

Abstract
Dynamic proteins perform critical roles in cellular machines, including those that control proteostasis, transcription, translation, and signaling. Thus, dynamic proteins are prime candidates for chemical probe and drug discovery but difficult targets because they do not conform to classical rules of design and screening. Selectivity is pivotal for candidate probe molecules due to the extensive interaction network of these dynamic hubs. Recognition that the traditional rules of probe discovery are not necessarily applicable to dynamic proteins and their complexes, as well as technological advances in screening, have produced remarkable results in the last 2–4 years. Particularly notable are the improvements in target selectivity for small molecule modulators of dynamic proteins, especially with techniques that increase the discovery likelihood of allosteric regulatory mechanisms. We focus on approaches to small molecule screening that appear to be more suitable for highly dynamic targets and have the potential to streamline identification of selective modulators.
Graphical Abstract
Introduction
Conformationally dynamic proteins underpin all biological processes, from signaling to gene expression (Barrios-Rodiles et al., 2005; Kholodenko, 2006; Yeger-Lotem et al., 2004). These proteins often form short-lived protein-protein interactions (PPIs) that allow cellular machines to assemble and disassemble as needed to regulate key pathways and events (Stein et al., 2009). Efforts to decipher the principles that govern PPI formation have been extensive (Heo et al., 2011; Jones and Thornton, 1996; Khan et al., 2011). However, most work has been focused on PPIs between stable, well-defined partners and the emergent rules governing assembly, molecular recognition, and modulation of PPIs are not generally applicable to dynamic proteins. Members of this protein class tend to have high flexibility and structural plasticity (Mittag et al., 2010). Malleable structures allow them to perform context-dependent regulatory functions and interact with a multitude of binding partners (Figure 1A). For example, the master coactivator CBP/p300 (Figure 1B) contains six conformationally dynamic domains that interact with hundreds of transcriptional activators, which are linked by intrinsically disordered regions, thus increasing the overall flexibility of the protein (Dyson and Wright, 2016).
Figure 1. Key attributes of dynamic proteins with representative examples.

(A) While no single definition can be used to identify dynamic proteins, useful metrics for successful classification include: 1) the structure and mobility of the protein and/or the individual domains; 2) the number of functional roles the protein can fulfill; and 3) the number binding interactions the protein can participate in. Dynamic proteins tend to be multifunctional, participating in many pathways and performing multiple cellular roles, either through the presence of multiple domains or by the formation of dynamic transient complexes that vary in function. The functional multiplicity can in part be attributed to the number of binding partners the protein engages with, which oftentimes leads to conformational changes either within a single domain or domain rearrangement as an entire protein to elicit variable functional effects. (B) The master coactivator CREB binding protein (CBP) analyzed as a representative dynamic protein. CBP is highly disordered, with more unstructured and dynamic regions than structured domains (Dyson and Wright, 2016). The protein has high conformational plasticity, as its domains are connected by long unstructured linkers. The individual domains within CBP allow it to perform multiple functions. For example, CBP has four activator binding domains, KIX, TAZ1, TAZ2, and IBiD, that interact with transcriptional activators to regulate gene expression. It also contains a histone acetyltransferase domain (HAT) and bromodomain (BD) that it utilizes for perform chromatin remodeling functions (Breen and Mapp, 2018). Each of these individual domains has its own network of interactions, summing up to 100s of potential PPIs made by CBP. The KIX domain is shown here as an example – it interacts with a suite of 15+ activator proteins, all which can be implicated in a variety of diseases (Mapp et al., 2015). (C) The chaperone HSP70 analyzed as a representative dynamic protein. HSP70 contains much more secondary structure than CBP. It is comprised of two main domains: the nucleotide binding domain (NBD) and substrate binding domain (SBD). These two domains are connected by a small dynamic linker (Fernández-Fernández and Valpuesta, 2018). Substrate binding can induce domain rearrangement to elicit different functional effects. HSP70 itself functions as an ATPase, however it is able to perform many diverse cellular functions by forming different multiprotein complexes. For example, Hsp70 in complex with cochaperones Hip and Hop acts as a protein folding complex, while Hsp70 in complex with BAG-1 and CHIP functions as a degradation complex (Muller et al., 2013). Finally, Hsp70 complexes also interact with many other proteins. Many regulatory proteins are known to be controlled via transient association with Hsp70. Thus, Hsp70 is a potential point of therapeutic intervention for conditions such as cancer, autoimmune and neurodegenerative diseases (A. Assimon et al., 2013).
Dynamic proteins are often observed in higher order cellular machines with a functional need to rapidly form and exist only transiently, such as the multiprotein complexes involved in chromatin remodeling (Euskirchen et al., 2011) or protein folding (Walter and Buchner, 2002). Thus, dynamic proteins are often hubs, interacting with subunits within a complex as well as other proteins and ligands (Batada et al., 2006). The composition of a dynamic protein complex can dictate enzymatic subunit activity or even change the function of the complex. For example, combinatorial assembly of Hsp70 (Figure 1C) allows the complex to switch between a folding system and a degradation complex (Muller et al., 2013). Transcription is another process that relies on dynamic proteins. It is important that individual components of the machinery, such as activators, coactivators, general transcription factors, and RNA polymerase, are able to assemble efficiently at one promotor and then move on to mediate expression of another gene (Fuxreiter et al., 2008). There are extensive reviews regarding the function and composition of these dynamic machines (Gavin and Superti-Furga, 2003; Kasahara et al., 2019; Marsh and Teichmann, 2015; Thompson et al., 2012).
The qualities that make dynamic proteins and their complexes effective cellular machines also make them challenging to target. Due to conformational flexibility, it is often difficult to obtain the structural and biophysical data useful for the rational design and/or optimization of chemical modulators. The PPIs of dynamic proteins are typically lower affinity, utilize a relatively large surface area, and have minimal topology, thus representing a significant challenge for orthosteric inhibition (Cossins and Lawson, 2015; Smith and Gestwicki, 2012). High throughput screening (HTS) with dynamic protein targets can also be challenging due to the difficult expression and isolation of the full-length proteins. Thus, in many cases, it is necessary to utilize simplified systems such as isolated domains that may not recapitulate the structure, dynamics, and/or activity of the full-length complexes. Finally, selective targeting of a particular PPI or specific binding interface is difficult because dynamic proteins typically interact with a multitude of binding partners, which is a challenge we focus upon in this review (Arkin et al., 2014; Cesa et al., 2015; Ran and Gestwicki, 2018).
Notably, dynamic proteins are often dysregulated in disease and dynamic multiprotein complexes often contain specific subunits that are attractive therapeutic targets. For example, the tumor suppressor BRCA1, an important target for breast cancer prevention and therapy, associates with the Swi/Snf chromatin remodeling complex (Semmler et al., 2019; Takaoka and Miki, 2018). CBP/p300, p53, and other transcription factors are all also dynamic proteins that are potential targets for a variety of diseases (Lee and Young, 2013; Wang et al., 2013). While dynamic PPIs do not conform to typical “rules” used for drug discovery, in recent years significant achievements have been made when it comes to modulating some of these challenging targets. Here we review key principles that govern selectivity in binding and function for dynamic proteins and their complexes. Additionally, we present notable recent success stories in small-molecule modulation of these proteins, examples that either intentionally or serendipitously mimic natural regulatory mechanisms of dynamic proteins. Finally, we highlight emerging strategies that facilitate the discovery of selective chemical modulators of conformationally dynamic proteins.
Controlling the interactions and function of dynamic proteins: Lessons from Nature
As hubs associated with many complex cellular machines, dynamic proteins are precisely regulated. These regulatory processes often control the access of binding surface(s) within the dynamic protein either directly or indirectly. This can occur through masking of binding surfaces via intra- or intermolecular interactions, through induced conformational changes of the protein, and, most commonly, combinations of both mechanisms.
Masking of binding surfaces within dynamic proteins occurs through intra- and intermolecular complexes that have a variety of functional outcomes. Masking interactions are common in transcriptional activators, where the highly disordered transcriptional activation domain is sequestered by a high affinity interaction until needed. Classic and well-studied examples include the Gal4•Gal80 (Gill and Ptashne, 1988; Wightman et al., 2008) and p53•Mdm2 (Zaika et al., 1999) complexes that provide temporal control of the transcription factors. Masking of dynamic proteins and/or protein domains is similarly important in many other cellular contexts. The scaffolding 14-3-3 protein, for example, regulates the subcellular localization and function of Caspase-2 through stabilizing two dynamic regions in individual subunits. 14-3-3 masks both the nuclear localization sequence of pro-Caspase2 and the dimerization interface, inhibiting pro-Caspase2 activation (Kalabova et al., 2020) (Figure 2A). 14-3-3 also regulates FOXO transcription factors by masking their nuclear localization sequences (Silhan et al., 2009). In all of these examples, the masking interaction provides fine temporal control of the dynamic protein. Obstruction of key binding surfaces in a dynamic protein is also associated with spatial control. For example, until the unfolded protein response is activated in a cell, the transcription factor ATF6 is sequestered at the ER membrane. Once the pathway is stimulated, ATF6 is cleaved from the ER membrane, enabling its translocation to the nucleus, where its potential binding partners reside (Wang et al., 2000).
Figure 2. Nature uses multiple mechanisms to dictate interaction and function of dynamic proteins.

(A) Masking particular sequences or binding surfaces on proteins provides control over function, localization, and ligand binding. 14-3-3 regulates activity of Caspase-2 by masking its dimerization interface. Without 14-3-3 bound, Caspase-2 dimerizes and proteolytically cleave other proteins (Kalabova et al., 2020). (B) Post-translational modifications can both orthosterically and allosterically regulate protein by covalent modification of amino acid residues. Palmitoylation of transcription factor TEAD4 at C367 acts as an allosteric switch to enhance interaction with the coactivator YAP1 (Chan et al., 2016). (C) Allostery, via intra or intermolecular interactions, regulates structure and function of proteins. The Polycomb group protein ASXL2 interacts with the deubiquitinase BAP1, stabilizing a distal dynamic loop and allowing ubiquitin to bind and be hydrolyzed. Without ASXL2 interaction, BAP1 is unable to perform its enzymatic function (Peng et al., 2018).
Emerging data regarding the composition and function of biomolecular condensates indicates that masking of dynamic proteins likely plays a crucial role in the compartmentalization that such condensates afford. More recent examples of compartmentalization focus on phase separation, which increases the strength and specificity of interactions that otherwise appear promiscuous, while still allowing the proteins involved to retain their dynamic nature (Hahn, 2018). The transcription factors TAZ, OCT4, GCN4, and the estrogen receptor (ER), have been shown to form phase-separated condensates with Mediator via its coactivator subunits (Boija et al., 2018). TAZ has also been observed to form nuclear condensates with its DNA binding cofactor TEAD4 as well as the coactivator BRD4 (Lu et al., 2020). In all of these cases, unmasked transcriptional activation domains appear necessary for condensate formation.
Posttranslational modifications (PTMs) and distal binding events that influence structure are also mechanisms of binding surface control. PTMs occur at many PPI interfaces to promote or inhibit binding. For example, phosphorylation of the coactivator p300 at S89 inhibits its interaction with peroxisome proliferator-activated receptor γ (PPAR-γ) and retinoic acid receptor (RAR) (Yang et al., 2001). PTMs can also alter stability, folding, or conformation as a mechanism to regulate dynamic protein interactions (Duan and Walther, 2015). PTMs, such as ubiquitination, acetylation, phosphorylation, and glycosylation, have been observed to be allosteric regulators (Nussinov et al., 2012). For example, N-linked glycosylation of interleukin-7 receptor α allosterically enhances binding to human IL-7 nearly 300-fold (Walsh, 2010). In another example, a conserved cysteine in the palmitate binding pocket of TEAD proteins undergoes palmitoylation that allosterically stabilizes the TEAD-YAP interaction (Chan et al., 2016) (Figure 2B). Nature often utilizes a combination of PTMs to fine-tune protein function, allowing a single protein to perform diverse cellular roles. A recent study of Hsp90, for example, identified a group of conserved PTMs that globally mediates dynamics and allosteric communication in the Hsp90 structures (Stetz et al., 2018).
Allosteric crosstalk via inter and intra-molecular interactions can lead to broader conformational changes, redistribution of conformers, and alterations in kinetic/thermodynamic barriers that also regulate protein interfaces (Swain and Gierasch, 2006; Tsai et al., 2009). For example, the binding of a cognate ligand or cofactor can lead to structural changes that influence protein function (Abrusán and Marsh, 2019). A classic example of this is G-protein coupled receptors (GPCRs). Ligand binding results in a conformational change in the GPCR, leading to activation of its associated G protein (Thal et al., 2018). PPIs can play a similar role. For example, the ubiquitinase BAP must interact with another protein, ASXL2, via its nonenzymatic ULD domain to allow ubiquitin to bind and be cleaved via its hydrolase (UCH) domain. Thus, allosteric changes induced in BAP1 via ASXL2 interaction with the ULD domain are critical for its enzymatic function. Biophysical studies suggest that a loop within the UCH domain of BAP1 is stabilized by ASXL2 interaction and this allows for ubiquitin to bind (Peng et al., 2018) (Figure 2C).
Allosteric sites often reside within the most flexible regions of any protein. Dynamic proteins and the complexes they form rely heavily on unstructured regions such as loops and linkers to influence interactions. Loops are often directing components of selectivity and linkers play an important role in crosstalk between multidomain proteins. Linkers also can play a key role in multiprotein complex assembly and intradomain interactions (Papaleo et al., 2016). A flexible glycine rich linker region in NFkB allows for the formation of an interaction between the p50 and SWI6/ANK domains to regulate intracellular transport (Henkel et al., 1992). Allosteric sites tend to be less conserved between closely related proteins, subfamily members, or homologues (Furnham et al., 2012; Lu et al., 2014; Nussinov et al., 2011). Thus, allosteric inhibitors are a potential route to selective targeting of challenging PPIs as well as control over multiprotein, dynamic complexes. Perhaps more than any other class of proteins, nature suggests that dynamic proteins are prime targets for allosteric regulation.
Achieving selectivity with chemical probes
Assessing the native mechanisms that dictate interaction between dynamic proteins and their binding partners can illuminate possible routes or methods to target such challenging interactions. Natural regulation of dynamic proteins suggests that a possible route to successful modulators of their interactions is via allosteric sites. Allosteric modulators have the ability to shift the conformational ensemble of proteins to favor the formation of specific complexes or toggle active/inactive conformers without the need to directly target the PPI interface. This strategy is particularly attractive when it comes to targeting large surface area interactions because orthosteric modulation, particularly with small molecules, is challenging due to the lack of topological characteristics that allow for high affinity ligand binding. Allosteric modulation can be achieved -even for these challenging targets- with small molecules that have more drug-like properties, such as lower MW (<600), greater structural rigidity, and hydrophobicity (Cossins and Lawson, 2015). Another advantage that comes with targeting allosteric regions of dynamic proteins is that they tend to be less conserved between the same protein family and homologous proteins, therefore, suggesting an avenue for selective modulation (Huang et al., 2013; Nussinov et al., 2011; Panjkovich and Daura, 2010). Figure 3 summarizes the allosteric compounds, all published since 2018, that will be discussed in this section and spotlights their selectivity profiles.
Figure 3. Recent allosteric modulators.

Quantitative affinity data for each target as well as any selectivity data from the literature is reported. Abbreviations: IP, inflection point of potentiation curve; EC50, half maximal effective concentration; IC50, half maximal inhibitory concentration; koff, off-rate constant; Ki, inhibitory constant – the concentration required to produce half maximum inhibition.
Allosteric regulators have demonstrated high selectivity for protein targets of many other classes, suggesting that similar results are possible with dynamic proteins. For example, the ability to selectively target kinases, GPCRS, and bromodomains (BRDs) has improved with our understanding of allostery in these systems (Fang et al., 2013; Filippakopoulos and Knapp, 2014; Wootten et al., 2013). Extensive research on selectivity within kinase families has shown that molecules that bind outside the catalytic domain display high selectivity on both a family and subtype level while still functioning as potent inhibitors of enzymatic activity (Fang et al., 2013). Allosteric modulation of GPCRs and the impact on selectivity is also well documented (Gao and Jacobson, 2013; Leach et al., 2007) For example, a group from Merck recently published work on MK-7622 a novel positive allosteric modulator of the muscarinic acetylcholine receptor M1 for the treatment of Alzheimer’s disease. This compound was highly selective for the M1 receptor, with no potentiation or agonism of M2–M4 subtypes in an overexpression model conducted in CHO cells (Beshore et al., 2018). Furthermore, compounds that are able to distinguish between the most homologous BRDs, BRD4/7/9, take advantage of subtle conformational differences in non-conserved regions distal to the acetyl lysine binding site (Karim et al., 2020; Olp et al., 2020).
Covalent small molecule modulators have been successful at allosterically modifying dynamic proteins in a manner that mimics covalent PTMs found in nature. In a study informed by a naturally occurring PTM, Bum-Erdene et al. targeted a conserved cysteine residue in TEAD family proteins known to undergo palmitoylation in order to stabilize interaction with YAP. Starting with the FDA approved compound flufenamic acid, shown to weakly bind TEAD2 non-covalently near the thiol of the conserved cysteine, a chloromethylketone moiety was incorporated to make it a covalent inhibitor. This compound, TED-347, allosterically inhibited YAP-TEAD PPIs both in vitro and in mammalian cells (Ki = 10 μM). TED-347 also demonstrated selectivity for the TEAD family over other related proteins (Bum-Erdene et al., 2019). The recently published compound MGH-CP1 covalently modifies and inhibits TEAD family proteins similarly to TED-347, but with increased potency. MGH-CP1 inhibits autopalmitoylation of TEAD2/4 but does not affect autopalmitoylation of ZDHCC family palmitoyl acetyltransferases, suggesting that the compound is selective for TEAD proteins (Li et al., 2020). The authors also find that TEAD2 adopts similar conformations when bound to palmitate or MGH-CP1, further suggesting that mimicking regulation by naturally occurring PTMs can be a successful strategy for selectively targeting dynamic proteins. In another example, covalent allosteric inhibitors were designed to selectively target isoforms of protein kinase Akt. There are three different isoforms of Akt, each with a unique intracellular location and function; thus, highly selective probe compounds are needed to study these proteins. The covalent compound borussertib alkylates a cysteine residue located within an interdomain pocket between the PH domain and kinase domain of Akt proteins, irreversibly stabilizing the inactive conformation. Quambusch et al. observed slight differences around this allosteric pocket between isoforms and designed a library of compounds based on the pyrazinone scaffold of borussertib that could engage these subtle sequence changes. With this approach, they were able to identify compounds with high selectivity for Akt1 and Akt2 (Quambusch et al., 2019). This study highlights paths to attain high selectivity and potency with allosteric modulators.
In the previous example, the structural differences exploited by selective small molecules at the allosteric site of Akt isoforms mainly occurred within a loop formed by residues 259–273. Often the most dynamic components of a protein structure, such as loops, linkers, and flexible helices, are effective sites for allosteric modulation and improved selectivity. Recent work targeting the dynamic coactivator Mediator subunit Med25 also highlights this point. Henderson et al. show that a small molecule, compound 22, can allosterically regulate binding of transcriptional activators to Med25 via covalent modification of a distal cysteine reside. This cysteine residue is located adjacent to regions predicted to be highly flexible via structural modelling. Additionally, compound 22 influences the flexible substructures within Med25, also known to be perturbed by interactions with transcriptional activation domains (Henderson et al., 2018). Thus, by targeting dynamic regions of a protein, even large PPI interfaces can be inhibited.
Another example illustrating that targeting loop modules can improve inhibitor selectivity is the discovery of LY3154207, a potent inhibitor of the Human Dopamine receptor, D1, presented by Lilly. Classical approaches to targeting D1 receptors have mainly been orthosteric, and candidate modulators bind similarly to the natural substrate dopamine (Zhang et al., 2009). However, as D1 and D5 receptors have high structural similarity, this approach leads to selectivity issues. The authors found that LYS3154207 and its analogs bound to a novel binding site in the intracellular loop 2 (ICL2), allosterically inhibiting binding at the orthosteric D1 site. Excitingly, this inhibitor was highly selective for the D1 receptor over the closely related D5 receptor (>2700-fold). LYS3154207 demonstrated over 1000-fold selectivity for the D1 receptor when tested against a panel of 40 additional targets (Hao et al., 2019). In another recent example targeting a transcription factor, fosfosal, a well-documented clinical prodrug, was found to inhibit the SH2 domain of STAT5b over its homologue STAT5a. Mutational analysis showed this selectivity was dictated by residues within an adjacent linker domain rather than within the SH2 domain itself (Gräb and Berg, 2020). This work illustrates the role that the STAT linker region in SH2 domain function, and targeting the linker with small molecules is thus an avenue for selective inhibition of STAT PPIs. This same group has identified additional molecules selective for both STAT5a and STAT5b utilizing the same concept (Elumalai et al., 2015; Gräb et al., 2019).
Emerging techniques for identification of selective dynamic protein complex modulators
The examples discussed above demonstrate the power and promise of allosteric regulators for modulating dynamic proteins, particularly in terms of selectivity. However, in the majority of these examples, the mechanism of action was not the result of the discovery method but rather serendipitous. Here we outline emerging techniques and strategies that facilitate screens more likely to produce allosteric and ultimately selective inhibitors.
Inhibitor identification from well-structured proteins allows for easier structural characterization and lead compound optimization. Dynamic proteins, by definition alone, provide a steep challenge, with many of the common experimental techniques requiring improvements before being applied to proteins that are highly mobile and lack definitive structure. Thus, advancements in structural biology and biophysics are key when it comes to understanding selectivity in dynamic protein complexes. Recent improvements in techniques such as mass spectrometry (Ishii et al., 2018), NMR (Huang and Kalodimos, 2017), and cryogenic electron microscopy (cryo-EM) (Merk et al., 2016; Schmidt and Urlaub, 2017) have allowed a high-resolution view of multiprotein complexes and aided in our understanding of composition as well as identification of key PPIs. For example, Khattabi et al. recently reported a 5.9 Å cryo-EM structure mapping the entire mammalian Mediator complex (~4 mDa). This included, for the first time, mapping of the exchangeable and conformationally dynamic proteins within the tail region of the complex (El Khattabi et al., 2019). Many of the tail proteins are the primary targets of transcription factors and these results provide key insights into how to target this class of proteins successfully. In another exciting example, time resolved cryoEM was used to obtain a near-atomic-resolution view of the conformational changes that drive and regulate subunit assembly, initiation factor dissociation, and fMet-tRNA positioning during the formation of the 70S elongation-competent complex in bacteria (Kaledhonkar et al., 2019). This type of approach allows dissection of the specific timing and order of conformational changes contributing to the mechanism and regulation of large and dynamic multiprotein systems. These examples highlight how improved techniques for understanding dynamic protein complexes will allow researchers to identify more promising regions to target.
Increased complexity of an assay system is characteristic of strategies that allow for enrichment of selective hits from screening. Grey box screening is a prominent example. In this method, multiprotein complexes are reconstituted in vitro and subjected to HTS with the goal of identifying compounds that effect biochemical properties, such as enzymatic activity, of the complex. This approach has identified specific chemical modulators of dynamic proteins within complexes such as Hsp70 (Cesa et al., 2013; Taylor et al., 2018), Hsp90 (Patwardhan et al., 2014), and regulators of G-protein signaling (Monroy et al., 2013). This method has been thoroughly reviewed in other sources (Cesa et al., 2015; Thompson et al., 2012). Mass spectrometry approaches can also allow for multiplexing of protein assays. Collision-induced unfolding (CIU) can distinguish individual protein and protein complex ions through their distinct unfolding pathways in the gas phase, allowing for analysis of multiprotein and protein-ligand complexes (Niu and Ruotolo, 2015) Combining CIU with collision induced dissociation (CID) mass spec analysis distinguished ATP competitive from allosteric kinase inhibitors of the tyrosine kinase Abl (Rabuck-Gibbons et al., 2018). There are also many other examples utilizing mass spectrometry to analyze intact multiprotein complexes (Ishii et al., 2018). Application of these techniques to dynamic targets in high throughput screens can streamline identification of selective modulators. Molecules displaying selectivity, as seen with the examples in the previous section, often act allosterically, and thus characterization of their effects on target proteins will provide insights into allosteric regions to focus on in future studies.
Classical approaches, such as Forster resonance energy transfer (FRET) and fluorescence polarization (FP), can be utilized for HTS of dynamic protein targets where allosteric sites are unknown. To assess the selectivity of lead compounds, secondary screening against a selected panel of alternate targets is necessary (Lea and Simeonov, 2011). This iterative screening process is highlighted in work from Majmudar et al. Hits from a FP-based HTS for inhibitors of CBP KIX were subjected to multiple rounds of secondary screening against increasingly relevant PPIs, from non-transcriptional PPIs to DNA-binding domain•DNA interactions to related coactivator•activator interactions. This led to the identification of the CBP KIX selective molecule lobaric acid (Majmudar et al., 2012). Library complexity is also key for screening dynamic proteins. Diversity oriented synthesis and DNA Encoded Libraries (DEL) have allowed for the generation of huge billion-trillion compound libraries that can be screened in a one pot format (Chan et al., 2015; Jr et al., 2016; Salamon et al., 2016). Other notable library developments with the ability to impact dynamic protein probe discovery include diverse natural product focused libraries, protein-protein interaction focused libraries, and cheminfomatic based design and optimization (Gong et al., 2017; Jin et al., 2018; Moret et al., 2019)
Targeted fragment approaches have been successful in discovering allosteric sites and modulators of diverse proteins. Fragment based screening does not rely on identification of a single high affinity hit, rather it provides the opportunity to diversify lead scaffolds into directed libraries or link together lower affinity binders, acting at unique sites on the target protein, to construct a more potent compound (Doak et al., 2016; Kirsch et al., 2019). The Cravatt lab has pioneered cell-based approaches to fragment screening, providing the advantage of being able to study a protein of interest in its native environment (Backus et al., 2016; Parker et al., 2017). This would be particularly useful for dynamic proteins that are challenging to work with in vitro. Tethering is a site-directed fragment technique that utilizes a disulfide moiety within each fragment to facilitate localization to protein binding sites via covalent bond formation with adjacent cysteine residues. Tethering has enabled the identification of allosteric modulators for many dynamic protein targets, from enzymes such as PDK1 and Ras to coactivators such as CBP and Med25 (Henderson et al., 2018; Ostrem et al., 2013; Sadowsky et al., 2011; Wang et al., 2013) In a recent example targeting the scaffolding protein 14-3-3 PPI, tethering was used to identify orthosteric stabilizers of the 14-3-3•ERα complex. These stabilizers were able to enhance the interaction by up to 40-fold while demonstrating selectivity for ERα over other 14-3-3 interacting proteins (Sijbesma et al., 2019).
Screening methods that provide mechanistic insight into small molecule binding have proven highly useful for conformationally dynamic proteins, providing the ability to select for molecules that influence desired regions of the target. NMR-based methods, including Protein Observed Fluorine (PrOF) and 1H15N HSQC can provide information on both binding site location and impact on protein dynamics and structure (Arntson and Pomerantz, 2016; Dalvit and Vulpetti, 2019; Furukawa et al., 2016). Thus, one can select for compounds during screening that show specific alterations in structure and/or dynamics. Because throughput in NMR based screening methods can be a limitation, this approach is extremely powerful when coupled with computational screening methods. Recent work by Gupta et al. combined computational and NMR screening approaches with the goal of targeting known allosteric sites on mutant and wildtype KRAS. Gupta et al. started from a virtual library of 76 million compounds and identified nine compounds that lead to 1H15N HSQC chemical shift perturbations of residues near the functionally responsive switch loop. Although the authors do not analyze the functional effect of their lead compound, E22, they find that it binds nearly 100-fold tighter to GNP-bound Ras compared to GDP bound Ras and hypothesize that the molecule may affect GEF-mediated GDP/GTP exchange, similar to other indole compounds that target KRAS (Gupta et al., 2019). Combinations of computational and experimental approaches, as this example illustrates, have and will continue to be important for dynamic protein probe discovery. Notably, computational studies of allostery, with a focus on identification of allosteric sites and discovery of potent allosteric inhibitors, have been extensively performed in recent years with many successes (De Vivo et al., 2016; Greener and Sternberg, 2018; Ma et al., 2016)
Thermal stability assays, such as differential scanning fluorimetry (DSF), are powerful screening options for dynamic protein targets(A. Senisterra and Patrick J. Finerty, 2009; Gao et al., 2020; Niesen et al., 2007). While DSF is a technique dating back to 1991, it recently has been used to screen some challenging targets such as STAT proteins (STAT1, STAT3, and STAT5) (Attarha et al., 2020; Desroses et al., 2018), nuclear receptors (DeSantis et al., 2012), and chaperone proteins (Mac Sweeney et al., 2018; Shao et al., 2020). Not only is this method simple, inexpensive, and amenable to HTS, it provides insights into the effect of a compound on the thermal stability of the target protein, making it possible to select for compounds with the desired stabilizing or destabilizing effect. Thermal stability assays have the added benefit of not requiring knowledge of binding partners. Recently, groups have been working to improve understanding of the theoretical underpinnings of DSF and further interrogate the potential information that can be obtained from DSF melting curves (Sun et al., 2020; Wu et al., 2020). This work has made it clear that more than just a melting temperature (Tm) emerges from these experiments. For example, the shape of the melt curve can provide some insight into ligand binding even if Tm shifts are not observed. Additionally, innovations to the technique suggest the ability to multiplex. DSF-GFP, a technique pioneered in 2012, involves labelling the target protein of interest with GFP and using the change in GFP fluorescence over a temperature gradient as a readout rather than solvatochromic dye (Sorenson and Schaeffer, 2020). This approach allows for the selective measurement of target protein Tm in the presence of other proteins.
The cellular thermal shift assay (CETSA) is another technique that interrogates changes in protein stability and can also be extremely useful for screening dynamic proteins (Molina et al., 2013). A benefit of this approach is the ability to screen the protein in a more native environment, such as in cell lysates. Recent advances to CETSA techniques have allowed for use in HTS. To date, there have been multiple HT-CETSA approaches published, all focusing on altering protein detection methods to allow for the classic CETSA western blot readout to be adapted to 384 or 1536 well format (Henderson et al., 2020). For example, Shaw et al. applied HT-CETSA via AlphaScreen technology to conduct a screen for modulators of BRAF and PARP1through screening 896 and 6288 compounds, respectively (Shaw et al., 2019). In a recent success story with a challenging target, a small molecule, NPD10084, was found to thermally destabilize PKM2, which can function as both a kinase and a transcriptional coactivator. This interaction with PKM2 leads to inhibition of PPIs with STAT3 and beta-catenin as well as altered expression of PKM2 regulated genes. Notably, the authors outlined a novel assay, which they termed 2D gel electrophoresis-based proteome-wide CETSA, or 2DE-CETSA, that allowed for proteome wide screening for small molecule target identification (Nagasawa et al., 2020)
The Outlook
As can be seen from the outlined examples, it is a particularly exciting time for those who study and/or target dynamic proteins. Discovery methods that are tailored for the distinct biophysical characteristics of dynamic proteins, as well as those that more closely recapitulate the native context, have been highly successful. In addition to these screening techniques, thoroughly interrogating the selectivity of lead compounds on a global scale will be integral in deriving effective biological probes of dynamic protein targets. Techniques such as thermal proteome profiling, activity based protein profiling and Drug Affinity Responsive Target Stability (DARTS) allow determination of potential off target effects for a chemical probe in a cellular context (Jessani and Cravatt, 2004; Pai et al., 2015; Savitski et al., 2014). The resulting generation of more selective and potent small molecule modulators now enable the chemical genetic dissection of critical cellular machines. For example, studies with full length p300 and other various multidomain constructs show BRD inhibitors can affect HAT activity, suggesting interdomain crosstalk does occur (Shrimp et al., 2018; Zucconi et al., 2016). Interdomain crosstalk has been observed in other dynamic systems, particularly in nuclear receptors such as the thyroid hormone receptor and ERα, where the DNA binding via the DNA binding domain (DBD) can be enhanced by changes in the ligand binding domain (LBD) (Huang et al., 2018). Further exciting studies focusing on the global effects of selective dynamic protein modulators on full length proteins or intact protein complexes are sure to come in the future and improve our understanding of interdomain communication in these systems.
Another interesting avenue for dynamic proteins that has yet to be fully explored is proteolysis targeting chimeras (PROTACs). PROTACs could be particularly advantageous in the field of dynamic protein complexes because the starting scaffold does not need to functionally affect any interactions, as it can be challenging to find modulators with low IC50s or EC50s for certain targets. Additionally, even non-specific inhibitors can be starting points for PROTACS. Recent work has shown specificity can be engineered into a PROTAC starting from a multi-target warhead by exploiting differences in the interface between the protein of interest and the E3 ligase (Bondeson et al., 2018; Gadd et al., 2017). It is possible that through PROTACS, some of the challenges that come with identifying selective modulators can be averted. With regards to dynamic protein complexes, one could imagine using a PROTAC to completely alter complex composition or skew complexes to favor one exchangeable subunit over others. These and other novel selective mechanisms to chemically target dynamic proteins are sure to be on the horizon and we look forward to the exciting discoveries to come.
Figure 4. Examples of techniques for identification of dynamic protein modulators.

(A) Collision induced unfolding (CIU) can be utilized as a medium-high throughput approach to determine binding profiles protein•ligand complexes. If the CIU profile is known for a certain type of protein•ligand complexes, for example an allosteric signature or orthosteric signature, a screen can be conducted and molecules can be sorted by distinct fingerprints (Dixit et al., 2018). (B) 1H15N HSQC NMR can be utilized as a low-medium throughput approach to determine structural effects of small molecule binding to proteins. If a particular amino acid can be associated with allostery, a library of molecules can be screened looking for perturbation of chemical shifts associated with that residue. This method is particularly powerful when used in combination with computation data (as seen in Gupta et al.). (C) Differential scanning fluorimetry (DSF) is a high throughput method for screening proteins. This method monitors protein unfolding as a function of temperature. As the protein unfolds, a solvatochromatic dye binds and fluorescence increases. The melting temperature (Tm) of the protein is interpreted as the inflection point of the resulting melt curve (Niesen et al., 2007). While allosteric effects of small molecules cannot be determined using this method, their effect on protein stability can be determined. In a HTS, molecules can be sorted by their ability to increase or decrease protein Tm.
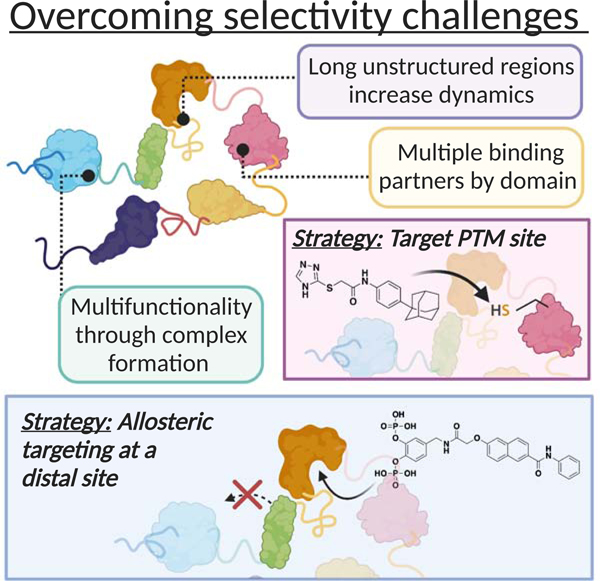
Garlick et al. summarizes the opportunities and difficulties of selectively targeting dynamic proteins. Reviewing natural mechanisms of regulation and highlighting recent examples of selective inhibitors from literature, the authors suggest avenues to overcome the challenge. Discussion of approaches with potential to streamline discovery of selective chemical modulators is also provided.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Assimon A, V., Gillies T, A., Rauch N, J., and Gestwicki E, J. (2013). Hsp70 Protein Complexes as Drug Targets. Curr. Pharm. Des 19, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrusán G, and Marsh JA (2019). Ligand-Binding-Site Structure Shapes Allosteric Signal Transduction and the Evolution of Allostery in Protein Complexes. Mol. Biol. Evol 36, 1711–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin MR, Tang Y, and Wells JA (2014). Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol 21, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arntson KE, and Pomerantz WCK (2016). Protein-Observed Fluorine NMR: A Bioorthogonal Approach for Small Molecule Discovery. J. Med. Chem 59, 5158–5171. [DOI] [PubMed] [Google Scholar]
- Senisterra A, G., and Finerty Patrick J., J. (2009). High throughput methods of assessing protein stability and aggregation. Mol. Biosyst 5, 217–223. [DOI] [PubMed] [Google Scholar]
- Attarha S, Reithmeier A, Busker S, Desroses M, and Page BDG (2020). Validating STAT Protein-inhibitor Interactions using Biochemical and Cellular Thermal Shift Assays. ACS Chem. Biol [DOI] [PubMed]
- Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González-Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. (2016). Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, et al. (2005). High-Throughput Mapping of a Dynamic Signaling Network in Mammalian Cells. Science 307, 1621–1625. [DOI] [PubMed] [Google Scholar]
- Batada NN, Hurst LD, and Tyers M (2006). Evolutionary and Physiological Importance of Hub Proteins. PLoS Comput. Biol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshore DC, Di Marco N, C., Chang RK, Greshock TJ, Ma L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Fuerst J, et al. (2018). MK-7622: A First-in-Class M1 Positive Allosteric Modulator Development Candidate. ACS Med. Chem. Lett 9, 652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, and Crews CM (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol 25, 78–87.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen ME, and Mapp AK (2018). Modulating the masters: chemical tools to dissect CBP and p300 function. Curr. Opin. Chem. Biol 45, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bum-Erdene K, Zhou D, Gonzalez-Gutierrez G, Ghozayel MK, Si Y, Xu D, Shannon HE, Bailey BJ, Corson TW, Pollok KE, et al. (2019). Small-Molecule Covalent Modification of Conserved Cysteine Leads to Allosteric Inhibition of the TEAD⋅Yap Protein-Protein Interaction. Cell Chem. Biol 26, 378–389.e13. [DOI] [PubMed] [Google Scholar]
- Cesa LC, Patury S, Komiyama T, Ahmad A, Zuiderweg ERP, and Gestwicki JE (2013). Inhibitors of difficult protein-protein interactions identified by high-throughput screening of multiprotein complexes. ACS Chem. Biol 8, 1988–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesa LC, Mapp AK, and Gestwicki JE (2015). Direct and Propagated Effects of Small Molecules on Protein–Protein Interaction Networks. Front. Bioeng. Biotechnol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AI, McGregor LM, and Liu DR (2015). Novel selection methods for DNA-encoded chemical libraries. Curr. Opin. Chem. Biol 26, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, Deng H, Pan D, Luo X, and Wu X (2016). Autopalmitoylation of TEAD Proteins Regulates Transcriptional Output of Hippo Pathway. Nat. Chem. Biol 12, 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossins BP, and Lawson ADG (2015). Small Molecule Targeting of Protein–Protein Interactions through Allosteric Modulation of Dynamics. Molecules 20, 16435–16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvit C, and Vulpetti A (2019). Ligand-Based Fluorine NMR Screening: Principles and Applications in Drug Discovery Projects. J. Med. Chem 62, 2218–2244. [DOI] [PubMed] [Google Scholar]
- De Vivo M, Masetti M, Bottegoni G, and Cavalli A (2016). Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem 59, 4035–4061. [DOI] [PubMed] [Google Scholar]
- DeSantis K, Reed A, Rahhal R, and Reinking J (2012). Use of Differential Scanning Fluorimetry as a High-Throughput Assay to Identify Nuclear Receptor Ligands: Nucl. Recept. Signal [DOI] [PMC free article] [PubMed]
- Desroses M, Busker S, Astorga-Wells J, Attarha S, Kolosenko I, Zubarev RA, Helleday T, Grandér D, and Page BDG (2018). STAT3 differential scanning fluorimetry and differential scanning light scattering assays: Addressing a missing link in the characterization of STAT3 inhibitor interactions. J. Pharm. Biomed. Anal 160, 80–88. [DOI] [PubMed] [Google Scholar]
- Dixit SM, Polasky DA, and Ruotolo BT (2018). Collision induced unfolding of isolated proteins in the gas phase: past, present, and future. Curr. Opin. Chem. Biol 42, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doak BC, Norton RS, and Scanlon MJ (2016). The ways and means of fragment-based drug design. Pharmacol. Ther 167, 28–37. [DOI] [PubMed] [Google Scholar]
- Duan G, and Walther D (2015). The Roles of Post-translational Modifications in the Context of Protein Interaction Networks. PLoS Comput. Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson HJ, and Wright PE (2016). Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J. Biol. Chem 291, 6714–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khattabi L, Zhao H, Kalchschmidt J, Young N, Jung S, Van Blerkom P, Kieffer-Kwon P, Kieffer-Kwon K-R, Park S, Wang X, et al. (2019). A Pliable Mediator Acts as a Functional Rather Than an Architectural Bridge between Promoters and Enhancers. Cell 178, 1145–1158.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elumalai N, Berg A, Natarajan K, Scharow A, and Berg T (2015). Nanomolar Inhibitors of the Transcription Factor STAT5b with High Selectivity over STAT5a. Angew. Chem. Int. Ed 54, 4758–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euskirchen GM, Auerbach RK, Davidov E, Gianoulis TA, Zhong G, Rozowsky J, Bhardwaj N, Gerstein MB, and Snyder M (2011). Diverse Roles and Interactions of the SWI/SNF Chromatin Remodeling Complex Revealed Using Global Approaches. PLoS Genet 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Grütter C, and Rauh D (2013). Strategies for the Selective Regulation of Kinases with Allosteric Modulators: Exploiting Exclusive Structural Features. ACS Chem. Biol 8, 58–70. [DOI] [PubMed] [Google Scholar]
- Fernández-Fernández MR, and Valpuesta JM (2018). Hsp70 chaperone: a master player in protein homeostasis. F1000Research 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, and Knapp S (2014). Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov 13, 337–356. [DOI] [PubMed] [Google Scholar]
- Furnham N, Sillitoe I, Holliday GL, Cuff AL, Laskowski RA, Orengo CA, and Thornton JM (2012). Exploring the evolution of novel enzyme functions within structurally defined protein superfamilies. PLoS Comput. Biol 8, e1002403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa A, Konuma T, Yanaka S, and Sugase K (2016). Quantitative analysis of protein–ligand interactions by NMR. Prog. Nucl. Magn. Reson. Spectrosc 96, 47–57. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M, Tompa P, Simon I, Uversky VN, Hansen JC, and Asturias FJ (2008). Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol 4, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd MS, Testa A, Lucas X, Chan K-H, Chen W, Lamont DJ, Zengerle M, and Ciulli A (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z-G, and Jacobson KA (2013). Allosteric modulation and functional selectivity of G protein-coupled receptors. Drug Discov. Today Technol 10, e237–e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K, Oerlemans R, and Groves MR (2020). Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev 12, 85–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A-C, and Superti-Furga G (2003). Protein complexes and proteome organization from yeast to man. Curr. Opin. Chem. Biol 7, 21–27. [DOI] [PubMed] [Google Scholar]
- Gill G, and Ptashne M (1988). Negative effect of the transcriptional activator GAL4. Nature 334, 721–724. [DOI] [PubMed] [Google Scholar]
- Gong Z, Hu G, Li Q, Liu Z, Wang F, Zhang X, Xiong J, Li P, Xu Y, Ma R, et al. (2017). Compound Libraries: Recent Advances and Their Applications in Drug Discovery. Curr. Drug Discov. Technol 14, 216–228. [DOI] [PubMed] [Google Scholar]
- Gräb J, and Berg T (2020). The selectivity of fosfosal for STAT5b over STAT5a is mediated by Arg566 in the linker domain. ChemBioChem n/a [DOI] [PMC free article] [PubMed]
- Gräb J, Berg A, Blechschmidt L, Klüver B, Rubner S, Fu DY, Meiler J, Gräber M, and Berg T (2019). The STAT5b Linker Domain Mediates the Selectivity of Catechol Bisphosphates for STAT5b over STAT5a. ACS Chem. Biol 14, 796–805. [DOI] [PubMed] [Google Scholar]
- Greener JG, and Sternberg MJ (2018). Structure-based prediction of protein allostery. Curr. Opin. Struct. Biol 50, 1–8. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Wang X, Pagba CV, Prakash P, Sarkar-Banerjee S, Putkey J, and Gorfe AA (2019). Multi-target, ensemble-based virtual screening yields novel allosteric KRAS inhibitors at high success rate. Chem. Biol. Drug Des 94, 1441–1456. [DOI] [PubMed] [Google Scholar]
- Hahn S (2018). Phase Separation, Protein Disorder, and Enhancer Function. Cell 175, 1723–1725. [DOI] [PubMed] [Google Scholar]
- Hao J, Beck JP, Schaus JM, Krushinski JH, Chen Q, Beadle CD, Vidal P, Reinhard MR, Dressman BA, Massey SM, et al. (2019). Synthesis and Pharmacological Characterization of 2-(2,6-Dichlorophenyl)-1-((1S,3R)-5-(3-hydroxy-3-methylbutyl)-3-(hydroxymethyl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (LY3154207), a Potent, Subtype Selective, and Orally Available Positive Allosteric Modulator of the Human Dopamine D1 Receptor. J. Med. Chem 62, 8711–8732. [DOI] [PubMed] [Google Scholar]
- Henderson AR, Henley MJ, Foster NJ, Peiffer AL, Beyersdorf MS, Stanford KD, Sturlis SM, Linhares BM, Hill ZB, Wells JA, et al. (2018). Conservation of coactivator engagement mechanism enables small-molecule allosteric modulators. Proc. Natl. Acad. Sci 115, 8960–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson MJ, Holbert MA, Simeonov A, and Kallal LA (2020). High-Throughput Cellular Thermal Shift Assays in Research and Drug Discovery. SLAS Discov. Adv. Sci. Drug Discov 25, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel T, Zabel U, van Zee K, Müller JM, Fanning E, and Baeuerle PA (1992). Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-kappa B subunit. Cell 68, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Heo M, Maslov S, and Shakhnovich E (2011). Topology of protein interaction network shapes protein abundances and strengths of their functional and nonspecific interactions. Proc. Natl. Acad. Sci 108, 4258–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, and Kalodimos CG (2017). Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu. Rev. Biophys 46, 317–336. [DOI] [PubMed] [Google Scholar]
- Huang W, Lu S, Huang Z, Liu X, Mou L, Luo Y, Zhao Y, Liu Y, Chen Z, Hou T, et al. (2013). Allosite: a method for predicting allosteric sites. Bioinformatics 29, 2357–2359. [DOI] [PubMed] [Google Scholar]
- Huang W, Peng Y, Kiselar J, Zhao X, Albaqami A, Mendez D, Chen Y, Chakravarthy S, Gupta S, Ralston C, et al. (2018). Multidomain architecture of estrogen receptor reveals interfacial cross-talk between its DNA-binding and ligand-binding domains. Nat. Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K, Zhou M, and Uchiyama S (2018). Native mass spectrometry for understanding dynamic protein complex. Biochim. Biophys. Acta BBA - Gen. Subj 1862, 275–286. [DOI] [PubMed] [Google Scholar]
- Jessani N, and Cravatt BF (2004). The development and application of methods for activity-based protein profiling. Curr. Opin. Chem. Biol 8, 54–59. [DOI] [PubMed] [Google Scholar]
- Jin X, Lee K, Kim NH, Kim HS, Yook JI, Choi J, and No KT (2018). Natural products used as a chemical library for protein–protein interaction targeted drug discovery. J. Mol. Graph. Model 79, 46–58. [DOI] [PubMed] [Google Scholar]
- Jones S, and Thornton JM (1996). Principles of protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jr RAG, Dumelin CE, and Keefe AD (2016). DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov 16, nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- Kalabova D, Filandr F, Alblova M, Petrvalska O, Horvath M, Man P, Obsil T, and Obsilova V (2020). 14-3-3 protein binding blocks the dimerization interface of caspase-2. FEBS J [DOI] [PubMed]
- Kaledhonkar S, Fu Z, Caban K, Li W, Chen B, Sun M, Gonzalez RL, and Frank J (2019). Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature 570, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim RM, Chan A, Zhu J-Y, and Schönbrunn E (2020). Structural Basis of Inhibitor Selectivity in the BRD7/9 Subfamily of Bromodomains. J. Med. Chem [DOI] [PMC free article] [PubMed]
- Kasahara K, Terazawa H, Takahashi T, and Higo J (2019). Studies on Molecular Dynamics of Intrinsically Disordered Proteins and Their Fuzzy Complexes: A Mini-Review. Comput. Struct. Biotechnol. J 17, 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SH, Ahmad F, Ahmad N, Flynn DC, and Kumar R (2011). Protein-Protein Interactions: Principles, Techniques, and their Potential Role in New Drug Development. J. Biomol. Struct. Dyn 28, 929–938. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN (2006). Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol 7, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch P, Hartman AM, Hirsch AKH, and Empting M (2019). Concepts and Core Principles of Fragment-Based Drug Design. Molecules 24, 4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea WA, and Simeonov A (2011). Fluorescence Polarization Assays in Small Molecule Screening. Expert Opin. Drug Discov 6, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Sexton PM, and Christopoulos A (2007). Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci 28, 382–389. [DOI] [PubMed] [Google Scholar]
- Lee TI, and Young RA (2013). Transcriptional Regulation and Its Misregulation in Disease. Cell 152, 1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Sun Y, Jarugumilli GK, Liu S, Dang K, Cotton JL, Xiol J, Chan PY, DeRan M, Ma L, et al. (2020). Lats1/2 Sustain Intestinal Stem Cells and Wnt Activation through TEAD-Dependent and Independent Transcription. Cell Stem Cell 26, 675–692.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Huang W, and Zhang J (2014). Recent computational advances in the identification of allosteric sites in proteins. Drug Discov. Today 19, 1595–1600. [DOI] [PubMed] [Google Scholar]
- Ma X, Meng H, and Lai L (2016). Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated. J. Chem. Inf. Model 56, 1725–1733. [DOI] [PubMed] [Google Scholar]
- Mac Sweeney A, Chambovey A, Wicki M, Müller M, Artico N, Lange R, Bijelic A, Breibeck J, and Rompel A (2018). The crystallization additive hexatungstotellurate promotes the crystallization of the HSP70 nucleotide binding domain into two different crystal forms. PLoS ONE 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmudar CY, Højfeldt JW, Arevang CJ, Pomerantz WC, Gagnon JK, Schultz PJ, Cesa LC, Doss CH, Rowe SP, Vásquez V, et al. (2012). Sekikaic Acid and Lobaric Acid Target a Dynamic Interface of the Coactivator CBP/p300. Angew. Chem. Int. Ed 51, 11258–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapp AK, Pricer R, and Sturlis S (2015). Targeting transcription is no longer a quixotic quest. Nat. Chem. Biol 11, 891–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JA, and Teichmann SA (2015). Structure, Dynamics, Assembly, and Evolution of Protein Complexes. Annu. Rev. Biochem 84, 551–575. [DOI] [PubMed] [Google Scholar]
- Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS, et al. (2016). Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 165, 1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T, Kay LE, and Forman-Kay JD (2010). Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. JMR 23, 105–116. [DOI] [PubMed] [Google Scholar]
- Molina DM, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, and Nordlund P (2013). Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 341, 84–87. [DOI] [PubMed] [Google Scholar]
- Monroy CA, Mackie DI, and Roman DL (2013). A High Throughput Screen for RGS Proteins Using Steady State Monitoring of Free Phosphate Formation. PLOS ONE 8, e62247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret N, Clark NA, Hafner M, Wang Y, Lounkine E, Medvedovic M, Wang J, Gray N, Jenkins J, and Sorger PK (2019). Cheminformatics Tools for Analyzing and Designing Optimized Small-Molecule Collections and Libraries. Cell Chem. Biol 26, 765–777.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, and Vojtesek B (2013). C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 32, 3101–3110. [DOI] [PubMed] [Google Scholar]
- Nagasawa I, Muroi M, Kawatani M, Ohishi T, Ohba S, Kawada M, and Osada H (2020). Identification of a Small Compound Targeting PKM2-Regulated Signaling Using 2D Gel Electrophoresis-Based Proteome-wide CETSA. Cell Chem. Biol 27, 186–196.e4. [DOI] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, and Vedadi M (2007). The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- Niu S, and Ruotolo BT (2015). Collisional unfolding of multiprotein complexes reveals cooperative stabilization upon ligand binding. Protein Sci 24, 1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai C-J, and Csermely P (2011). Allo-network drugs: harnessing allostery in cellular networks. Trends Pharmacol. Sci 32, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai C-J, Xin F, and Radivojac P (2012). Allosteric post-translational modification codes. Trends Biochem. Sci 37, 447–455. [DOI] [PubMed] [Google Scholar]
- Olp MD, Sprague DJ, Goetz CJ, Kathman SG, Wynia-Smith SL, Shishodia S, Summers SB, Xu Z, Statsyuk AV, and Smith BC (2020). Covalent-Fragment Screening of BRD4 Identifies a Ligandable Site Orthogonal to the Acetyl-Lysine Binding Sites. ACS Chem. Biol [DOI] [PMC free article] [PubMed]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai MY, Lomenick B, Hwang H, Schiestl R, McBride W, Loo JA, and Huang J (2015). Drug Affinity Responsive Target Stability (DARTS) for Small Molecule Target Identification. Methods Mol. Biol. Clifton NJ 1263, 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjkovich A, and Daura X (2010). Assessing the structural conservation of protein pockets to study functional and allosteric sites: implications for drug discovery. BMC Struct. Biol 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, and Nussinov R (2016). The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chem. Rev 116, 6391–6423. [DOI] [PubMed] [Google Scholar]
- Parker CG, Galmozzi A, Wang Y, Correia BE, Sasaki K, Joslyn CM, Kim AS, Cavallaro CL, Lawrence RM, Johnson SR, et al. (2017). Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 168, 527–541.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan CA, Alfa E, Lu S, and Chadli A (2014). Progesterone Receptor Chaperone Complex–Based High-Throughput Screening Assay: Identification of Capsaicin as an Inhibitor of the Hsp90 Machine. J. Biomol. Screen [DOI] [PMC free article] [PubMed]
- Peng H, Prokop J, Karar J, Park K, Cao L, Harbour JW, Bowcock AM, Malkowicz SB, Cheung M, Testa JR, et al. (2018). Familial and Somatic BAP1 Mutations Inactivate ASXL1/2-Mediated Allosteric Regulation of BAP1 Deubiquitinase by Targeting Multiple Independent Domains. Cancer Res 78, 1200–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quambusch L, Landel I, Depta L, Weisner J, Uhlenbrock N, Müller MP, Glanemann F, Althoff K, Siveke JT, and Rauh D (2019). Covalent-Allosteric Inhibitors to Achieve Akt Isoform-Selectivity. Angew. Chem 131, 18999–19005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabuck-Gibbons JN, Keating JE, and Ruotolo BT (2018). Collision induced unfolding and dissociation differentiates ATP-competitive from allosteric protein tyrosine kinase inhibitors. Int. J. Mass Spectrom 427, 151–156. [Google Scholar]
- Ran X, and Gestwicki JE (2018). Inhibitors of protein–protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol 44, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, and Wells JA (2011). Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. U. S. A 108, 6056–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamon H, Klika Škopić M, Jung K, Bugain O, and Brunschweiger A (2016). Chemical Biology Probes from Advanced DNA-encoded Libraries. ACS Chem. Biol 11, 296–307. [DOI] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Molina DM, Jafari R, Dovega RB, Klaeger S, et al. (2014). Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346. [DOI] [PubMed] [Google Scholar]
- Schmidt C, and Urlaub H (2017). Combining cryo-electron microscopy (cryo-EM) and cross-linking mass spectrometry (CX-MS) for structural elucidation of large protein assemblies. Curr. Opin. Struct. Biol 46, 157–168. [DOI] [PubMed] [Google Scholar]
- Semmler L, Reiter-Brennan C, and Klein A (2019). BRCA1 and Breast Cancer: a Review of the Underlying Mechanisms Resulting in the Tissue-Specific Tumorigenesis in Mutation Carriers. J. Breast Cancer 22, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Oltion K, Wu T, and Gestwicki E, J. (2020). Differential scanning fluorimetry (DSF) screen to identify inhibitors of Hsp60 protein–protein interactions. Org. Biomol. Chem [DOI] [PubMed]
- Shaw J, Dale I, Hemsley P, Leach L, Dekki N, Orme JP, Talbot V, Narvaez AJ, Bista M, Martinez Molina D, et al. (2019). Positioning High-Throughput CETSA in Early Drug Discovery through Screening against B-Raf and PARP1. Slas Discov 24, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimp JH, Grose C, Widmeyer SRT, Thorpe AL, Jadhav A, and Meier JL (2018). Chemical Control of a CRISPR-Cas9 Acetyltransferase. ACS Chem. Biol 13, 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijbesma E, Hallenbeck KK, Leysen S, de Vink PJ, Skóra L, Jahnke W, Brunsveld L, Arkin MR, and Ottmann C (2019). Site-Directed Fragment-Based Screening for the Discovery of Protein-Protein Interaction Stabilizers. J. Am. Chem. Soc 141, 3524–3531. [DOI] [PubMed] [Google Scholar]
- Silhan J, Vacha P, Strnadova P, Vecer J, Herman P, Sulc M, Teisinger J, Obsilova V, and Obsil T (2009). 14-3-3 protein masks the DNA binding interface of forkhead transcription factor FOXO4. J. Biol. Chem 284, 19349–19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MC, and Gestwicki JE (2012). Features of protein–protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev. Mol. Med 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson AE, and Schaeffer PM (2020). High-Throughput Differential Scanning Fluorimetry of GFP-Tagged Proteins In Targeting Enzymes for Pharmaceutical Development: Methods and Protocols, Labrou NE, ed. (New York, NY: Springer US; ), pp. 69–85. [DOI] [PubMed] [Google Scholar]
- Stein A, Pache RA, Bernadó P, Pons M, and Aloy P (2009). Dynamic interactions of proteins in complex networks: a more structured view. FEBS J 5390–5405. [DOI] [PubMed]
- Stetz G, Tse A, and Verkhivker GM (2018). Dissecting Structure-Encoded Determinants of Allosteric Cross-Talk between Post-Translational Modification Sites in the Hsp90 Chaperones. Sci. Rep 8, 6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Li Y, Yates EA, and Fernig DG (2020). SimpleDSFviewer: A tool to analyze and view differential scanning fluorimetry data for characterizing protein thermal stability and interactions. Protein Sci 29, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain JF, and Gierasch LM (2006). The changing landscape of protein allostery. Curr. Opin. Struct. Biol 16, 102–108. [DOI] [PubMed] [Google Scholar]
- Takaoka M, and Miki Y (2018). BRCA1 gene: function and deficiency. Int. J. Clin. Oncol 23, 36–44. [DOI] [PubMed] [Google Scholar]
- Taylor IR, Dunyak BM, Komiyama T, Shao H, Ran X, Assimon VA, Kalyanaraman C, Rauch JN, Jacobson MP, Zuiderweg ERP, et al. (2018). High-throughput screen for inhibitors of protein–protein interactions in a reconstituted heat shock protein 70 (Hsp70) complex. J. Biol. Chem 293, 4014–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Glukhova A, Sexton PM, and Christopoulos A (2018). Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53. [DOI] [PubMed] [Google Scholar]
- Thompson AD, Dugan A, Gestwicki JE, and Mapp AK (2012). Fine-Tuning Multiprotein Complexes Using Small Molecules. ACS Chem. Biol 7, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C-J, Sol A del, and Nussinov R (2009). Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Mol. Biosyst 5, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh STR (2010). A biosensor study indicating that entropy, electrostatics, and receptor glycosylation drive the binding interaction between interleukin-7 and its receptor. Biochemistry 49, 8766–8778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S, and Buchner J (2002). Molecular Chaperones—Cellular Machines for Protein Folding. Angew. Chem. Int. Ed 41, 1098–1113. [DOI] [PubMed] [Google Scholar]
- Wang N, Majmudar CY, Pomerantz WC, Gagnon JK, Sadowsky JD, Meagher JL, Johnson TK, Stuckey JA, Brooks CL, Wells JA, et al. (2013). Ordering a dynamic protein via a small-molecule stabilizer. J. Am. Chem. Soc 135, 3363–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, and Prywes R (2000). Activation of ATF6 and an ATF6 DNA Binding Site by the Endoplasmic Reticulum Stress Response. J. Biol. Chem 275, 27013–27020. [DOI] [PubMed] [Google Scholar]
- Wightman R, Bell R, and Reece RJ (2008). Localization and Interaction of the Proteins Constituting the GAL Genetic Switch in Saccharomyces cerevisiae. Eukaryot. Cell 7, 2061–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, and Sexton PM (2013). Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov 12, 630–644. [DOI] [PubMed] [Google Scholar]
- Wu T, Yu J, Gale-Day Z, Woo A, Suresh A, Hornsby M, and Gestwicki JE (2020). Three Essential Resources to Improve Differential Scanning Fluorimetry (DSF) Experiments. BioRxiv 2020.03.22.002543.
- Yang W, Hong YH, Shen X-Q, Frankowski C, Camp HS, and Leff T (2001). Regulation of Transcription by AMP-activated Protein Kinase PHOSPHORYLATION OF p300 BLOCKS ITS INTERACTION WITH NUCLEAR RECEPTORS. J. Biol. Chem 276, 38341–38344. [DOI] [PubMed] [Google Scholar]
- Yeger-Lotem E, Sattath S, Kashtan N, Itzkovitz S, Milo R, Pinter RY, Alon U, and Margalit H (2004). Network motifs in integrated cellular networks of transcription–regulation and protein–protein interaction. Proc. Natl. Acad. Sci 101, 5934–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika A, Marchenko N, and Moll UM (1999). Cytoplasmically “Sequestered” Wild Type p53 Protein Is Resistant to Mdm2-mediated Degradation. J. Biol. Chem 274, 27474–27480. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xiong B, Zhen X, and Zhang A (2009). Dopamine D1 receptor ligands: where are we now and where are we going. Med. Res. Rev 29, 272–294. [DOI] [PubMed] [Google Scholar]
- Zucconi BE, Luef B, Xu W, Henry RA, Nodelman IM, Bowman GD, Andrews AJ, and Cole PA (2016). Modulation of p300/CBP Acetylation of Nucleosomes by Bromodomain Ligand I-CBP112. Biochemistry 55, 3727–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]