

ABSTRACT
Reduced autophagy has been implicated in aging, yet whether its loss can promote aging phenotypes and pathologies in mammals, and how reversible this process is, has never been fully explored. Using inducible short hairpin RNA (shRNA) mouse models, we have recently shown that autophagy inhibition accelerates aging, and that even a temporary block in autophagy can create irreversible damage that increases a cancer risk.
KEYWORDS: Autophagy, senescence, aging, cancer
Aging is associated with a loss of proteostasis, organelle homeostasis, as well as an accumulation of damaged and misfolded aggregates1 Thus, it is no surprise that increasing age is associated with a corresponding decrease in autophagy. Whilst conventional and conditional knockout mouse models of essential autophagy genes have provided important insights into the role of autophagy in development and disease, aging studies have been difficult to perform either due to the associated neonatal lethality, or the rapid adult neurotoxicity that inevitably occurs with systemic autophagy loss.2,3
Despite the technical limitations to explore autophagy and aging in mammalian systems, numerous studies have provided supportive evidence that regimens intended to promote autophagy, both through lifestyle (dietary restriction) and pharmacological modulation, can promote health-span.4,5 However, these approaches are pleiotropic in nature, often promoting autophagy through upstream mechanisms that impinge upon additional signaling cascades. Recently, two genetic studies have shown that by promoting autophagy constitutively, mice display an increased health-span and reduced rates of spontaneous tumorigenesis.6,7 Together these data provide compelling evidence that pharmacological modulation of autophagy may have broad-spectrum health benefits.
In our recent publication, we attempted to provide answers to some of the outstanding questions that remain in the field of autophagy and mammalian aging. The first was simply, is a systemic reduction in autophagic flux enough to promote aging and age-related phenotypes in a mammalian system? The second was, if yes then are these effects reversible by restoration of autophagy? This latter point is of importance as it argues for the correct timepoint of intervention. Is it possible to reverse pathological phenotypes, or is it more effective to prevent the damage from ever accruing?
To circumvent the limitations imposed by traditional (systemic) knockout mice, we employed doxycycline-inducible short hairpin RNA (shRNA) models to achieve body-wide temporal control of autophagy through the targeting of Autophagy-related 5 (Atg5), an essential autophagy component (termed ‘Atg5i’ mice). A major caveat of our inducible system is that doxycycline is poorly permeable across the blood-brain-barrier and as such the system shows no activation
in the brain, leaving autophagy intact in this organ.8 We realized that this discrepancy of system activation provided us with a unique opportunity to perform long-term, ‘systemic’ autophagy experiments without the associated neurotoxic effects seen in whole-body knockout models (e.g. systemic Autophagy-related 7 [Atg7] knockout in adult mice leads to death between 2 to 3 months3). Furthermore, we have developed two models with shRNAs of different inhibition efficiencies. The original Atg5i mice provided a knockdown efficiency that generated gross phenotypes akin to those seen in knockout mice (hepatomegaly and splenomegaly),8 whilst the second hairpin mouse line (termed Atg5i-2) was hypomorphic, leading to a reduction in autophagy without these obvious associated pathologies.9
In our latest work, Atg5i mice were aged until eight-weeks old, to separate out developmental from tissues homeostatic effects, before being placed on a doxycycline-containing diet to inhibit autophagy.9 As expected, although they survive premature death due to the neuronal toxicity, mice with autophagy inhibition still displayed dramatically reduced life-spans compared to normal mice in both the shRNAs tested (Atg5i median survival 185 days). Furthermore, they displayed evidence of phenotypes that occur with increasing age such as graying, kyphosis, loss of muscle mass, and evidence of autoimmunity. Molecular markers of aging, including inflammatory cytokines, telomere-associated DNA damage, and markers of senescence in various tissues were also elevated in our autophagy inhibited mice. Together, the study provides evidence that loss of autophagy systemically induces molecular, cellular, and organismal phenotypes that are strongly associated with increasing age. Whilst we were unable to determine a singular cause of death to contributed to mortality in our Atg5i mice (as the cause of death appears to be non-uniform), we view this as a positive outcome for the model. Aging is a complex and multifaceted process that is associated with some degree of stochasticity, this process at least appears to be recapitulated in our Atg5i mice. Note similar aging phenotypes were observed in Atg5i-2 (median survival 240 days), suggesting that a modest decrease in autophagy reduction is sufficient for age acceleration.
Using the Atg5i mice, we next approached our second question and inhibited autophagy in mice for four months, wherein Atg5i mice would universally display aging phenotypes and an increase in frailty. At this timepoint, we removed doxycycline from the diet and restored autophagy levels. Strikingly, mice showed a gross recovery in frailty and a dramatic recovery in their health- and life-span. However, we also noted that kyphosis and graying phenotypes were largely, if not completely, irreversible, whilst the molecular damage that had accumulated (such as telomere-associated DNA damage) was also irremediable. Thus, although encouraging from a translatable aspect, it is important to note that not all damage induced by autophagy loss was reversible in this model. Furthermore, we noted that this temporal period of reduced autophagy was associated with an increased incidence of spontaneous tumor formation, suggesting that this irreversible damage can be tumorigenic in nature. We speculate that preventing damage through the promotion of autophagy would be more beneficial than late-stage restoration.
Overall our study provides missing data further linking autophagy and mammalian aging, whilst at the same time exploring the timing and efficacy of autophagy restoration therapies on aging pathologies. Importantly our work also provides supportive evidence for a biphasic role of autophagy in cancer development (Figure 1). Wherein a reduction in autophagy is associated with the accumulation of pro-tumorigenic damage and possibly the establishment of premalignant disease without further progression, at least in the liver as previously reported.10 Once the irreversible damage has accumulated, a tumorous cell that has reestablished autophagic flux would undoubtedly have a fitness advantage, as autophagy is important for basic cellular homeostasis. Although it should be noted that our work does not conclusively prove this and further studies would be required. These results also raise the specter of long-term cancer risk in the setting of early life autophagy inhibition, such as childhood obesity. Further studies, both in mice and epidemiologically in humans, would add welcome real-world data to support or refute these possibilities.
Figure 1.
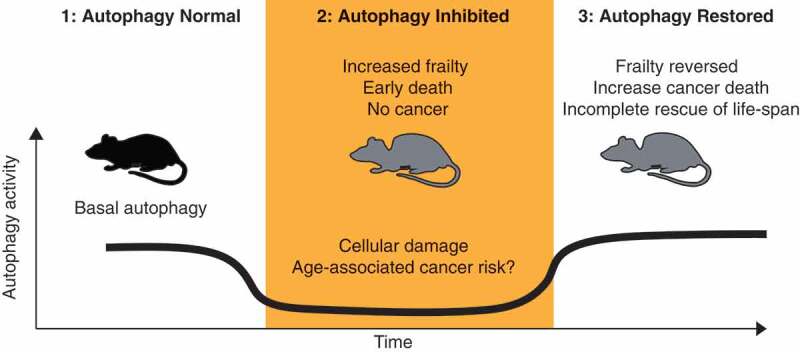
Potential impact of autophagy modulation on aging and tumorigenesis. (1) Autophagy is required to maintain normal tissue and organismal homeostasis. (2) Loss of autophagy induces premature aging phenotypes, which are associated with cellular dysfunction. (3) The restoration of autophagy leads to the gross recovery of cellular and organismal homeostasis. However, some effects are irreversible, such as genomic damage. We speculate that the accumulation of this irreversible damage may accelerate the development of spontaneous tumor formation in the presence of the intact autophagy flux.
Funding Statement
This work was funded by the Cancer Research UK Cambridge Institute Core Grant [C9545/A29580].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.López-Otín C, Galluzzi L, Freije JMP, Madeo F, Kroemer G.. Metabolic control of longevity. Cell. 2016;166:1–3. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 2.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 3.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 5.Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125:85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyo J-O, Yoo S-M, Ahn -H-H, Nah J, Hong S-H, Kam T-I, Jung S, Jung Y-K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4(1):2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, He C, Ting T, Liu Y, Chiang W-C, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–140. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassidy LD, Young AR, Pérez-Mancera PA, Nimmervoll B, Jaulim A, Chen H-C, McIntyre DJO, Brais R, Ricketts T, Pacey S, et al. A novel Atg5-shRNA mouse model enables temporal control of Autophagy in vivo. Autophagy. 2018;14:1256–1266. doi: 10.1080/15548627.2018.1458172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassidy LD, Young ARJ, Young CNJ, Soilleux EJ, Fielder E, Weigand BM, Lagnado A, Brais R, Ktistakis NT, Wiggins KA, et al. Temporal inhibition of autophagy reveals segmental reversal of ageing with increased cancer risk. Nat Commun. 2020;11(1):307. doi: 10.1038/s41467-019-14187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]