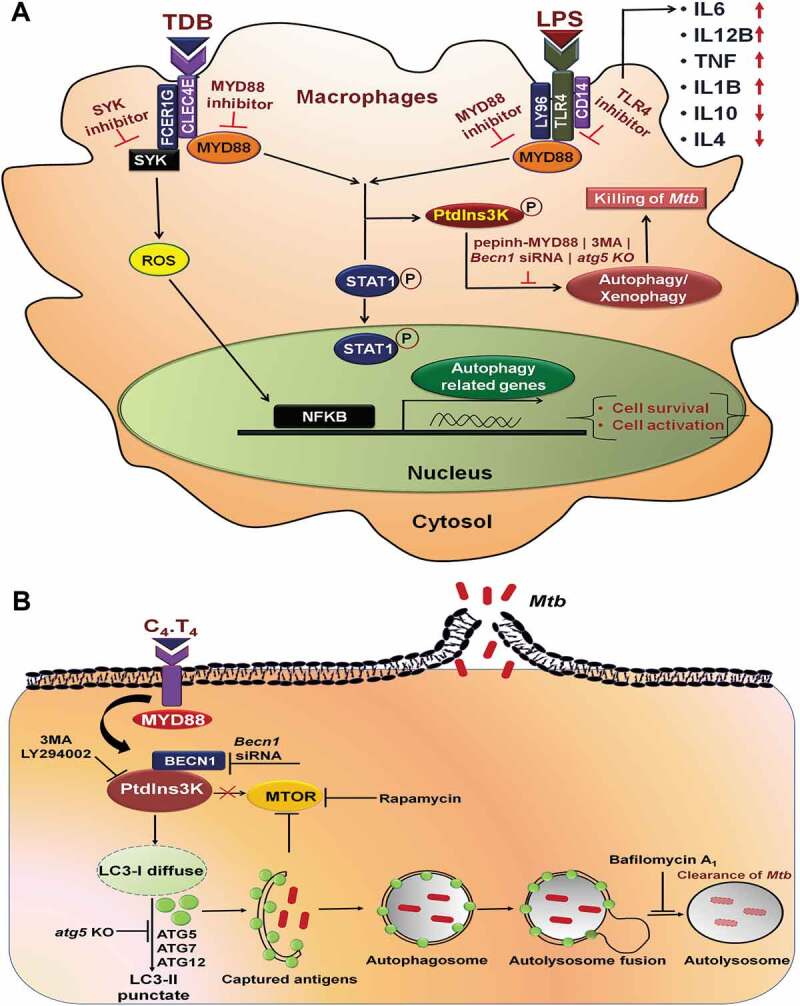
Proposed mechanism of signaling pathways initiated by C4.T4 in macrophages. (A) C4.T4 signaling activated RELA/NFKB through MYD88 pathway and enhanced cytokine secretion, which received positive feedback through phosphorylated PtdIns3K, STAT1 and activated BCL2L1. Consequently, these macrophages induced autophagy, activated cells, and increased cell survival. This mechanism played a key role in controlling the growth of Mtb. (B) Schematic representation of C4.T4-mediated clearance of Mtb through modulation of autophagy and lysosomal biogenesis.
Discussion
Modulation of host immunity can be an efficacious approach to successfully eliminate pathogens. Several innate immune receptors such as TLRs, CLRs, NLRs and RLRs can effectively bolster immunity against an array of pathogens [48,49,77–80]. Among the molecules that stimulate innate immunity, a TLR4 agonist has been considered a potential immunotherapeutic due to its ability to stimulate the immune system against several pathogens [32,33]. Additionally, CLEC4E is a promising vaccine adjuvant in modulating immunity against chronic Mtb infection [77,78]. There are no studies demonstrating the mechanistic role of CLEC4E and TLR4 agonist-mediated combinatorial signaling in imparting immunity against Mtb. The role of CLEC4E in inducing autophagy is totally unexplored. Therefore, we studied both in vitro and in vivo efficacy of CLEC4E and TLR4 on the activation of Mtb-infected macrophages. The macrophages exhibited the following major changes: i) enhanced release of cytokines, up-regulated expression of the costimulatory molecules such as CD40, CD86 and H2/MHC-II, and down-regulated inhibitory molecules such as FAS, HAVCR2/TIM-3, and CD274/PD-L1; ii) the phenotype of MφC4.T4 (IL6hi, IL12Bhi, TNFhi, IL1Bhi, IFNGhi, IL17Ahi, NOhi, IL10lo) suggested their enhanced capability to kill Mtb; iii) reduced dose and increased potency of anti-TB drugs to kill Mtb; iv) improved ability to activate Mtb-specific T cells and generate Th1 cells and Th17 cells; v) expanded pool of memory T cells; vi) induced MYD88, PtdIns3K, STAT1, and RELA/NFKB pathways; vii) restricted survival of Mtb through autophagy. These data provide sufficient evidence for a novel role of CLEC4E and TLR4 in boosting the function of macrophages to kill Mtb.
The currently available regime to treat TB patients is highly effective but the major drawbacks are its adverse side-effects, immune-suppression, long-duration of treatment and emergence of drug resistance in Mtb [81–84]. Our study showed that adjunct therapy of anti-TB drugs (RIF and INH) with C4.T4 agonists dramatically improved their potency to kill Mtb. Notably, we achieved better killing of the bacterium with 10-fold lower dose of rifampicin along with C4.T4, as compared to the drug alone. This observation suggested an immunomodulatory role of C4.T4 in reducing the dose as well as duration of anti-TB drugs. C4.T4 will bolster host immunity against the pathogen and thereby provide an added advantage for the drug to act more efficiently in killing the bacterium, and consequently, may minimize the opportunity for the bacterium to develop drug resistance.
Both CD4- and CD8A-positive T cells protect against Mtb [85]. We observed that C4.T4 therapy enhanced CD4-positive T cells and CD8A-positive T cells proliferation. Furthermore, Th1 cells and Th17 cells played an important role in protection against Mtb. In contrast, Th2 cells support the propagation of Mtb [86]. Excitingly, an expanded pool of Th1 cells and Th17 cells but not Th2 cells was noted in the animals that were treated with C4.T4. The same group of animals showed significant clearance of Mtb from the lungs and reduced its dissemination to the spleen and liver. The presence of polyfunctional Th1 and Th17 cells, which provide better protection against pathogens when compared to their counterparts secreting a single cytokine [53–55], further supports the role of C4.T4 therapy against Mtb. Memory T cells induce long-lasting protection [87,88]. C4.T4 significantly enhanced the pool of memory T cells in the same animals that exhibited decreased Mtb burden, signifying the generation of immunological memory to protect from subsequent Mtb infection.
There are growing evidences that autophagy helps to protect against many diseases [47,60,61,89]. The contribution of autophagy during Mtb infection has been well-defined as autophagy induction or inhibition controls and enhances Mtb growth, respectively, and Mtb co-localizes with autophagy factors ATG5, ATG12, SQSTM1, BECN1 and LC3 [90–93]. In contrast, a previous report shows autophagy-independent functions of ATG5 in different experimental settings in vivo [94–96]. Autophagy can control Mtb through multiple mechanisms: it targets the antigen for lysosomal degradation and improves antigen processing and presentation to T cells [97]. Concurrently, it inhibits the excessive inflammatory response [37,60]. Notably, C4.T4-treated mice showed enhanced autophagy and autophagic flux. Additionally, expression of autophagy-related genes was increased, which correlated with a decreased number of Mtb. Blocking of autophagy (through reduced BECN1 or ATG5 expression) rescued Mtb growth. These results provided strong support for the involvement of autophagy in combating Mtb in mice that were administered C4.T4.
Following ligand binding, CLEC4E interacts with FCER1G/FcRγ chain, then phosphorylates and activates SYK [72]. This activation induces ROS generation and CARD9-mediated RELA/NFKB activation; which leads to IL6, TNF and IL1B secretion [72–74]. TLR4 signaling activates the conventional MYD88-mediated pathway, leading to RELA/NFKB activation and cytokine production, which may contribute to a positive feedback loop. Consequently, PtdIns3K becomes activated, which induces a survival signal and autophagy. We observed involvement of the MYD88 pathway and induction of autophagy in CLEC4E signaling. Activation of PtdIns3K delivered a signal through RELA/NFKB and BCL2L1. Interestingly, we noticed that signaling through C4.T4 activates RELA/NFKB and augments production of IL6, IL12B, TNF, and IL1B, consequently delivering a feedback mechanism through STAT1 phosphorylation that supported the activation and survival of MφC4.T4.
Overall, we have elucidated a novel signaling network operating through C4.T4 (Figure 11). Signaling via C4.T4 activated RELA/NFKB through MYD88-mediated phosphorylation of PtdIns3K, STAT1 and release of NO. Signaling through C4.T4 activated macrophages and induced autophagy to effectively control Mtb infection. In the future, treatment with C4.T4 agonists alone or as an adjunct therapy with anti-TB drugs may be an important new host-directed strategy to treat TB patients.
Materials and methods
Animals
C57BL/6 mice, 6–8 weeks were obtained from the institute’s animal facility. Guinea pigs (6–8 weeks) were purchased from the CCS Haryana Agricultural University, Hisar, India. Atg5fl/fl and atg5fl/fl-Lyz2/LysM-Cre mice bones were kindly provided by Dr. Christina L. Stallings, Department of Molecular Microbiology, Washington University School of Medicine, St Louis, MO.
Ethics statement
All the experiments were approved by the Institutional Animal Ethics Committees (IAEC) of the Institute of Microbial Technology (IMTECH), Chandigarh and International Center for Genetic Engineering and Biotechnology (ICGEB), New Delhi. The experiments were accomplished according to the National Regulatory Guideline issued by Committee for the Purpose of Control and Supervision of Experiments on Animals (No. 55/1999/CPCSEA), Ministry of Environment and Forest, Government of India. Mice experiments were performed at IMTECH and guinea pigs at the ICGEB. The atg5 knockout experiments were performed at Texas Biomedical Research Institute, San Antonio, TX, USA. The experiments were approved by the Texas Biomedical Research Institute, Institutional Animal Care and Use Committee (IACUC).
Antibodies and reagents
Agonist of CLEC4E (TDB, tlrl-tdb), TLR4 (LPS EK ultrapure, tlrl-peklps), SYK inhibitor (Piceatannol, tlrl-pct), TLR4 signaling inhibitor (CLI-095, tlrl-cli95) and MYD88 inhibitor (Pepinh-MYD, tlrl-pimyd) were purchased from InvivoGen.
Antibodies (Abs) and reagents used in western blotting: phospho-PI3 kinase p85 (Cell Signaling Technology, 4228S), PI3 kinase p85 (Cell Signaling Technology, 4257S), BCL2L1/Bcl-xL (Cell Signaling Technology, 2762S), pMTOR (Ser2448; Cell Signaling Technology, 5536S), MTOR (Cell Signaling Technology, 2983S), p-STAT6 (Cell Signaling Technology, 56554S), STAT6 (Cell Signaling Technology, 5397S), RELA/NFKB-p65 (Cell Signaling Technology, 8242S) and MYD88 (Cell Signaling Technology, 4283S), SQSTM1/p62 (Santa Cruz Biotechnology, sc-48402), NOS2/iNOS (Abcam, ab3523). Other reagents were procured from the specified companies: inhibitors against PtdIns3K (Ly294002; Sigma Aldrich, 440202) and NOS2 [NM] (Calbiochem, 475886), FlexiTube siRNA-specific for mouse Becn1 (Qiagen, GS56208), acridine orange (Sigma Aldrich, A6014), monodansylcadaverine (Sigma Aldrich, D4008), rabbit polyclonal Ab against LC3 (Sigma Aldrich, L8918), anti-ACTB antibody (Sigma Aldrich, A1978), p-STAT1 (BD Pharmingen, 612232), STAT1 (BD Pharmingen, 610115), LysoTracker Red (Life Technologies, L-7528), anti-fade reagent (Life Technologies, P36934), dihydrorhodamine 123 (Sigma Aldrich, D1054), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma Aldrich, D8417), 3-methyladenine (3MA; Sigma Aldrich, M9281), rapamycin (Sigma Aldrich, B1793), 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE; Sigma Aldrich, 21888F), phorbol 12-myristate 13-acetate (PMA; Calbiochem, 524400), DMSO (Sigma Aldrich, D8418), RELA/NFKB consensus oligonucleotide (Promega, E3292), paraformaldehyde (Sigma Aldrich, P6148), Griess reagent (Sigma Aldrich, G4410), poly-L-lysine solution (Sigma Aldrich, P8920), HBSS (GIBCO, 14185052), isoniazid (Sigma Aldrich, 13377), rifampicin (Sigma Aldrich, R3501), bovine serum albumin (BSA) (Sigma Aldrich, A0281), Triton™ X-100 solution (Sigma Aldrich, 93443), glycerol (Sigma Aldrich, G5516), TWEEN 20 (Sigma Aldrich, P9416), TWEEN 80 (Sigma Aldrich, P4780), avidin–peroxidase (Sigma Aldrich, A7419), o-Phenylenediamine (Sigma Aldrich, P9029), Middlebrook 7H9 broth (Difco-Becton Dickinson, 271310), Middlebrook 7H11 agar (Difco-Becton Dickinson, 283810), Middlebrook OADC growth supplement (Sigma Aldrich, M0678), saponin (Sigma Aldrich, 84510), PVDF western blotting membranes (Sigma Aldrich, 3010040001), amikacin hydrate (Sigma Aldrich, A3650), sodium nitrite (Sigma Aldrich, 237213), phenylmethanesulfonyl fluoride (PMSF; Sigma Aldrich, 78830), protease and phosphatase inhibitor cocktail (Sigma Aldrich, PPC1010).
All other reagents and chemicals were procured from the following companies. Free or fluorochrome-conjugated Abs: Purified anti-mouse FCGR3/FCGR2B Ab (eBioscience, 14-0161-86), CD80-FITC (BD Pharmingen, 553768), ADGRE1/F4/80-APC (eBioscience, 14-4801-82), F4/80-PerCP-Cyanine5.5 (eBioscience, 45-4801-82), CD40-PE-Cyanine5 (eBioscience, 15-0401-82), CD86-PE (eBioscience, 12-0862-85), H2/MHC-II-PerCPefluor710 (eBioscience, 46-5321-82), FAS/CD95-FITC (BD Pharmingen, 561979), CD274/PD-L1-PE (eBioscience, 12-5982-83), HAVCR2/CD366/TIM3-APC (eBioscience, 17-5871-82), LAMP1/CD107a-Alexa Fluor 647 (BioLegend, 121610), ANXA5 (annexin A5)-FITC (BioLegend, 640906), propidium iodide (Sigma Aldrich, P4170), CD4-Pacific Blue (BD Pharmingen, 558107), CD8A-APC-Cy7 (BD Pharmingen, 557654), IFNG-PE-Cyanine7 (eBioscience, 25-7311-82), IL17A-PE (BioLegend, 506904), SELL/CD62L-FITC (BD Pharmingen, 553150), IL7R/CD127-PE-Cyanine5 (eBioscience, 15-1271-83), CD44-PerCP/Cyanine5.5 (eBioscience, 45-0441-82), CCR7-PE-Cyanine7 (eBioscience, 25-1971-82), TNF-PerCP-Cyanine5.5 (BioLegend, 506322), Alexa fluor 633-anti-rabbit secondary Ab (Invitrogen, A-21071), Alexa fluor 488-anti-rabbit secondary Ab (Invitrogen, A-11034). Other reagents: Dulbecco’s phosphate-buffered saline (DPBS/PBS) (GIBCO, 14190-144), Roswell Park Memorial Institute (RPMI)-1640 (GIBCO, 11875-093) and Fetal bovine serum (FBS; GIBCO, 10082-147), L-pyruvate (SERVA, 15220), L-glutamine (SERVA, 22942), streptomycin (SERVA, 35500), penicillin (SERVA, 31749). ELISA Abs: purified anti-mouse IL6 (BD Pharmingen, 554400), biotin anti-mouse IL6 (BD Pharmingen, 554402), recombinant mouse IL6 (BD Pharmingen, 554582), purified anti-mouse IL12B (BD Pharmingen, 551219), biotin anti-mouse IL12B (BD Pharmingen, 554476), recombinant mouse IL12B (BD Pharmingen, 554592), purified anti-mouse TNF (BD Pharmingen, 551225), biotin anti-mouse TNF (BD Pharmingen, 554415), recombinant mouse TNF (BD Pharmingen, 554589), purified anti-mouse/rat IL1B (BioLegend, 503502), biotin anti-mouse TNF (BioLegend, 515801), recombinant mouse IL1B (BioLegend, 575109), purified rat anti-mouse IL10 (BD Pharmingen, 551215), biotin rat anti-mouse IL10 (BD Pharmingen, 554465), recombinant mouse IL10 (BD Pharmingen, 550070). The primer sequences for RT-qPCR were purchased from Sigma Aldrich. All tissue culture grade plasticware was procured from BD Biosciences, Thermo Fisher Scientific, Corning™ Costar™ or Sigma Aldrich.
Culture of bone marrow derived macrophages (BMDMs)
Briefly, bone marrow cells (BMCs) from the femurs and tibia of C57BL/6 wild type, Atg5fl/fl and atg5fl/fl-Lys2/LysM-Cre mice were flushed aseptically. BMCs were cultured in RPMI-1640 (GIBCO, Invitrogen Life Technologies, 11875-093) containing FBS (10%) with L-glutamine (100 mM), penicillin (100 U/ml), streptomycin (100 mg/ml) and replenished with L929 cells culture supernatant (20%), as a source of macrophage colony-stimulating factor (M-CSF). The cultures were kept in a humidified atmosphere at 37ºC (5% CO2). The medium was replenished after 3 d. After 7 d, macrophages (BMDMs) (2.5 × 105/well) were harvested and cultured in 48 well tissue culture grade plates with the agonists C4/TDB (24 µg/ml), T4/ultra-pure LPS (10 ng/ml) or C4.T4 for 48 h with or without Mtb infection. For western blot and RT-qPCR experiments, BMDMs (2.5 × 106/well) were cultured in 6 well tissue culture grade plates. The cells were then infected and stimulated with C4 (24 µg/ml), T4 (10 ng/ml) or C4.T4 for 4–8 h.
THP-1 macrophage differentiation
THP-1 monocytes (3 × 105/well) were treated with PMA (20 ng/ml) for 16 h in 48 well plates in RPMI containing FBS (10%) at 37ºC (5% CO2). The adherent macrophages were washed with RPMI twice and then rested for another 24 h prior to stimulation with C4 (24 µg/ml), T4 (10 ng/ml) or C4.T4 for 48 h.
Phenotype of BMDMs
The phenotype of Mtb-infected BMDMs was assessed by flow cytometry. Infected macrophages were triggered with CLEC4E and TLR4 agonist (C4.T4) for 48 h. BMDMs harvested at 48 h were re-suspended in FACS buffer (2% FCS in PBS). The cells were treated with anti-FCGR3/FCGR2B Ab for 30 min at 4ºC to block non-specific binding of Abs to FCGRs. Cells were then stained with fluorochrome-labeled Abs specific for mouse CD40, CD86, H2/MHC-II, FAS, CD274/PD-L1, and HAVCR3/TIM-3 or isotype-matched control Abs. Further, the cells were then fixed with paraformaldehyde (1%). Cells were washed between each step. The samples were analyzed using a FACS Aria and data analyzed with BD DIVA (BD Biosciences, San Jose, CA) and FlowJo software (Williamson Way, Ashland, OR).
Propidium iodide (PI) and ANXA5 assays
Mtb-infected BMDMs were cultured with the agonists of CLEC4E (C4/TDB) or TLR4 (T4/ultra-pure LPS) or CLEC4E-TLR4 (C4.T4) at 37ºC (5% CO2) for 48 h. This was followed by re-suspending cells in binding buffer (0.01 M HEPES [pH 7.4], 0.14 M NaCl and 2.5 mM CaCl2). FITC-labeled ANXA5 (5 μl/tube) and 2 μl of PI (50 μg/ml) were added to the samples and then incubated in the dark at RT. After 15 min, 1X binding buffer (400 μl) was added and cells were analyzed immediately employing a BD FACS Aria flow cytometer and data analysis was performed by DIVA software.
Scanning electron microscopy (SEM)
BMDMs were stimulated with agonists of CLEC4E (24 µg/ml) and TLR4 (10 ng/ml) for 48 h. The cells were fixed on Poly-L-Lysine pre-coated coverslips with modified Karnovsky’s fixative (MKF) at 4ºC. After 3 h, cells were washed 3 x with PBS at 4ºC. Dehydration of samples was done with 30%, 50% and 70% ethanol at the interval of 30 min at 4ºC. Samples were treated with 90%, 100%-I, and 100%-II ethanol at intervals of 30 min at RT and dehydrated with tetra-butyl alcohol (TBA) twice at intervals of 30 min at RT. TBA was removed and freeze drying was performed at −120ºC for 3 h and cells observed under SEM at 15 KX magnification.
Cytokine estimation
Cytokine release was measured in culture supernatants (SNs) after 48 h of C4.T4 stimulation by standard ELISA method, according to manufacturer’s instructions. Macrophages (2.5 x 105/well) were incubated in 48 well plates and stimulated with the agonists C4 (24 µg/ml), T4 (10 ng/ml) or C4.T4. The SNs were collected after 48 h to measure IL6, IL12B, TNF, IL1B, and IL10. Briefly, 96 well plates were coated with Abs to mouse IL6 (2 μg/ml), IL12B (2 μg/ml), IL1B (2 μg/ml) and TNF (4 μg/ml), IL10 (4 μg/ml) in phosphate buffer (0.01 M-pH 9.2 and pH 6 respectively) overnight (O.N.) at 4°C. Blocking with BSA (1%) in PBS was done for 3 h at RT. Afterward, the corresponding recombinant cytokines were used as standard, and culture SNs (50 µl volume) were added and then incubated O.N. at 4°C. Biotinylated anti-mouse IL6 (2 μg/ml), IL12B (2 μg/ml), IL10 (2 μg/ml) and TNF (1 μg/ml), IL1B (1 μg/ml) Abs in BSA (1%)-PBS-TWEEN 20 buffers were added into the ELISA plates and incubated for 2 h at RT. Subsequently, avidin-HRP (1:10,000) was added and then incubated at 37°C for 45 min. The regular procedures of incubation and washing with PBST (1XPBS-0.05% TWEEN 20) were achieved at each step. Later, the color was developed using H2O2-OPD substrate-chromogen. The reaction was stopped by 7% H2SO4 after color development. The plates were read in an ELISA reader at 492 nm. The results are expressed as pg/ml. The secreted cytokines were determined using serial log2 dilutions of standard curve drawn using rIL6, rIL12B, rTNF, rIL1B, and rIL10.
Culturing of mycobacteria
Mycobacterium strains (H37Rv, H37Ra) were a kind gift from Dr. V.M. Katoch (National JALMA Institute for Leprosy and other Mycobacterial Diseases, Agra, India) and GFP-H37Ra, H37Rv were kindly provided by Dr. P. Gupta (IMTECH, Chandigarh, India). Unless otherwise indicated, Mtb H37Rv was used for all experiments. Mycobacterium strains were cultivated in Middlebrook 7H9 broth with glycerol (0.2%) and TWEEN-80 (0.05%), complemented with dextrose, catalase and albumin. The viability of Mtb was confirmed by colony-forming units (CFUs) on Middlebrook 7H11 medium supplemented with albumin, oleic acid, dextrose as well as catalase. CFUs were enumerated 20 d after plating.
In vitro infection with mycobacteria
BMDMs were infected with M. smegmatis (MOI 1:5) or Mtb-H37Rv/H37Ra (MOI 1:5) and harvested after 3 h and 4 h, respectively. The extracellular bacteria were removed by treating cultures with amikacin (2 µg/ml) followed by extensive washing with PBS. M. smegmatis and Mtb-infected BMDMs were treated with the agonists C4.T4 (C4/TDB: 24 µg/ml, T4/ultra-pure LPS: 10 ng/ml) for 16 h and 48 h, respectively, in 48 well plate. Where indicated, INH (isoniazid) (2.5 µg/ml) and RIF (rifampicin) (0.5 µg/ml) were incubated along with the C4.T4 agonists.
Determination of CFUs
Intracellular Mtb growth in BMDMs and THP-1 macrophages were enumerated after 4 h of Mtb infection followed by 48 h of stimulation with C4.T4. The cell SNs were collected for ELISA and 0.1% saponin (in water) was added to lyse the cells, followed by 10-fold serial dilutions and plating on 7H11 agar to enumerate CFUs. The colonies were counted 3 days (M. smegmatis) and 3 wks (H37Rv and H37Ra) after incubation at 37ºC (5% CO2).
Nitric oxide (NO) production
Culture SNs of Mtb-infected BMDMs or lung cells from Mtb-infected mice were harvested after 48 h and NO was measured by the Griess method. In brief, SNs were added to an equal volume (50 µl) of Griess reagent for 5 min at RT and absorbance at 550 nm was assessed. The measurement of NO was performed by comparing with sodium nitrite (NaNO2) as a standard (µM).
Antigen uptake
BMDMs (2.5 × 105/well) were stimulated in either the presence or the absence of CLEC4E/C4 (24 µg/ml) and TLR4/T4 (10 ng/ml) agonists for 48 h. The cells were then infected with GFP-Mtb (H37Ra) at MOI 1:3 for 4 h. BMDMs were washed vigorously (3 X) with ice cold PBS. The cells were then treated with amikacin (2 µg/ml) and incubated for 1h to remove extracellular Mtb and fixed with paraformaldehyde (1%). The cells were washed and placed on poly-L-lysine coated coverslips and imaged using confocal microscopy (Nikon A1; Nikon, Tokyo, Japan). Z-stacks were taken to exclude the interference of extracellular bacteria. Analysis was performed using Nikon NIS-AR 4.1 image analysis software (Nikon, Melville, NY). Further, the confocal results were authenticated by flow cytometry. The analysis was done by FACS Suite software (BD Biosciences, San Jose, CA).
The enumeration of bacterial uptake was done through CFUs. BMDMs (2.5 × 105/well) were stimulated with CLEC4E/C4 (24 µg/ml) and TLR4/T4 (10 ng/ml) agonists for 48 h. BMDMs were harvested and infected with H37Rv at MOI (1:5) for 4 h. The cells were then treated with amikacin (2 µg/ml) and incubated for 1 h to remove extracellular Mtb. Later, the cells were lysed with saponin (0.1% in water) and subsequent plating was performed with 10-fold serial dilutions on 7H11 plates. The colonies were counted 3 weeks after incubation at 37ºC (5% CO2).
Western blot
Mtb-(H37Rv and H37Ra) infected BMDMs (2.5 X 106 cells/well) were treated with CLEC4E/C4 (24 µg/ml) and TLR4/T4 (10 ng/ml) agonists for 15–30 min (STAT1, STAT6, PtdIns3K), 1 h (MYD88, Phospho MTOR), 4 h (LC3B, SQSTM1), 18 h (NOS2), and 24 h (BCL2L1) in 12 well plates. The cells were then harvested, washed and lysed in cytosolic extraction lysis buffer (with PMSF, protease and phosphatase inhibitor cocktail). Cytosolic protein was then quantified and subjected to SDS-PAGE electrophoresis. After transfer to PVDF membranes and following blocking with BSA (2%), the membranes were immunoblotted with phosphorylated and non-phosphorylated Abs against STAT1, STAT6, PtdIns3K, MYD88, MTOR, LC3B, NOS2, BCL2L1. ACTB was used as a loading control. Regular washing was performed in each step. Blots were developed using a chemiluminescence kit (Amersham Pharmacia Biotech, Buckinghamshire, UK and ECL; Pharmacia-Amersham, Freiburg, Germany). Scanning of the blots was completed with ImageQuant LAS 4000 (GE Healthcare, Pittsburgh, PA). The image analysis was achieved with MultiGuage and ImageJ analysis software. Cells stimulated with LPS (2 μg/ml) were used as a positive control.
Expression of LC3, LAMP1, RELA/NFKB p65 by confocal microscopy
BMDMs were placed on poly-L-lysine pre-coated coverslips for 3 h. The cells were stimulated with C4.T4 for 4 h (LC3), 8–10 h (LAMP1) and 30 min (RELA/NFKB p65). The cells were fixed with 2% PFA (paraformaldehyde) for 10 min. This was followed by treatment with Triton X-100 (0.1%) for 2 min. To block non-specific binding, macrophages were incubated with BSA (2%) for 2 h, followed by addition of primary Abs for 2 h. Afterward, cells were incubated with secondary Ab for 1 h. The cells were stained with DAPI for 10 min at RT. Cells were washed in between each step. The cells on coverslips were mounted onto slides with Fluoromount-G. The cells were imaged using confocal (NIKON A1) microscopy (Nikon, Tokyo, Japan). Analysis was achieved by Nikon-NIS-AR 4.1 image analysis software (Nikon, Melville, NY). Positive controls for LC3 and RELA/NFKB p65 labeling involved starving macrophages in HBSS medium and LPS, respectively. For co-localization of Mtb with autophagosomes (LC3) and lysosomes (LAMP1), BMDMs were infected with GFP-Mtb for 4 h, followed by stimulation with C4.T4. Cells having 5 or more LC3 puncta were recorded as LC3-positive cells. The formation of LC3 puncta was manually counted in at least 50 cells for each group and plotted as number of puncta per cells with fluorescent LC3 puncta.
Acridine orange, MDC and LysoTracker Red staining
BMDMs were treated with the autophagy inhibitor 3MA (10 mM) for 1 h, prior to C4.T4 stimulation for 4 h (acridine orange and MDC staining), followed by incubation at 37°C with acridine orange (1 μg/ml) or 0.05 mM MDC for 20 min. The cells were fixed with 2% PFA at RT and observed immediately with an immunofluorescence microscope (Olympus IX71/TH4 200, Hamburg, Germany). For LysoTracker Red staining, BMDMs were infected with GFP-Mtb for 4 h and treated with C4.T4 for 8–10 h. The cells were then fixed with PFA (2%) for 10 min and treated with Triton X-100 (0.1%) for 2 min. The cells were stained with 100 nM LysoTracker Red for 30 min at 37°C and then observed either through confocal microscope (Nikon A1R, Nikon, Yokohama, Japan) or flow cytometer (BD FACS ARIA, BD Biosciences, CA). For flow cytometry experiments, the BMDMs were treated with 3MA for 1 h prior C4.T4 stimulation.
Demonstration of RELA/NFKB by EMSA
BMDMs were washed and treated with CLEC4E/C4 (24 µg/ml) and TLR4/T4 (10 ng/ml) for 30 min. The cells were then harvested, and the nuclear extract was collected. EMSA was performed after P32 probe labeling. In brief, an equal concentration of nuclear protein (3 μg) from each sample was incubated for 20 min at 37°C in a water bath with [P32] end labeled duplex oligonucleotides that contain the binding site for RELA/NFKB. The DNA–protein complexes were resolved on a native PAGE-gel (7%) by electrophoresis. The gel was dried at 80°C in a vacuum gel dryer for 2 h and exposed to screen at RT for 6–12 h and scanned by phosphor-imager scanning screen (Fujifilm, FLA-5000, Tokyo, Japan).
RELA/NFKB nuclear translocation by confocal microscopy
BMDMs were placed on poly-L-lysine pre-coated coverslips for 2 h. The cells were then stimulated with C4.T4 for 30 min at 37°C. Cells were then fixed with PFA (2%) for 5–10 min, followed by treatment with Triton X-100 (0.1%) for 2 min. The samples were incubated with BSA (2%) to block the non-specific sites for 2 h. The cells were then incubated with anti-mouse RELA/NFKB p65 Ab (1:400) for 2 h. Afterward, cells were treated with Alexa fluor 633-anti-rabbit Ab for 1 h and then stained with DAPI for 10 min. Regular washings with 1XPBS were conducted at each step. The samples were mounted on slides with Fluoromount-G and cells were imaged with a Nikon A1 confocal laser microscope (Nikon, Tokyo, Japan). The data were analyzed using image analysis software Nikon NIS-AR 4.1 (Nikon, Melville, NY).
The quantification of autophagy-related genes by real time PCR
Total RNA was isolated from the lungs of mice immunized with C4.T4 using trizol reagent, according to the manufacturer’s instruction (Invitrogen, Carlsbad, CA). RNA was quantified through NanoDrop machine (BioTek, Winooski, VT). Purity of all RNA samples was measured by A260/A280 ratio and was in the range of 1.9 to 2.0. All RNA samples (3 μg) were treated with amplification grade DNase 1 (3 U) (Sigma Aldrich, AMPD1-1KT) for 15 min in 1 X reaction buffer to avoid DNA contamination. DNase activity was stopped with stop solution (Sigma Aldrich, AMPD1-1KT) and then incubating the samples at 70°C for 10 min. The cDNA was then prepared by maxima first strand cDNA synthesis kit for RT-qPCR (Thermo Fisher Scientific, K1642). Real time PCR and data analysis was done by the ABI 7500 Fast Real-time PCR system (Applied Biosystems, Chromos, Singapore), with ACTB as a loading control. Analysis was done by the comparative Ct method. Results are presented as relative expression (fold change). The primer sequences for RT-qPCR are in Figure S15.
siRNA transfection of Becn1 in BMDMs
BMDMs were cultured in 48 well plate and transient knockdown of Becn1, 120 nM of FlexiTube siRNA GeneSolution (Qiagen, GS56208), comprising of 4 different target sequences or a scrambled siRNA control were taken to transfect 2.5 × 105 cells, as per manufacturer’s instructions. After 72 h, the cells were infected with Mtb-H37Rv (MOI 1:5) and harvested after 4 h. The extracellular bacteria were removed by treating cultures with amikacin (2 µg/ml) followed by extensive washing with PBS. Then Mtb-infected BMDMs were treated with the C4.T4 agonists (C4/TDB: 24 µg/ml, T4/ultra-pure LPS: 10 ng/ml) for 8 h (RT-qPCR) and 48 h (CFU assay).
Treatment of Mtb-infected mice with C4.T4
Mice (100 CFUs) and guinea pigs (30 CFUs) were aerosol challenged with Mtb and then treated with C4.T4 in the footpad for a total of 2 injections with an interval of 10 d. The control groups were treated with either CLEC4E (50 µg/100 µl/mouse; 200 μg/200 µl/guinea pig) or TLR4 (0.1 µg/100 µl/mouse; 1 μg/200 µl/guinea pig) agonists or PBS. Anti-TB drugs (rifampicin: 0.5, 1, 10 mg/kg bw of mice; 6 mg/kg bw guinea pig and isoniazid: 5 mg/kg bw of guinea pig) were orally administrated twice with 0.1% CMC (carboxymethylcellulose) (Sigma Aldrich, C5013). After 45 d of aerosol challenge, animals were sacrificed, and then bacterial burden was enumerated in the lung, liver and spleen by CFUs. CFUs were calculated taking into consideration weight of the organs.
Isolation of T lymphocytes, enumeration of cell surface and intracellular expression
Mice were aerosol challenged with Mtb. After 21 d, experimental groups were administered C4.T4 and controls with PBS twice at an interval of 10 d. Forty-five days later, animals were sacrificed. The mice and guinea pigs were perfused with PBS-heparin and spleen, lymph nodes (mediastinal) and lungs were harvested, and then single cell suspensions were made. Briefly, RBCs were removed with ACK (NH4Cl, 0.15 M, KHCO3 10 mM, EDTA 88 mM) lysis buffer, washed 3X with PBS and lymphocytes isolated from spleen and lungs were re-suspended in RPMI-1640-FBS-10%. Cell viability was measured by trypan blue dye exclusion. In vitro experiments were conducted in the presence of purified protein derivative (PPD) to detect intracellular expression of cytokines and frequency of effector/central memory populations in Mtb-specific T cells.
Histopathology
Mice and guinea pigs were sacrificed, and lung and spleen tissues were fixed in buffered formalin (10%). Histological sections were stained using hematoxylin and eosin. The microscopic photographs were taken using an Olympus IX71 microscope and displayed at 10X and 40X magnifications.
Statistical analysis
Analysis of all data was done by Student’s “t” test, non-parametric Mann-Whitney two tailed and repeated measure ANOVA with post Student-Newman-Keuls multiple comparison test using Graph Pad InStat 3 and Graph Pad Prism 6 software. The statistical differences were considered significant at a level of p < 0.05.
Supplementary Material
Acknowledgments
We are thankful to Prof. B. N. Datta (Medicos Center, Chandigarh, India) for the analysis of histopathology sections, Council of Scientific & Industrial Research (CSIR) for financial support. SP was the recipient of the fellowship of CSIR; SN from Department of Biotechnology (DBT); MA from Department of Science and Technology (DST). We appreciate Dr. Chrissy Leopold Wager (Texas Biomedical Research Institute, Texas, USA), and Dr. Ansu Louis, Dr. Gurpreet Kaur (Indian Institute of Technology, Ropar, India) for critically evaluating the manuscript.
Funding Statement
We thank the Council of Scientific & Industrial Research (CSIR) for financial support (Project Number: OLP 088).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material:
Supplemental data for this article can be accessed here.
References
- [1].World Health Organization (WHO) . Global tuberculosis report 2017. Geneva: WHO; 2017. [cited 2017 November14]. Available from http://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- [2].World Health Organization (WHO) . Global tuberculosis report. Geneva: World Health Organization; 2018. Available from http://www.who.int/iris/handle/10665/274453 [Google Scholar]
- [3].Lawn SD, Zumla AI.. Tuberculosis. Lancet. 2011. July 02;378(9785):57–72. PubMed PMID: 21420161. [DOI] [PubMed] [Google Scholar]
- [4].Prabowo SA, Groschel MI, Schmidt ED, et al. Targeting multidrug-resistant tuberculosis (MDR-TB) by therapeutic vaccines. Med Microbiol Immunol. 2013. April;202(2):95–104. PubMed PMID: 23143437. [DOI] [PubMed] [Google Scholar]
- [5].Kaufmann SH. Tuberculosis vaccines: time to think about the next generation. Semin Immunol. 2013. April;25(2):172–181. PubMed PMID: 23706597. [DOI] [PubMed] [Google Scholar]
- [6].Howitt MR, Garrett WS. A complex microworld in the gut: gut microbiota and cardiovascular disease connectivity. Nat Med. 2012. August;18(8):1188–1189. PubMed PMID: 22869188. [DOI] [PubMed] [Google Scholar]
- [7].Mukaida N. Intestinal microbiota: unexpected alliance with tumor therapy. Immunotherapy. 2014;63:231–233. PubMed PMID: 24762069 [DOI] [PubMed] [Google Scholar]
- [8].Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013. October;19(10):1264–1272. PubMed PMID: 24056773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Organization WH . WHO report 2007: global tuberculosis control: surveillance, planning, financing. Geneva: World health organization; 2007. [Google Scholar]
- [10].Pahari S, Kaur G, Aqdas M, et al. Bolstering immunity through pattern recognition receptors: a unique approach to control tuberculosis. Front Immunol. 2017;8:906. PubMed PMID: 28824632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schoenen H, Bodendorfer B, Hitchens K, et al. Cutting edge: mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol. 2010. March 15;184(6):2756–2760. PubMed PMID: 20164423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yamasaki S, Ishikawa E, Sakuma M, et al. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol. 2008. October;9(10):1179–1188. PubMed PMID: 18776906. [DOI] [PubMed] [Google Scholar]
- [13].Suzuki Y, Nakano Y, Mishiro K, et al. Involvement of mincle and syk in the changes to innate immunity after ischemic stroke. Sci Rep. 2013. November 11;3:3177. PubMed PMID: 24212132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].de Rivero Vaccari JC, Brand FJ 3rd, Berti AF, et al. Mincle signaling in the innate immune response after traumatic brain injury. J Neurotrauma. 2015. February 15;32(4):228–236. PubMed PMID: 25111533. [DOI] [PubMed] [Google Scholar]
- [15].Wells CA, Salvage-Jones JA, Li X, et al. The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol. 2008. June 1;180(11):7404–7413. PubMed PMID: 18490740. [DOI] [PubMed] [Google Scholar]
- [16].Bugarcic A, Hitchens K, Beckhouse AG, et al. Human and mouse macrophage-inducible C-type lectin (Mincle) bind Candida albicans. Glycobiology. 2008. September;18(9):679–685. PubMed PMID: 18509109. [DOI] [PubMed] [Google Scholar]
- [17].Vijayan D, Radford KJ, Beckhouse AG, et al. Mincle polarizes human monocyte and neutrophil responses to Candida albicans. Immunol Cell Biol. 2012. October;90(9):889–895. PubMed PMID: 22641025. [DOI] [PubMed] [Google Scholar]
- [18].Yamasaki S, Matsumoto M, Takeuchi O, et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci U S A. 2009. February 10;106(6):1897–1902. PubMed PMID: 19171887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ishikawa T, Itoh F, Yoshida S, et al. Identification of distinct ligands for the C-type lectin receptors Mincle and Dectin-2 in the pathogenic fungus Malassezia. Cell Host Microbe. 2013. April 17;13(4):477–488. PubMed PMID: 23601109. [DOI] [PubMed] [Google Scholar]
- [20].Wevers BA, Kaptein TM, Zijlstra-Willems EM, et al. Fungal engagement of the C-type lectin mincle suppresses Dectin-1-induced antifungal immunity. Cell Host Microbe. 2014. April 09;15(4):494–505. PubMed PMID: 24721577. [DOI] [PubMed] [Google Scholar]
- [21].Bekierkunst A, Yarkoni E, Flechner I, et al. Immune response to sheep red blood cells in mice pretreated with mycobacterial fractions. Infect Immun. 1971. September;4(3):256–263. PubMed PMID: 4949490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Geisel RE, Sakamoto K, Russell DG, et al. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guerin is due principally to trehalose mycolates. J Immunol. 2005. April 15;174(8):5007–5015. PubMed PMID: 15814731. [DOI] [PubMed] [Google Scholar]
- [23].van Dissel JT, Joosten SA, Hoff ST, et al. A novel liposomal adjuvant system, CAF01, promotes long-lived Mycobacterium tuberculosis-specific T-cell responses in human. Vaccine. 2014. December 12;32(52):7098–7107. PubMed PMID: 25454872. [DOI] [PubMed] [Google Scholar]
- [24].Agger EM, Rosenkrands I, Hansen J, et al. Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): a versatile adjuvant for vaccines with different immunological requirements. PLoS One. 2008. September 08;3(9):e3116. PubMed PMID: 18776936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pimm MV, Baldwin RW, Polonsky J, et al. Immunotherapy of an ascitic rat hepatoma with cord factor (trehalose-6, 6ʹ-dimycolate) and synthetic analogues. Int J Cancer. 1979. December 15;24(6):780–785. PubMed PMID: 544531. [DOI] [PubMed] [Google Scholar]
- [26].Olds GR, Chedid L, Lederer E, et al. Induction of resistance to Schistosoma mansoni by natural cord factor and synthetic lower homologues. J Infect Dis. 1980. April;141(4):473–478. PubMed PMID: 7373082. [DOI] [PubMed] [Google Scholar]
- [27].Davidsen J, Rosenkrands I, Christensen D, et al. Characterization of cationic liposomes based on dimethyldioctadecylammonium and synthetic cord factor from M. tuberculosis (trehalose 6,6ʹ-dibehenate)-a novel adjuvant inducing both strong CMI and antibody responses. Biochim Biophys Acta. 2005. December 10;1718(1–2):22–31. PubMed PMID: 16321607. [DOI] [PubMed] [Google Scholar]
- [28].Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 2004. December;6(15):1382–1387. . PubMed PMID: 15596124. [DOI] [PubMed] [Google Scholar]
- [29].Hemmi H, Akira S. TLR signalling and the function of dendritic cells. Chem Immunol Allergy. 2005;86:120–135. PubMed PMID: 15976491. [DOI] [PubMed] [Google Scholar]
- [30].Holscher C, Reiling N, Schaible UE, et al. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, −4 and −9. Eur J Immunol. 2008. March;38(3):680–694. PubMed PMID: 18266299. [DOI] [PubMed] [Google Scholar]
- [31].Vacchelli E, Galluzzi L, Eggermont A, et al. Trial watch: FDA-approved toll-like receptor agonists for cancer therapy. Oncoimmunology. 2012. September 1;1(6):894–907. PubMed PMID: 23162757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Needham BD, Carroll SM, Giles DK, et al. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc Natl Acad Sci U S A. 2013. January 22;110(4):1464–1469. PubMed PMID: 23297218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bohannon JK, Hernandez A, Enkhbaatar P, et al. The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock. 2013. December;40(6):451–462. PubMed PMID: 23989337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gantner BN, Simmons RM, Canavera SJ, et al. Collaborative induction of inflammatory responses by dectin-1 and toll-like receptor 2. J Exp Med. 2003. May 05;197(9):1107–1117. PubMed PMID: 12719479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Loures FV, Araujo EF, Feriotti C, et al. TLR-4 cooperates with Dectin-1 and mannose receptor to expand Th17 and Tc17 cells induced by Paracoccidioides brasiliensis stimulated dendritic cells. Front Microbiol. 2015;6:261. PubMed PMID: 25873917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004. December 17;119(6):753–766. PubMed PMID: 15607973. [DOI] [PubMed] [Google Scholar]
- [37].Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012. November 13;109(46):E3168–76. PubMed PMID: 23093667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Klein L, Munz C, Lunemann JD. Autophagy-mediated antigen processing in CD4(+) T cell tolerance and immunity. FEBS Lett. 2010. April 02;584(7):1405–1410. PubMed PMID: 20074571. [DOI] [PubMed] [Google Scholar]
- [39].Lunemann JD, Munz C. Autophagy in CD4+ T-cell immunity and tolerance. Cell Death Differ. 2009. January;16(1):79–86. . PubMed PMID: 18636073. [DOI] [PubMed] [Google Scholar]
- [40].Xu Y, Jagannath C, Liu XD, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007. July;27(1):135–144. PubMed PMID: 17658277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shi CS, Kehrl JH. MyD88 and trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008. November 28;283(48):33175–33182. . PubMed PMID: 18772134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007. October 18;449(7164):819–826. . PubMed PMID: 17943118. [DOI] [PubMed] [Google Scholar]
- [43].LeibundGut-Landmann S, Gross O, Robinson MJ, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007. June;8(6):630–638. PubMed PMID: 17450144. [DOI] [PubMed] [Google Scholar]
- [44].Ostrop J, Jozefowski K, Zimmermann S, et al. Contribution of MINCLE-SYK signaling to activation of primary human APCs by mycobacterial cord factor and the novel adjuvant TDB. J Immunol. 2015. September 01;195(5):2417–2428. PubMed PMID: 26202982. [DOI] [PubMed] [Google Scholar]
- [45].Domingo-Gonzalez R, Prince O, Cooper A, et al. Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol Spectr. 2016. October;4(5). PubMed PMID: 27763255. DOI: 10.1128/microbiolspec.TBTB2-0018-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stockinger B. Capacity of antigen uptake by B cells, fibroblasts or macrophages determines efficiency of presentation of a soluble self antigen (C5) to T lymphocytes. Eur J Immunol. 1992. May;22(5):1271–1278. PubMed PMID: 1577067. [DOI] [PubMed] [Google Scholar]
- [47].Pahari S, Khan N, Aqdas M, et al. Infergen stimulated macrophages restrict Mycobacterium tuberculosis growth by autophagy and release of nitric oxide. Sci Rep. 2016. December 21;6:39492. PubMed PMID: 28000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Khan N, Pahari S, Vidyarthi A, et al. Stimulation through CD40 and TLR-4 is an effective host directed therapy against Mycobacterium tuberculosis. Front Immunol. 2016;7:386. PubMed PMID: 27729911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Khan N, Pahari S, Vidyarthi A, et al. NOD-2 and TLR-4 signaling reinforces the efficacy of dendritic cells and reduces the dose of tb drugs against Mycobacterium tuberculosis. J Innate Immun. 2016;8(3):228–242. PubMed PMID: 26613532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Quenelle DC, Staas JK, Winchester GA, et al. Efficacy of microencapsulated rifampin in Mycobacterium tuberculosis-infected mice. Antimicrob Agents Chemother. 1999. May;43(5):1144–1151. PubMed PMID: 10223927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Flynn JL, Chan J, Triebold KJ, et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993. December 01;178(6):2249–2254. PubMed PMID: 7504064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Monin L, Griffiths KL, Slight S, et al. Immune requirements for protective Th17 recall responses to Mycobacterium tuberculosis challenge. Mucosal Immunol. 2015. September;8(5):1099–1109. PubMed PMID: 25627812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Caccamo N, Guggino G, Joosten SA, et al. Multifunctional CD4(+) T cells correlate with active Mycobacterium tuberculosis infection. Eur J Immunol. 2010. August;40(8):2211–2220. PubMed PMID: 20540114. [DOI] [PubMed] [Google Scholar]
- [54].Rai PK, Chodisetti SB, Nadeem S, et al. A novel therapeutic strategy of lipidated promiscuous peptide against Mycobacterium tuberculosis by eliciting Th1 and Th17 immunity of host. Sci Rep. 2016. April 07;6:23917. PubMed PMID: 27052185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Forbes EK, Sander C, Ronan EO, et al. Multifunctional, high-level cytokine-producing Th1 cells in the lung, but not spleen, correlate with protection against Mycobacterium tuberculosis aerosol challenge in mice. J Immunol. 2008. October 01;181(7):4955–4964. PubMed PMID: 18802099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ni Cheallaigh C, Keane J, Lavelle EC, et al. Autophagy in the immune response to tuberculosis: clinical perspectives. Clin Exp Immunol. 2011. June;164(3):291–300. PubMed PMID: 21438870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Khader SA, Bell GK, Pearl JE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007. April;8(4):369–377. PubMed PMID: 17351619. [DOI] [PubMed] [Google Scholar]
- [58].Riol-Blanco L, Sanchez-Sanchez N, Torres A, et al. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J Immunol. 2005. April 1;174(7):4070–4080. PubMed PMID: 15778365. [DOI] [PubMed] [Google Scholar]
- [59].Seth S, Oberdorfer L, Hyde R, et al. CCR7 essentially contributes to the homing of plasmacytoid dendritic cells to lymph nodes under steady-state as well as inflammatory conditions. J Immunol. 2011. March 15;186(6):3364–3372. PubMed PMID: 21296980. [DOI] [PubMed] [Google Scholar]
- [60].Jo EK. Innate immunity to mycobacteria: vitamin D and autophagy. Cell Microbiol. 2010. August;12(8):1026–1035. PubMed PMID: 20557314. [DOI] [PubMed] [Google Scholar]
- [61].Khan N, Vidyarthi A, Pahari S, et al. Signaling through NOD-2 and TLR-4 bolsters the T cell priming capability of dendritic cells by inducing autophagy. Sci Rep. 2016. January 12;6:19084. PubMed PMID: 26754352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Harris J, De Haro SA, Master SS, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007. September;27(3):505–517. PubMed PMID: 17892853. [DOI] [PubMed] [Google Scholar]
- [63].Vergne I, Singh S, Roberts E, et al. Autophagy in immune defense against Mycobacterium tuberculosis. Autophagy. 2006. Jul–Sep;2(3):175–178. PubMed PMID: 16874111. [DOI] [PubMed] [Google Scholar]
- [64].Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol. 2010. June;221(2):117–124. PubMed PMID: 20225337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol. 2012. July 10;22(13):R540–5. PubMed PMID: 22790007. [DOI] [PubMed] [Google Scholar]
- [66].Tapper H, Sundler R. Bafilomycin A1 inhibits lysosomal, phagosomal, and plasma membrane H(+)-ATPase and induces lysosomal enzyme secretion in macrophages. J Cell Physiol. 1995. April;163(1):137–144. PubMed PMID: 7896890. [DOI] [PubMed] [Google Scholar]
- [67].Drose S, Altendorf K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J Exp Biol. 1997. January;200(Pt 1):1–8. PubMed PMID: 9023991. [DOI] [PubMed] [Google Scholar]
- [68].Desel C, Werninghaus K, Ritter M, et al. The Mincle-activating adjuvant TDB induces MyD88-dependent Th1 and Th17 responses through IL-1R signaling. PLoS One. 2013;8(1):e53531. PubMed PMID: 23308247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kleinnijenhuis J, Oosting M, Joosten LA, et al. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol. 2011;2011:405310. PubMed PMID: 21603213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhou ZQ, Wang ZK, Zhang L, et al. Role of EAST-6 in renal injury by regulating microRNA-155 expression via TLR4/MyD88 signaling pathway in mice with Mycobacterium tuberculosis infection. Biosci Rep. 2017. June 27 PubMed PMID: 28655852. DOI: 10.1042/BSR20170021 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [71].Paglin S, Hollister T, Delohery T, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001. January 15;61(2):439–444. PubMed PMID: 11212227. [PubMed] [Google Scholar]
- [72].Brungs S, Kolanus W, Hemmersbach R. Syk phosphorylation - a gravisensitive step in macrophage signalling. Cell Commun Signal. 2015. February 03;13:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang J, Wang X, Vikash V, et al. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev. 2016;2016:4350965. PubMed PMID: 26998193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nakajima S, Kitamura M. Bidirectional regulation of NF-kappaB by reactive oxygen species: a role of unfolded protein response. Free Radic Biol Med. 2013. December;65:162–174. PubMed PMID: 23792277. [DOI] [PubMed] [Google Scholar]
- [75].Chan J, Tanaka K, Carroll D, et al. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun. 1995;63(2):736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chan J, Xing Y, Magliozzo RS, et al. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med. 1992;175(4):1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Abel B, Thieblemont N, Quesniaux VJ, et al. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J Immunol. 2002. September 15;169(6):3155–3162. PubMed PMID: 12218133. [DOI] [PubMed] [Google Scholar]
- [78].Miyake Y, Toyonaga K, Mori D, et al. C-type lectin MCL is an FcRgamma-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity. 2013. May 23;38(5):1050–1062. PubMed PMID: 23602766. [DOI] [PubMed] [Google Scholar]
- [79].Ishikawa E, Ishikawa T, Morita YS, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med. 2009. December 21;206(13):2879–2888. PubMed PMID: 20008526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Arnett E, Krishnan N, BD R, et al. Host pathogen biology for airborne Mycobacterium tuberculosis. In: Hickey A, Misra A, Fourie Peditors. Drug delivery systems for tuberculosis preveniton and treatment. Cellular and molecular events in the lung. Chichester, UK: John Wiley & Sons, Ltd; 2016. p. 11–47. [Google Scholar]
- [81].Paunescu E. In vivo and in vitro suppression of humoral and cellular immunological response by rifampicin. Nature. 1970. December 19;228(5277):1188–1190. PubMed PMID: 5487244. [DOI] [PubMed] [Google Scholar]
- [82].Bellahsene A, Forsgren A. Effect of rifampin on the immune response in mice. Infect Immun. 1980. January;27(1):15–20. PubMed PMID: 6987166; PubMed Central PMCID: PMCPMC550714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Floersheim GL. Suppression of cellular immunity in vivo by rifampicin. Experientia. 1973. December;29(12):1545–1546. PubMed PMID: 4589325. [DOI] [PubMed] [Google Scholar]
- [84].Manabe YC, Bishai WR. Latent Mycobacterium tuberculosis-persistence, patience, and winning by waiting. Nat Med. 2000. December;6(12):1327–1329. . PubMed PMID: 11100115. [DOI] [PubMed] [Google Scholar]
- [85].Canaday DH, Wilkinson RJ, Li Q, et al. CD4(+) and CD8(+) T cells kill intracellular Mycobacterium tuberculosis by a perforin and Fas/Fas ligand-independent mechanism. J Immunol. 2001. September 01;167(5):2734–2742. PubMed PMID: 11509617. [DOI] [PubMed] [Google Scholar]
- [86].He XY, Xiao L, Chen HB, et al. T regulatory cells and Th1/Th2 cytokines in peripheral blood from tuberculosis patients. Eur J Clin Microbiol Infect Dis. 2010. June;29(6):643–650. PubMed PMID: 20306324. [DOI] [PubMed] [Google Scholar]
- [87].Singh V, Gowthaman U, Jain S, et al. Coadministration of interleukins 7 and 15 with bacille Calmette-Guerin mounts enduring T cell memory response against Mycobacterium tuberculosis. J Infect Dis. 2010. August 15;202(3):480–489. PubMed PMID: 20569158. [DOI] [PubMed] [Google Scholar]
- [88].Lindenstrom T, Agger EM, Korsholm KS, et al. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol. 2009. June 15;182(12):8047–8055. PubMed PMID: 19494330. [DOI] [PubMed] [Google Scholar]
- [89].Pahari S, Kaur G, Negi S, et al. Reinforcing the functionality of mononuclear phagocyte system to control tuberculosis. Front Immunol. 2018;9:193. PubMed PMID: 29479353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Deretic V. Autophagy in tuberculosis. Cold Spring Harb Perspect Med. 2014. August 28;4(11):a018481. . PubMed PMID: 25167980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012. August 17;150(4):803–815. . PubMed PMID: 22901810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Seto S, Tsujimura K, Horii T, et al. Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome formation in response to Mycobacterium tuberculosis infection in dendritic cells. PLoS One. 2013;8(12):e86017. PubMed PMID: 24376899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Franco LH, Nair VR, Scharn CR, et al. The ubiquitin ligase smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. 2017. January 11;21(1):59–72. PubMed PMID: 28017659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kimmey JM, Huynh JP, Weiss LA, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015. December 24;528(7583):565–569. PubMed PMID: 26649827; PubMed Central PMCID: PMCPMC4842313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Behar SM, Baehrecke EH. Tuberculosis: autophagy is not the answer. Nature. 2015. December 24;528(7583):482–483. . PubMed PMID: 26649822. [DOI] [PubMed] [Google Scholar]
- [96].Dallenga T, Repnik U, Corleis B, et al. M. tuberculosis-induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host Microbe. 2017. October 11;22(4):519–530 e3. PubMed PMID: 29024644. [DOI] [PubMed] [Google Scholar]
- [97].Jagannath C, Lindsey DR, Dhandayuthapani S, et al. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009. March;15(3):267–276. PubMed PMID: 19252503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.