

ABSTRACT
Meiosis is the process by which haploid gametes are produced from diploid precursor cells. We used stable isotope labeling by amino acids in cell culture (SILAC) to characterize the meiotic proteome in the fission yeast Schizosaccharomyces pombe. We compared relative levels of proteins extracted from cells harvested around meiosis I with those of meiosis II, and proteins from premeiotic S phase with the interval between meiotic divisions, when S phase is absent. Our proteome datasets revealed peptides corresponding to short open reading frames (sORFs) that have been previously identified by ribosome profiling as new translated regions. We verified expression of selected sORFs by Western blotting and analyzed the phenotype of deletion mutants. Our data provide a resource for studying meiosis that may help understand differences between meiosis I and meiosis II and how S phase is suppressed between the two meiotic divisions.
KEYWORDS: Meiosis, fission yeast Schizosaccharomyces pombe, silac, short open reading frames
Introduction
Sexual reproduction depends on meiosis, a process that generates haploid gametes from a diploid precursor cell. The fission yeast Schizosaccharomyces pombe is a useful model organism for studying meiosis. One of the advantages of using fission yeast is that highly synchronous meiosis can be induced by inactivation of the Pat1 protein kinase [1–4]. Moreover, a broad spectrum of genomic and proteomic tools is available. Progression of meiosis is accompanied by complex changes of gene expression [5]. These changes in fission yeast meiosis have been studied by various approaches including transcriptional profiling using DNA microarrays and ribosome profiling to investigate the translational landscape [6,7].
A comprehensive study analyzing changes of the S. pombe meiotic proteome using stable isotope labeling by amino acids in cell culture (SILAC) was published during the course of our work [8]. Krapp et al. quantified 3268 proteins throughout fission yeast meiosis induced by the inactivation of a temperature-sensitive allele of the Pat1 kinase (pat1-114) and found that the levels of 880 proteins changed at least 2-fold. Their study revealed a high degree of post-transcriptional regulation of protein levels and a global switch from anabolic to catabolic processes during meiosis [8].
In our current work, we performed SILAC based quantitative analysis of the S. pombe proteome during meiosis. In addition to pat1-114-induced meiosis, we used an improved synchronization protocol based on chemical inactivation of an ATP analog–sensitive form of the Pat1 kinase (pat1-as2), which eliminates negative effects of the higher temperature needed to inactivate the Pat1-114 kinase. We not only analyzed standard proteins, but also proteins encoded by short open reading frames (sORFs), which are usually defined as proteins smaller than 100 amino acids. Such sORFs were often ignored during genome annotations to minimize false positive ORFs [9–11]. However, recent analyses have revealed numerous examples of proteins encoded by sORFs that have important cellular functions [12,13]. We searched our SILAC based mass-spectrometry data and found peptides corresponding to novel sORFs that have been previously identified by ribosome profiling. We verified expression of selected sORFs by Western blot analysis and performed phenotypical characterization of deletion mutants. Finally, we discuss gene organization at the corresponding genomic regions and relevant refinements in the annotation of the fission yeast genome.
Results and discussion
SILAC based analysis of the meiotic proteome
There are several important differences between meiosis I (MI) and meiosis II (MII). Chiasma formation, mono-orientation of sister kinetochores, and protection of centromeric cohesion are key aspects of MI chromosomes that are absent during MII. In addition, MI is preceded by an S phase during which DNA is replicated but there is no S phase between MI and MII [14,15]. Quantitative comparison of the proteomes of various meiotic stages allows identification of proteins whose levels are differentially regulated. Such proteins that are specifically present or absent during a particular stage of meiosis may be important regulators of meiosis-specific processes. To identify such regulators, we used SILAC based proteome analysis to compare relative levels of proteins present during various stages of meiosis. SILAC labeling combined with high-resolution mass spectrometry is one of the key methods for quantitative proteomics [16] that is also available for fission yeast [8,17].
We used SILAC based proteome analysis in synchronous meiotic cultures induced by inactivation of a temperature-sensitive allele pat1-114 to compare relative levels of proteins present during premeiotic S phase (meiS) with the interval between meiotic nuclear divisions (MI-II), when S phase is absent (Figure 1, Table S1). Although this synchronization protocol based on the inactivation of a temperature-sensitive allele of the Pat1 kinase (pat1-114) has been widely used to study meiosis in the fission yeast S. pombe, it is not ideal for studying meiotic divisions because of chromosome missegregation defect [18,19]. Previous studies showed that pat1-114-induced meiosis differs from wild-type meiosis in some aspects, such as chromosome segregation. Whereas in wild-type cells sister centromeres segregate to the same pole in anaphase I, in meiosis induced by inactivation of Pat1-114 by elevated temperature sister centromeres segregate to the same pole very inefficiently in anaphase I cells [18,19]. In order to overcome this obstacle, we have developed a synchronization protocol based on pat1-as2. Chemical inactivation of an ATP analog–sensitive form of the Pat1 kinase (pat1-as2) by adding the ATP analog 1-NM-PP1 allows the induction of synchronous meiosis without the need of elevated temperature. In pat1-as2-induced meiosis, chromosomes segregate with higher fidelity and spore viability is higher than in pat1-114 meiosis [2–4]. We used pat1-as2-induced meiotic cultures to compare relative levels of proteins extracted from cells harvested around MI with those of MII (Figure 1, Table S1).
Figure 1.
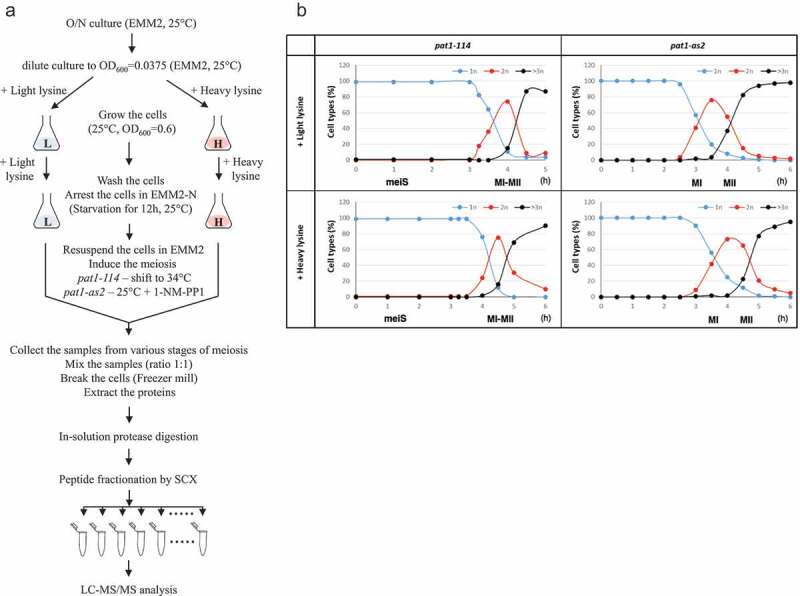
Flowchart of the SILAC based proteome analysis and progression of pat1-induced meiosis.
(a) Unlabeled (+ light lysine) and heavy lysine (+ heavy lysine) labeled diploid S. pombe cells were arrested by nitrogen starvation and meiosis was induced by shifting the cells to 34°C (pat1-114) or by adding 1-NM-PP1 (pat1-as2). Unlabeled and heavy lysine labeled cells from various stages of meiosis were mixed in equal amounts and protein extracts were prepared. After digestion with lysC protease, peptides were fractionated by strong cation exchange (SCX) and analyzed by mass spectrometry (LC-MS/MS). (b) Meiotic cells as described in (a) were fixed, stained with DAPI and nuclei were counted in 100 cells per time point. Shown are the fractions of cells that contained one nucleus (1n), two nuclei (2n) or more than two nuclei (3n or more) at the indicated time points after meiosis induction (hours). MeiS, MI-MII, MI and MII indicate when cells were harvested for the SILAC based proteome analysis.
To exclude possible isotope effects of the heavy 13C6 lysine and differences between batches of labeled amino acids, we performed experimental replicates with reversed labels. Unlabeled meiS was analyzed with heavy lysine labeled MI-MII transition (meiS (L) + MI-MII (H)) and heavy lysine labeled meiS was analyzed with unlabeled MI-MII transition (meiS (H) + MI-MII (L)). Similarly, unlabeled MI was analyzed with heavy lysine labeled MII (MI (L) + MII (H)) and heavy lysine labeled MI was analyzed with unlabeled MII (MI (H) + MII (L)) (Table S1). Normalized ratios (heavy/light) of peptides corresponding to selected proteins involved in DNA replication and chromosome segregation are shown in Table 1.
Table 1.
Normalized ratios (heavy/light) of peptides corresponding to selected proteins involved in DNA replication and chromosome segregation. Nda2 and Act1 are used as controls.
| Proteins involved in DNA replication | MI-MII (heavy lysine)/meiS (light lysine) | meiS (heavy lysine)/MI-MII (light lysine) |
|---|---|---|
| Pcn1 (proliferating cell nuclear antigen PCNA) | 0,91 | 1,10 |
| Cdc45 (DNA replication pre-initiation complex subunit) | 0,94 | 1,11 |
| Mcm4 (MCM replicative helicase complex subunit) | 0,97 | 1,02 |
| Mcm7 (MCM replicative helicase complex subunit) | 0,70 | 1,68 |
| Pol1 (DNA polymerase alpha catalytic subunit) | 0,78 | 1,37 |
| Mrc1 (mediator of replication checkpoint) | 0,16 | 3,41 |
| Tos4 (FHA-containing DNA binding protein) | 0,07 | 6,02 |
| Dfp1 (Hsk1-Dfp1 kinase complex regulatory subunit) | 8,98 | 0,16 |
| Nda2 (alpha tubulin) | 1,06 | 0,92 |
| Act1 (actin) | 0,98 | 0,99 |
| Proteins involved in chromosome segregation |
MII (heavy lysine)/ MI (light lysine) |
MI (heavy lysine)/MII (light lysine) |
| Top2 (DNA topoisomerase II) | 0,97 | 1,10 |
| Sgo2 (shugoshin) | 0,95 | 0,94 |
| Mad1 (spindle assembly checkpoint protein) | 1,01 | 1,09 |
| Cnd2 (condensin complex subunit) | 1,43 | 0,76 |
| Rec8 (cohesin complex subunit) | 0,13 | 5,10 |
| Spo4 (protein kinase) | 14,90 | 0,11 |
| Nda2 (alpha tubulin) | 1,01 | 0,95 |
| Act1 (actin) | 1,00 | 0,96 |
Verification of proteins encoded by sORFs identified by ribosome profiling
Ribosome profiling of S. pombe diploid cells undergoing meiosis identified 373 sORFs encoding short proteins that were at least 30 amino acids long that have not been previously described (Table S2) [7]. These included short proteins in ncRNAs, unannotated regions and 5ʹ-UTRs (encoded in uORFs). We searched our SILAC based mass-spectrometry results for peptides corresponding to these 373 novel sORFs identified by ribosome profiling and found unique peptides corresponding to nine sORFs (Figure 2(a), Figure S1, Table S3).
Figure 2.
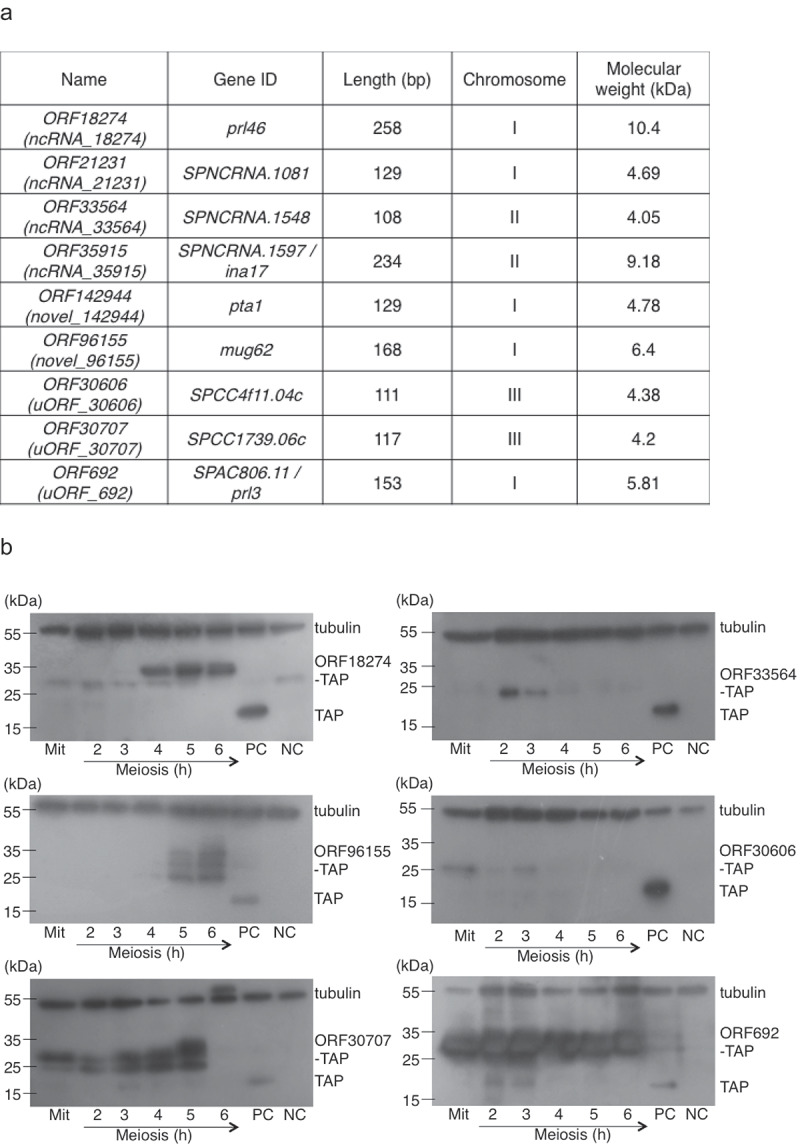
Verification of proteins encoded by novel sORFs identified by ribosome profiling.
(a) List of sORFs for which corresponding peptides were identified by mass-spectrometry. (b) pat1-114 cells expressing indicated TAP-tagged proteins were arrested by nitrogen starvation and released into meiosis at 34°C (Figure S2). Cells were harvested at the indicated time points (hours) after meiosis induction and protein extracts were analyzed by Western blotting. Protein extracts were also prepared from cycling vegetative cells (Mit). As a positive control (PC), protein extracts were prepared from a pool of cells expressing TAP tag alone, harvested at 4, 5 and 6 hours after meiosis induction. As a negative control (NC), protein extracts were prepared from a pool of wild-type cells, harvested 2–6 hours after meiosis induction. TAP tag was detected using rabbit antiperoxidase antibody linked to peroxidase and tubulin was detected using mouse-anti-α-tubulin antibody. Molecular weight marker (kDa) is indicated on the left. Additional Western blots are shown in Figure S3.
Next, we constructed strains expressing C-terminally TAP-tagged ORF18274, ORF33564, ORF96155, ORF30606, ORF30707 and ORF692. Western blot analyses revealed bands of expected sizes but also additional bands (Figure 2(b), Figure S2, Figure S3). Further experiments are needed to clarify what these additional bands represent. While all six TAP-tagged proteins were detected in meiotic extracts, ORF30606-TAP, ORF30707-TAP and ORF692-TAP were present also in extracts from vegetative cells (Figure 2(b), Figure S2, Figure S3). ORF18274-TAP (Prl46-TAP) and ORF692-TAP (Prl3-TAP) were independently constructed and detected by Western blotting by Duncan and Mata [7]. However, we noticed that ORF18274-TAP is larger than originally described [7]. This is probably due to extended N-terminus, which starts already before the beginning of the non-coding RNA prl46.
During the course of this work, there were changes in annotations of three sORFs [11]. ORF35915 has been annotated as the second exon of the ina17 gene [20] and ORF96155 as the first exon of the mug62 gene [7]. Detailed analysis of the sequence variants identified an indel error that affected the gene structure annotation of pta1, whose coding sequence was extended at the 3ʹ-end and included ORF142944 [21]. Thus, it is likely that ORF35915, ORF142944 and ORF96155 do not encode independent short proteins but they are part of larger genes (Figure S1).
Phenotypical characterization of sORFs deletion mutants
We analyzed the consequences of deleting ORF18274, ORF35915, ORF142944, ORF30707 and ORF692. In a haploid S. pombe strain, we were able to delete ORF18274, ORF35915, ORF30707, ORF692 but not ORF142944. Tetrad analysis of a diploid strain heterozygous for ORF142944 deletion showed that spores carrying ORF142944 deletion germinated but did not form colonies (data not shown). This result is consistent with the finding that ORF142944 is part of the pta1 gene, which is essential for cell growth [10,21].
We next analyzed phenotypes of ORF18274Δ, ORF35915Δ, ORF30707Δ and ORF692Δ deletion strains. Mutant vegetative cells showed no apparent growth defects or altered cell morphology (data not shown). The growth of mutant cells was similar to wild type in the presence of DNA damaging agents such as methyl methanesulfonate, camptothecin, hydroxyurea, zeocin and menadione (Figure 3(a)). Chromosome segregation in mutant cells, as scored by GFP labeled centromere of chromosome I, was similar to wild type during both mitosis and meiosis (data not shown). Spore viability in mutant strains was similar to wild type, suggesting that there is no major meiotic defect in mutant cells (Figure 3(b,c)). We conclude that ORF18274, ORF35915, ORF30707 and ORF692 are dispensable for vegetative growth under all tested conditions and production of viable spores.
Figure 3.
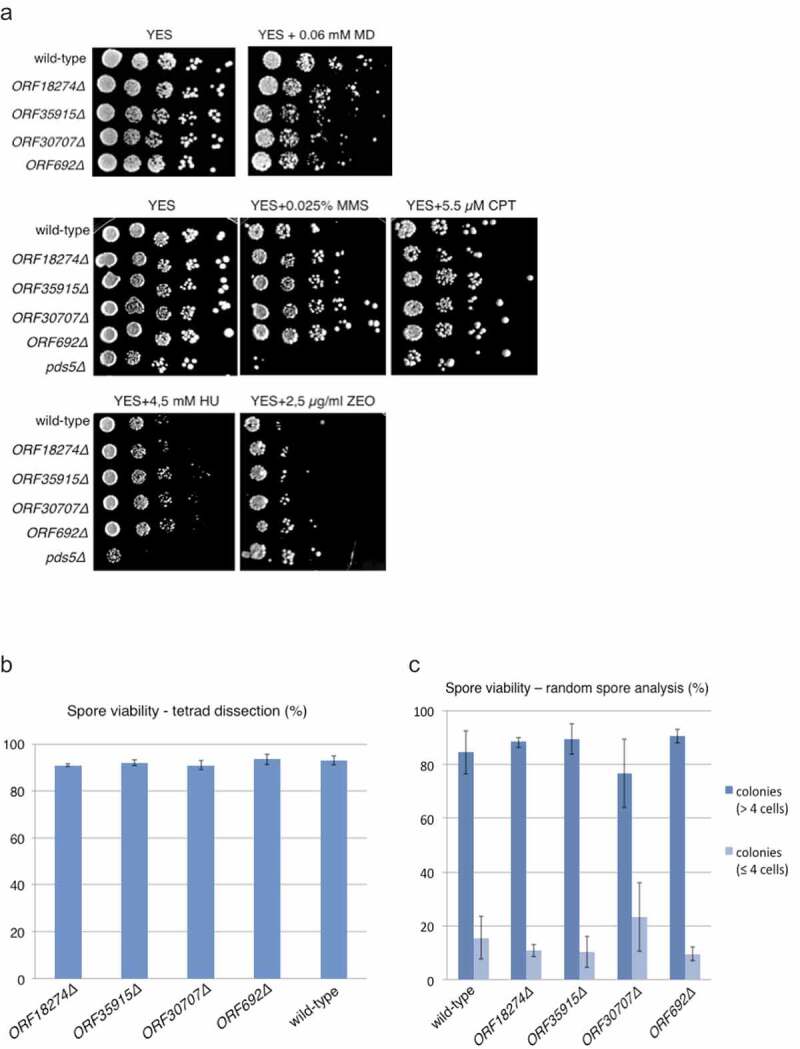
ORF18274Δ, ORF35915Δ, ORF30707Δ and ORF692Δ cells are not sensitive to DNA damaging agents and produce viable spores.
(a) Cells were grown on YES medium for one day, diluted and spotted onto YES plates containing the indicated amounts of menadione (MD), methyl methanesulfonate (MMS), camptothecin (CPT), hydroxyurea (HU) or zeocin (ZEO). Plates were incubated for 3 days at 32°C. pds5Δ was used as a control that is known to be sensitive to DNA damaging agents. (b) Spore viability of the indicated strains was measured by tetrad dissection in two independent experiments. 80 tetrads were dissected for each strain. (c) Spore viability of the indicated strains was measured by random spore analysis. 100,000 spores were plated per plate, incubated for 30 hours at 32°C and 200 spores/colonies were scored in at least two independent experiments. Microcolonies consisting of up to four cells and colonies containing more than four cells were scored.
Taken together, we performed SILAC based quantitative analysis of the S. pombe proteome during meiosis. Our results provide a resource for studying meiosis that may help understand differences between MI and MII and how S phase is suppressed between the two meiotic divisions. Our meiotic proteome datasets revealed unique peptides corresponding to only nine sORFs, out of 373 sORFs that have been previously identified by ribosome profiling as new translated regions. It is possible that more of these short proteins are present in meiotic and/or vegetative S. pombe cells, however their reliable detection will require more sensitive analyses including enrichment for short proteins before the mass-spectrometry analysis. Our results are consistent with previous findings that ORF35915, ORF142944 and ORF96155 do not encode independent short proteins but they are part of larger genes. They are also consistent with the notion that ORF21231, ORF18274, ORF33564, ORF30606, ORF30707 and ORF692 encode short proteins. However, we cannot exclude the possibility that these six sORFs are also part of larger genes. While we detected ORF21231 only by mass-spectrometry, ORF18274, ORF33564, ORF30606, ORF30707 and ORF692 were detected by both mass-spectrometry and Western blotting. The role of these short proteins remains unknown. Future experiments should include detailed analyses of mutant phenotypes and sensitive in silico searches to assess possible conservation of identified sORFs during evolution. Identification of sORFs that encode proteins and deciphering their roles are important future goals arising from our current work and other proteomic and ribosome profiling studies.
Materials and methods
Strain construction
We constructed TAP-tagging plasmids containing long regions homologous to the target gene according to our protocol described in Cipak et al. [22] for all nine sORFs (ORF18274, ORF21231, ORF33564, ORF35915, ORF142944, ORF96155, ORF30606, ORF30707 and ORF692). We transformed these plasmids into a haploid S. pombe strain JG12017 and verified successful tagging in yeast transformants by PCR. We constructed strains expressing C-terminally TAP-tagged ORF18274, ORF33564, ORF96155, ORF30606, ORF30707 and ORF692 but not ORF142944, where no yeast transformants were obtained. Western blot analyses revealed bands of expected sizes but also additional bands (Figure 2(b), Figure S2, Figure S3). No bands were observed in extracts prepared from strains carrying ORF21231-TAP and ORF35915-TAP (data not shown).
Genotypes of strains and the figures and tables in which each was used are in Table S4. Genes were deleted as described in Gregan et al. [23]. Spore viability was determined as described in Phadnis et al. [24].
Meiotic synchronization and SILAC labeling
Diploid S. pombe strains carrying temperature sensitive pat1-114 (JG16328) or ATP analog-sensitive pat1-as2 (JG16419) were incubated at 25°C over-night in EMM2 liquid medium (3.0 g/l potassium hydrogen phthalate, 2.2 g/l Na2HPO4, 5.0 g/l NH4Cl, 1.0% (w/v) glucose, 75 mg/l lysine, supplemented with salts, vitamins and minerals) [2,3]. The cells were collected by centrifugation, diluted in fresh EMM2 medium supplemented with 75 mg/l lysine (light sample) or 75 mg/l heavy lysine (heavy sample) into OD600 = 0.0375 and grown at 25°C until OD600 = 0.5–0.6. After centrifugation the cells were washed 3 times with deionized water, resuspended in EMM2-N medium (3.0 g/l potassium hydrogen phthalate, 2.2 g/l Na2HPO4, 1.0% (w/v) glucose, supplemented with salts, vitamins and minerals) and incubated at 25°C for 12 h to arrest the cells in G1 phase. Arrested cells were centrifuged and resuspended in the same volume of EMM2 medium supplemented with 75 mg/l lysine (light sample) or 75 mg/l heavy lysine (heavy lysine labeled sample). Meiosis was induced by shifting the cells to 34°C (pat1-114) or by adding 1-NM-PP1 (Toronto Research Chemicals) to 25 μM and incubated at 25°C (pat1-as2). Unlabeled and heavy lysine labeled cells from various stages of meiosis were collected by filtration through 0.45 μm membrane disc filter (Pall Corporation). Cells were frozen in liquid nitrogen and disrupted by Cryogenic Grinder (6775 Freezer/Mill Cryogenic Grinder, SPEX SamplePrep).
Heavy lysine was purchased from Cambridge Isotope Laboratories (U-13C6, CLM-2247-0.25), TRIzole Reagent from Invitrogen (15,596–026, 100 ml), GN-6 Metricel MCE Membrane Disc Filters from Pall Corporation (66,265, 47 mm, plain, sterile) and Magnetic Filter Funnels from Pall Corporation (4242, 47 mm, 300 mL capacity).
Protein extraction
Yeast powders from light sample (0.1 g) and heavy lysine labeled sample (0.1 g) isolated from particular stages of meiosis were mixed and resuspended in 6 ml of TRIzol reagent. Proteins were extracted by vigorous shaking at 4°C for 15 min. The sample was centrifuged at 12,000 g for 15 min at 4°C to remove insoluble material. Supernatant was incubated for 5 min at RT, extracted with chloroform (ratio TRIzol to chloroform was 5:1) and centrifuged at 12,000 g for 15 min at 4°C. Organic phase containing DNA and proteins was collected and DNA was precipitated by mixing the supernatant with 1.8 ml 100% ethanol and centrifuged at 2000 g for 5 min at 4°C. Phenol-ethanol supernatant was collected and mixed with 9 ml of isopropanol to precipitate the proteins. The proteins were collected by centrifugation at 12,000 g for 10 min at 4°C and washed 3 times by 0.3 M guanidine hydrochloride in 95% ethanol and 1 time in 95% ethanol. Vacuum dried proteins were dissolved in 1 ml of 8 M urea supplemented with 0.5 M NH4HCO3 for 1 h at RT.
Mass spectrometry analysis
Protein extracts were reduced with DTT and then alkylated with iodoacetamide. Protein solution was diluted with water to 6 M urea and then digested with LysC (Wako) at 1:30 ratio at 37°C overnight. The digests were desalted and lyophilized, then dissolved and chromatographically separated on a strong cationic exchanger (SCX) with a mixed salt- and pH-gradient in 15% acetonitrile (ACN). Up to 70 fractions were collected and ACN was removed by sample concentration in the speed vac. The peptide fractions were separated in a second dimension on a C18 column on a nano HPLC (Dionex, Thermo Scientific) applying 1 hour gradient. Eluting peptides were analyzed on a QExactive Orbitrap (Thermo Scientific) in a data-dependent mode. The 12 most intense peptides in the survey scan recorded at 70,000 resolution at 200 m/z, were subjected to CID fragmentation with 30% collision energy. CID spectra were recorded at 17,500 resolution and an AGC target value of 5E4. The MS data were searched with MaxQuant 1.4 [25]. against the S. pombe reference database (https://www.pombase.org, 2013–03-19) and the sequences of the sORFs (Table S2) with the following settings: LysC specificity, carabamidomethylation on Cys as fixed, oxidation of methionin and acetylation of protein N-termini as variable modification. The SILAC quan node was selected with 13C6 lysine as the heavy label. All other parameters were set to default. Results were filtered on protein and peptide level for a 1% FDR.
Western blot analysis
Proteins were separated by electrophoresis through 12% polyacrylamide gels containing SDS (0.1%) and transferred to a PVDF membrane (Immobilon-P membrane with 0.45 μm pore size from Millipore). The membrane was blocked with 2% (w/v) milk-PBS-T (phosphate buffer saline buffer with 0.1% (v/v) Tween-20) and probed with antibodies. TAP-tagged proteins were detected using rabbit antiperoxidase antibody linked to peroxidase (PAP, Dako; 1:10,000 dilution) in 0.1% PBS-T (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.1% (v/v) Tween-20). Tubulin was detected using mouse-anti-α-tubulin antibody (Sigma-Aldrich T5168; 1:10,000 dilution) and rabbit anti-mouse IgG-HRP secondary antibody (Santa Cruz Biotechnology; 1:5000 dilution) in 2% (w/v) milk PBS-T.
Supplementary Material
Acknowledgments
We thank K. Gaplovska-Kysela, L. Molnarova, S. Kearsey, B. Brejova, T. Vinar, K. Elsayad and J. Loidl for their help.
Funding Statement
This work was supported by the Slovak Grant Agency VEGA (1/0450/18, 2/0039/19, 2/0034/19 and 2/0026/18), Slovak Research and Development Agency (APVV-17-0130, APVV-18-0219 and APVV-16-0120), Biotechnology and Biological Sciences Research Council Grant BB/M021483/1 and the Austrian Science Fund (FWF): P30516; Agentúra na Podporu Výskumu a Vývoja [APVV-18-0219]; Agentúra na Podporu Výskumu a Vývoja [APVV-17-0130]; Agentúra na Podporu Výskumu a Vývoja [APVV-16-0120]; Austrian Science Fund [P30516]; Vedecká Grantová Agentúra MŠVVaŠ SR a SAV [2/0039/19]; Vedecká Grantová Agentúra MŠVVaŠ SR a SAV [1/0450/18]; Vedecká Grantová Agentúra MŠVVaŠ SR a SAV [2/0026/18].
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
B.H., J.K., S.B.P., L.C. and D.A. performed experiments. C.D., J.M., G.A., A.S., Z.B., S.B.P., L.C. and J.G. analysed the data and prepared the manuscript.
Supplementary material
Supplementary data for this article can be accessed here.
References
- [1].Bahler J, Schuchert P, Grimm C, et al. Synchronized meiosis and recombination in fission yeast: observations with pat1-114 diploid cells. Curr Genet. 1991;19:445–451. [DOI] [PubMed] [Google Scholar]
- [2].Cipak L, Polakova S, Hyppa RW, et al. Synchronized fission yeast meiosis using an ATP analog-sensitive Pat1 protein kinase. Nat Protoc. 2014;9:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cipak L, Hyppa RW, Smith GR, et al. ATP analog-sensitive Pat1 protein kinase for synchronous fission yeast meiosis at physiological temperature. Cell Cycle. 2012;11:1626–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guerra-Moreno A, Alves-Rodrigues I, Hidalgo E, et al. Chemical genetic induction of meiosis in Schizosaccharomyces pombe. Cell Cycle. 2012;11:1621–1625. [DOI] [PubMed] [Google Scholar]
- [5].Otto GM, Brar GA.. Seq-ing answers: uncovering the unexpected in global gene regulation. Curr Genet. 2018;64:1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mata J, Lyne R, Burns G, et al. The transcriptional program of meiosis and sporulation in fission yeast. Nat Genet. 2002;32:143–147. [DOI] [PubMed] [Google Scholar]
- [7].Duncan CD, Mata J. The translational landscape of fission-yeast meiosis and sporulation. Nat Struct Mol Biol. 2014;21:641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Krapp A, Hamelin R, Armand F, et al. Analysis of the S. pombe meiotic proteome reveals a switch from anabolic to catabolic processes and extensive post-transcriptional regulation. Cell Rep. 2019;26:1044–1058 e1045. [DOI] [PubMed] [Google Scholar]
- [9].Spirek M, Benko Z, Carnecka M, et al. S. pombe genome deletion project: an update. Cell Cycle. 2010;9:2399–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kim DU, Hayles J, Kim D, et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2010;28:617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lock A, Rutherford K, Harris MA, et al. PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res. 2019;47:D821–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hollerer I, Higdon A, Brar GA. Strategies and challenges in identifying function for thousands of sORF-encoded peptides in meiosis. Proteomics. 2018;18:e1700274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Herberg S, Gert KR, Schleiffer A, et al. The Ly6/uPAR protein bouncer is necessary and sufficient for species-specific fertilization. Science. 2018;361:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marston AL, Amon A. Meiosis: cell-cycle controls shuffle and deal. Nat Rev Mol Cell Biol. 2004;5:983–997. [DOI] [PubMed] [Google Scholar]
- [15].Hua H, Namdar M, Ganier O, et al. Sequential steps in DNA replication are inhibited to ensure reduction of ploidy in meiosis. Mol Biol Cell. 2013;24:578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. [DOI] [PubMed] [Google Scholar]
- [17].Macek B, Carpy A, Koch A, et al. Stable isotope labeling by amino acids in cell culture (SILAC) technology in fission yeast. Cold Spring Harb Protoc. 2017;2017:pdb top079814. [DOI] [PubMed] [Google Scholar]
- [18].Yamamoto A, Hiraoka Y. Monopolar spindle attachment of sister chromatids is ensured by two distinct mechanisms at the first meiotic division in fission yeast. Embo J. 2003;22:2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kakui Y, Sato M, Tanaka K, et al. A novel fission yeast mei4 mutant that allows efficient synchronization of telomere dispersal and the first meiotic division. Yeast. 2011;28:467–479. [DOI] [PubMed] [Google Scholar]
- [20].Jimenez J, Duncan CD, Gallardo M, et al. AnABlast: a new in silico strategy for the genome-wide search of novel genes and fossil regions. DNA Res. 2015;22:439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hu W, Suo F, Du LL. Bulk segregant analysis reveals the genetic basis of a natural trait variation in fission yeast. Genome Biol Evol. 2015;7:3496–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cipak L, Spirek M, Novatchkova M, et al. An improved strategy for tandem affinity purification-tagging of Schizosaccharomyces pombe genes. Proteomics. 2009;9:4825–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gregan J, Rabitsch PK, Rumpf C, et al. High-throughput knockout screen in fission yeast. Nat Protoc. 2006;1:2457–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Phadnis N, Cipak L, Polakova S, et al. Casein kinase 1 and phosphorylation of cohesin subunit rec11 (SA3) promote meiotic recombination through linear element formation. PLoS Genet. 2015;11:e1005225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.