

ABSTRACT
Growing evidence shows that autophagy is deficient in neurodegenerative and psychiatric diseases, and that its induction may have beneficial effects in these conditions. However, as autophagy shares signaling pathways with cell death and interferes with protein synthesis, prolonged use of autophagy inducers available nowadays is considered unwise. The search for novel autophagy inducers indicates that DRD2 (dopamine receptor 2)-DRD3 ligands may also activate autophagy, though critical aspects of the action mechanisms and effects of dopamine ligands on autophagy are still unknown. In order to shed light on this issue, DRD2- and DRD3-overexpressing cells and drd2 KO, drd3 KO and wild-type mice were treated with the DRD2-DRD3 agonist pramipexole. The results revealed that pramipexole induces autophagy through MTOR inhibition and a DRD3-dependent but DRD2-independent mechanism. DRD3 activated AMPK followed by inhibitory phosphorylation of RPTOR, MTORC1 and RPS6KB1 inhibition and ULK1 activation. Interestingly, despite RPS6KB1 inhibition, the activity of RPS6 was maintained through activation of the MAPK1/3-RPS6KA pathway, and the activity of MTORC1 kinase target EIF4EBP1 along with protein synthesis and cell viability, were also preserved. This pattern of autophagy through MTORC1 inhibition without suppression of protein synthesis, contrasts with that of direct allosteric and catalytic MTOR inhibitors and opens up new opportunities for G protein-coupled receptor ligands as autophagy inducers in the treatment of neurodegenerative and psychiatric diseases.
Abbreviations
AKT/Protein kinase B: thymoma viral proto-oncogene 1; AMPK: AMP-activated protein kinase; BECN1: beclin 1; EGFP: enhanced green fluorescent protein; EIF4EBP1/4E-BP1: eukaryotic translation initiation factor 4E binding protein 1; GPCR; G protein-coupled receptor; GFP: green fluorescent protein; HEK: human embryonic kidney; MAP1LC3/LC3: microtubule-associated protein 1 light chain 3; MAP2K/MEK: mitogen-activated protein kinase kinase; MAPK1/ERK2: mitogen-activated protein kinase 1; MAPK3/ERK1: mitogen-activated protein kinase 3; MDA: malonildialdehyde; MTOR: mechanistic target of rapamycin kinase; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PPX: pramipexole; RPTOR/raptor: regulatory associated protein of MTOR, complex 1; RPS6: ribosomal protein S6; RPS6KA/p90S6K: ribosomal protein S6 kinase A; RPS6KB1/p70S6K: ribosomal protein S6 kinase B1; SQSTM1/p62: sequestosome 1; ULK1: unc-51 like autophagy activating kinase 1; WT: wild type.
KEYWORDS: AMPK, dopamine agonists, EIF4EBP1/4E-BP1, G protein-coupled receptors, MTORC1, neuropsychiatric diseases, protein synthesis, ribosomal protein S6, RPS6KA/p90S6K
Introduction
Autophagy is a lysosomal degradation process essential for preserving cellular homeostasis that works in three ways: macroautophagy, chaperone-mediated autophagy and microautophagy. Macroautophagy/autophagy is responsible for the degradation of misfolded proteins through successive steps regulated by MTOR (mechanistic target of rapamycin kinase)-dependent or MTOR-independent mechanisms [1]. Autophagic activity is particularly important in the nervous system because neurons are post-mitotic cells and cannot reduce the toxicity of anomalous proteins through cell division. Studies in the last decade indicate that autophagy disruption is a central player in the formation of aggregate-prone proteins that cause neuronal death in neurodegenerative diseases [2–4]. More recently, autophagy dysfunction has also been related to psychiatric conditions such as schizophrenia and depression [5,6]. So, autophagy activation is regarded as a promising therapeutic strategy in neurodegenerative and psychiatric disorders. The benefit of autophagy activation has been demonstrated in different models of neurodegenerative diseases using the MTOR inhibitor rapamycin and MTOR-independent autophagy inducers [7–9]. However, as autophagy shares signaling pathways with apoptosis, autophagy may also lead to cell death under particular circumstances [10].
The search for novel autophagy inducers indicates that compounds with affinity for DRD2 (dopamine receptor D2) and DRD3 (dopamine receptor D3) can regulate autophagy [11]. These findings open up the possibility of inducing autophagy in selective neuronal populations using DRD2-DRD3 ligands. However, data about the effects of DRD2-DRD3 ligands on autophagy are still fragmentary and, in some aspects, contradictory. For example, autophagy activation has been reported after treatment with DRD2-DRD3 agonists [12,13] but also with antagonists [11,14]. Furthermore, DRD2-DRD3-induced autophagy has been associated with MTOR inhibition in some studies [13] but not in others [12,14]. These discrepancies may arise in part from differences in the experimental design. For example, different doses and times of treatment have been used, and some studies were performed in cells where DRD2-DRD3 expression has not been appropriately established. It should also be noted that DRD2-DRD3 agonists can act through dopamine receptor-independent mechanisms [15], and that some molecules can either induce or inhibit autophagy in a dose-dependent manner [16,17].
DRD2 and DRD3 are expressed in midbrain dopaminergic neurons, medium spiny neurons of the striatum, and different neuronal populations in the cerebral cortex [18–20]. This condition makes them a key target in the treatment of motor symptoms derived from the loss of striatal dopamine in Parkinson disease, and hallucinations and maniac episodes in schizophrenia and psychosis, using agonists or antagonists, respectively [21–23]. Pramipexole (PPX) is a DRD2-DRD3 agonist used as first line treatment in Parkinson disease [24] that has also demonstrated efficacy as an antidepressant in bipolar and depressive patients [25,26]. Some data indicate that PPX may be an autophagy modulator. Li and coworkers [27] firstly found an increase in the number of autophagic vacuoles in the brain of mice treated with PPX. Thereafter, in vitro experiments proposed that autophagy can be activated by PPX through DRD2 and DRD3 [12]. In order to shed further light on the relationship between dopamine DRD2 and DRD3 and autophagy, here drd2 KO, drd3 KO and wild-type (WT) mice, and DRD2- and DRD3-transfected cells were treated with PPX at the lowest dose signaling through DRD2-DRD3. The results show that prolonged PPX treatment activates autophagy through a DRD3-dependent, but a DRD2-independent mechanism, and MTORC1 inhibition. However, MTORC1 downstream targets involved in protein synthesis, RPS6 (ribosomal protein S6) and EIF4EBP1/4E-BP1, were not affected.
Results
Pramipexole induces autophagy through a DRD3-dependent DRD2-independent mechanism
In order to establish the lowest dose of PPX having dopaminergic effects, a range of them were tested in mice and cells. WT mice received a single intraperitoneal injection of 0.01–1.5 mg/kg PPX and were sacrificed 10 min later (Figure S1A and B), and DRD2- and DRD3-HEK cells were incubated in 0.1–100 μM PPX for 5 min after 30 min serum starvation (Figure S1C and D). Following previous studies in rodents [28,29], AKT phosphorylation at Thr308 was used as a readout of DRD2-DRD3 activation in mouse striata. However, since AKT is constitutively activated in HEK cells, MAPK1/3 phosphorylation is more useful for monitoring activation of dopamine receptors in these cells [29,30]. AKT phosphorylation was detected in mouse striata at doses of 0.15 mg/kg and 1.5 mg/kg, and the effect was blocked by pre-treatment with the DRD2-DRD3 antagonist raclopride (1 mg/kg, i.p., 30 min before PPX; Figure S1A). MAPK1/3 phosphorylation was detected at 10 μM and 100 μM PPX in DRD2- and DRD3-transfected cells (Figure S1C). So, 0.15 mg/kg and 10 μM were considered as the lowest PPX doses having dopaminergic effects in mice and HEK cells, respectively, and were used throughout the study.
Autophagy was first assessed by western blotting for LC3 and SQSTM1/p62. MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) is detected in two bands in western-blots corresponding to LC3-I (cytosolic), and LC3-II (conjugated to phosphatidylethanolamine and present in the inner and outer membranes of autophagosomes). LC3-II levels are considered an index of the autophagosome number, but in high autophagy flux conditions, enhanced autophagosome degradation is paralleled by a decrease in LC3-II levels [31,32], making it necessary to block the autophagosome-lysosome fusion to reliably detect an increase in autophagosome formation using LC3-II as readout [31–34]. In addition, the demand of LC3-II for the synthesis of new autophagosomes may use LC3-I thereby reducing its levels. Therefore, although LC3-I is not a marker of autophagy, a decrease of both LC3-I and LC3-II in western-blot reinforces evidence for autophagy activation [31–34]. SQSTM1 (sequestosome1 [35]) is an ubiquitin-binding scaffold protein that serves as a link between LC3 and ubiquitinated substrates. Since SQSTM1 is incorporated into autophagosomes and degraded with its ubiquitinated substrates by lysosomes, the decrease in SQSTM1 is used as a marker of autophagy activation [31,35].
LC3 and SQSTM1 expressions were not altered by PPX after a single injection in mice or 5 min incubation in DRD2- and DRD3-cells (Figure S1B and D). However, when PPX treatment (0.15 mg/kg/d) was prolonged for 6 d, LC3-I and LC3-II fractions and SQSTM1 levels were significantly reduced in the striata. These changes were observed in WT and drd2 KO mice but not in drd3 KO mice Figure 1(a,b), suggesting that the effect is DRD3-dependent. Consistent with this idea, when PPX treatment was prolonged in HEK cells (10 µM, 3 h), both LC3 fractions and SQSTM1 levels were also reduced in DRD3-HEK cells, but not in DRD2- and non-transfected HEK cells Figure 1(c,d). No changes were detected in DRD2 and DRD3 expression in HEK transfected cells following 10 min and 3 h of PPX treatment (Figure S1E).
Figure 1.
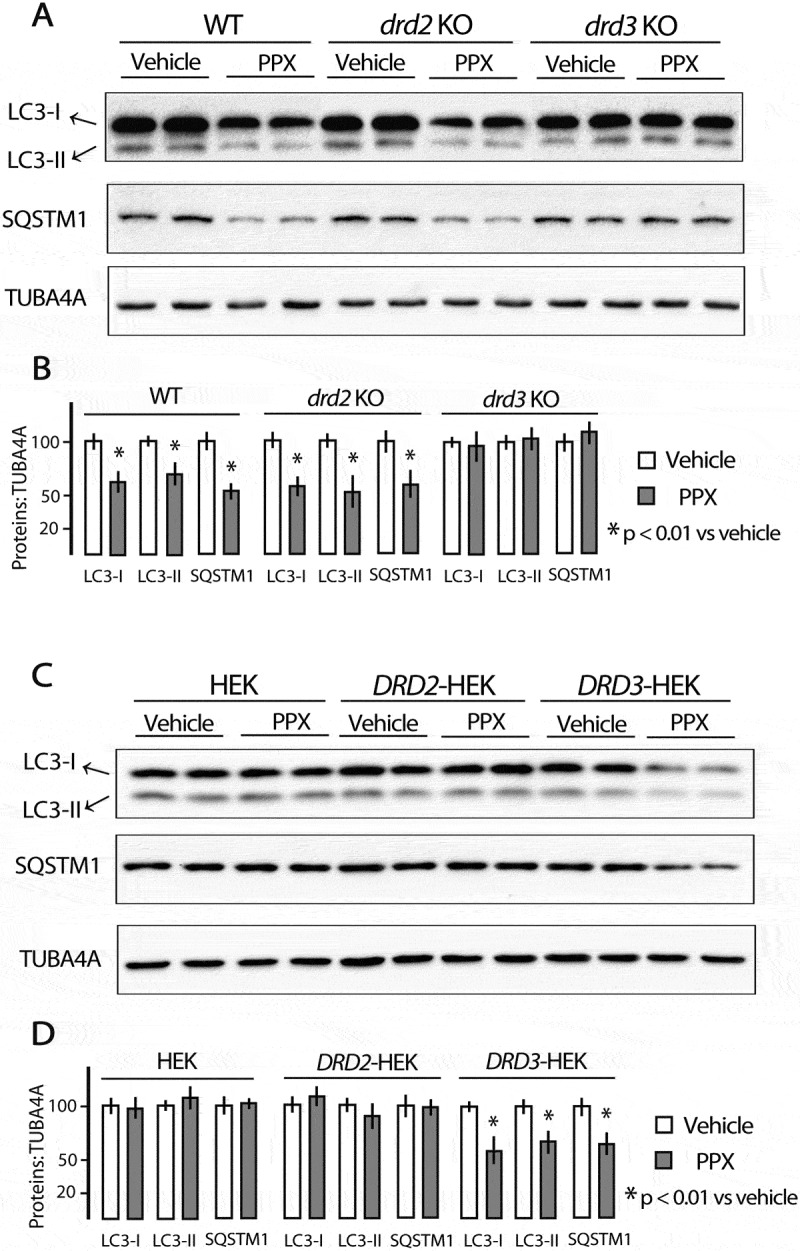
Prolonged PPX treatment induces DRD3-dependent decrease of LC3 and SQSTM1. Western blot for LC3 and SQSTM1 in the striatum of WT, drd2 KO and drd3 KO mice treated for 6 d and sacrificed at day 7 (A and B) and in untransfected and DRD2- and DRD3-transfected HEK cells (C and D). PPX treatment (0.15 mg/Kg/d/6 d) induced a decrease in LC3-I and LC3-II, and SQSTM1 in the striatum of WT and drd2 KO mice but not in drd3 KO mice. PPX (10 µM, 3 h) induces a decrease in LC3-I and LC3-II, and SQSTM1 in HEK cells expressing DRD3 but not in untransfected and DRD2-transfected cells.
Since the decrease in LC3-II may reflect an increase in autophagy flux or a decrease in autophagosome synthesis [31,34], additional experiments including autophagy blockade were performed in striatal neurons and transfected HEK cells. PPX treatment (10 µM, 3 h) promoted a decrease in both LC3 fractions in striatal neurons of WT but not of drd3 KO mice (Figure 2(a) [top and bottom], lanes 3 and 4, 2B). Blockade of autophagosome-lysosome fusion by chloroquine (20 µM, 1 h) led to an increase in the LC3-II fraction in striatal cells of both WT and drd3 KO mice (Figure 2(a) [top and bottom], lanes 5 and 6, 2B). However, the addition of 10 µM PPX 2 h, promoted an additional increase in the LC3-II fraction in WT mice (Figure 2(a) [top], lanes 7 and 8, 2B) but not in drd3 KO mice (Figure 2(a) [bottom], lanes 7 and 8, 2B). Similarly, the increase in LC3-II levels induced by PPX in WT striatal neurons pretreated with chloroquine was reversed by the DRD3 antagonist NGB2904 (1μM, 30 min before PPX) but not by the DRD2 antagonist L741,626 (1μM, 30 min before PPX; Figure S2A), indicating that PPX increases the autophagy flux in a DRD3-dependent manner. Consistent with these findings, chloroquine also increased LC3-II levels in DRD2- and DRD3-HEK cells (Figure 2(a) [top and bottom], lanes 5 and 6, 2D) but the addition of PPX (10 µM, 2 h) provoked an additional increase in LC3-II in DRD3-HEK cells but not in DRD2-HEK cells (Figure 2(c) [top and bottom], lanes 7 and 8; 2D). Furthermore, as shown in the Cyto-ID assay Figure 2(e,f), after blockade of autophagosome-lysosome fusion, PPX treatment produced a substantial increase in the number and size of autophagic vesicles only in DRD3-HEK cells (p < 0.001 vs. CQ, Figure 2(e,f) and Figure S2C). The large sized dots probably correspond to two or more autophagic vesicles localized close to one another.
Figure 2.
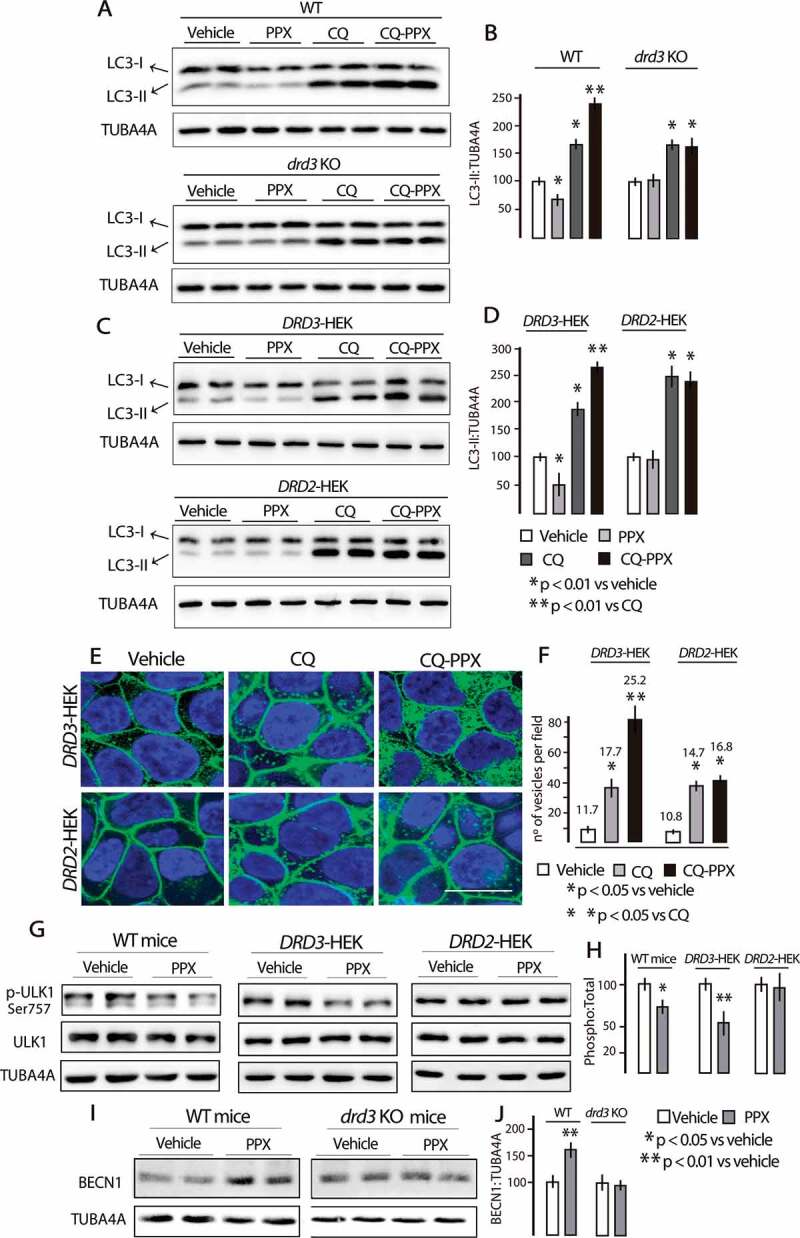
PPX promotes a DRD3-dependent increase in autophagy flux. (A-D) Western blot for LC3 in striatal cells of WT and drd3 KO mice (A and B) and in DRD3- and DRD2-HEK cells (C and D). PPX treatment (10 µM, 3 h) reduces the labeling intensity in both fractions of LC3 in striatal neurons of WT mice (lanes 3 and 4, A [top] and B) and DRD3-HEK cells (lanes 3 and 4, C [top] and D) but not in drd3 KO mice (lanes 3 and 4, A [bottom] and B) and DRD2-HEK cells (lanes 3 and 4, C [bottom] and D). Preincubation with chloroquine (CQ, 20 µM, 1 h) increases the labeling intensity of LC3-II in striatal cells of WT and drd3 KO mice (lanes 5 and 6, A [top and bottom] and B), and in DRD3- and DRD2-HEK cells (lanes 5 and 6, C [top and bottom] and D). The addition of PPX (10 µM, 2 h) promotes an additional increase of LC3-II in striatal cells of WT mice (lanes 7 and 8, A [top] and B) and DRD3-HEK cells (lines 7 and 8, C [top] and D), but not in striatal cells of drd3 KO mice (lanes 7 and 8, A [bottom] and B) and DRD2-HEK cells (lanes 7 and 8, C [bottom] and D). (E and F) Cyto-ID assay in DRD3- and DRD2-HEK cells. The number and size of autophagy vesicles also increased after PPX treatment in DRD3- but not in DRD2-HEK cells (compare CQ and CQ-PPX in E and F; see also Figure S2). Green fluorescent labeling in the plasma membrane of HEK-cells corresponds to EGFP-DRD3 and GFP-DRD2 expression. The numbers in F indicate the average size (pixels) of the cytosolic vesicles. Bar in E, 10 µm. (G and H) Western blot for ULK1 and pSer757-ULK1 in WT mice treated for 6 d (left panel) and in DRD3- and DRD2-HEK cells (10 µM, 3 h, middle and right panels). Ser757-ULK1 was dephosphorylated in WT mice and DRD3- but not in DRD2-HEK cells. (I and J) Western blot for BECN1 in WT- and drd3 KO mice. BECN1 levels increased in WT mice but not in drd3 KO mice.
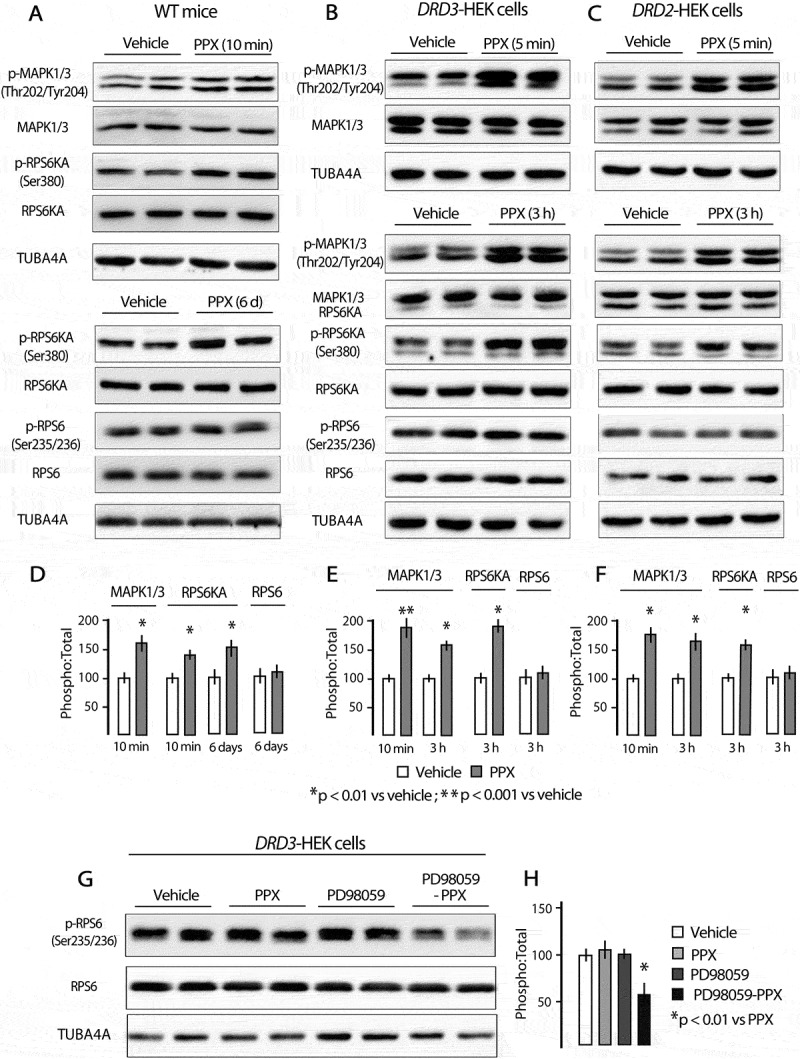
ULK1 (unc-51 like autophagy activating kinase 1) and BECN1 (beclin1) are the mammalian orthologs of ATG1 and ATG6, respectively, and two essential proteins for initiating autophagosome formation. Phosphorylation at Ser757 by MTOR maintains ULK1 inactive [36], and BECN1 is used as a marker of autophagy due to its role in the recruitment of other ATGs involved in autophagosome formation [31,37]. As shown in Figure 2(g,h), Ser757-ULK1 phosphorylation was significantly reduced after PPX treatment in WT mice (p < 0.05 vs. vehicle) and DRD3-HEK cells (p < 0.01 vs. vehicle) but not in DRD2-HEK cells. On the other hand, BECN1 levels were increased in WT (p < 0.01 vs. vehicle) but not in drd3 KO mice Figure 2(i,j). Taken together, these findings indicate that prolonged PPX treatment induces autophagy through a DRD3-dependent yet DRD2-independent mechanism.
Pramipexole inhibits MTORC1 through DRD3 and AMPK
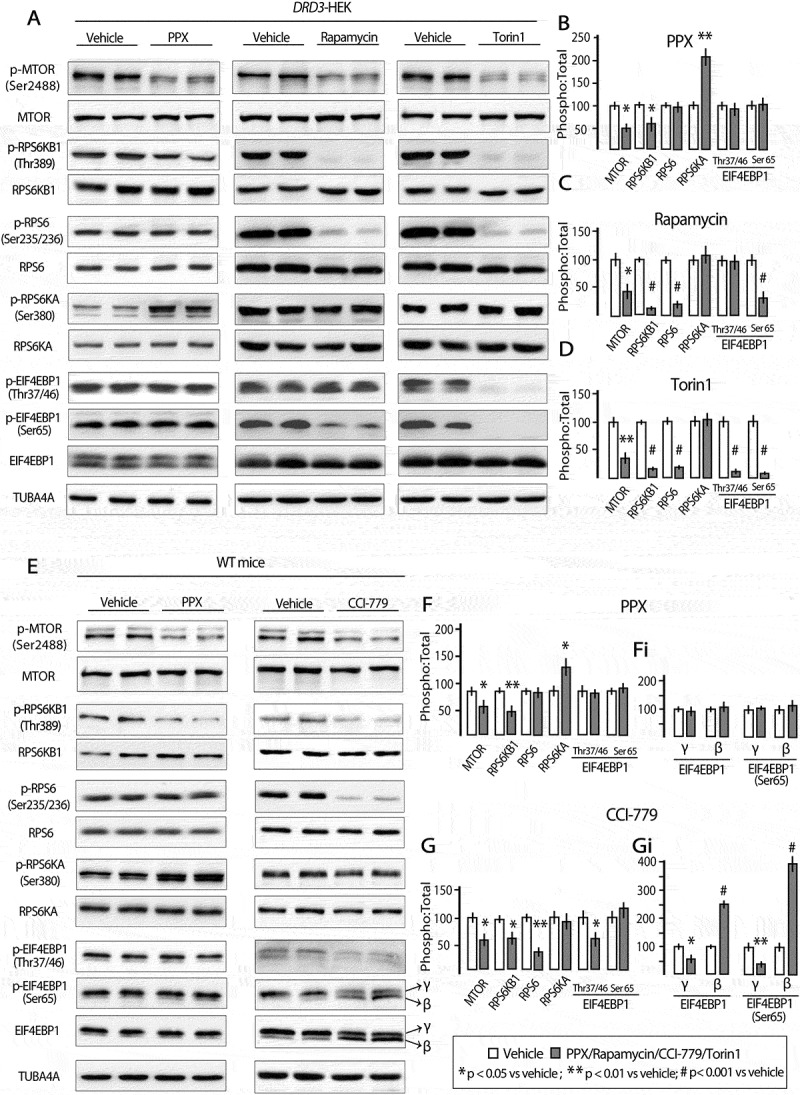
As mentioned above, autophagy may be activated by MTOR-dependent and MTOR-independent mechanisms. MTOR is the catalytic core of two functionally different multi-protein complexes known as MTORC1 and MTORC2 [38]. The classical pathway of autophagy is under the negative control of MTORC1 [1]. In starvation and rapamycin treatment, the MTOR kinase activity of MTORC1 is inhibited initiating the downstream signaling of autophagy through Ser757-ULK1 dephosphorylation [1,36]. The fact that Ser757-ULK1 is dephosphorylated in WT mice and DRD3-HEK cells after PPX treatment strongly suggests that PPX-induced autophagy is MTOR-dependent. As MTORC1 activity may be assessed by monitoring the phosphorylation status of MTOR at Ser2488 and its effector RPS6KB1/p70S6K (ribosomal protein S6 kinase-1) at Thr389 [39,40], these phosphorylations were investigated in PPX-treated mice and HEK cells. PPX (0.15 mg/kg/d/6d) promoted a significant decrease in MTOR and RPS6KB1 phosphorylation (pSer2488-MTOR:MTOR and pThr389-RPS6KB1:RPS6KB1 ratios; p < 0.01 vs. vehicle) in the striatum of WT and drd2 KO mice, but not in that of drd3 KO mice Figure 3(a,b). In addition, phosphorylation of MTOR and RPS6KB1 were also reduced after PPX treatment (10 µM, 3 h) in DRD3-HEK cells (p < 0.01 vs. vehicle) but not in DRD2- and non-transfected HEK cells Figure 3(c,d), confirming that MTORC1 inhibition is DRD3-dependent.
Figure 3.
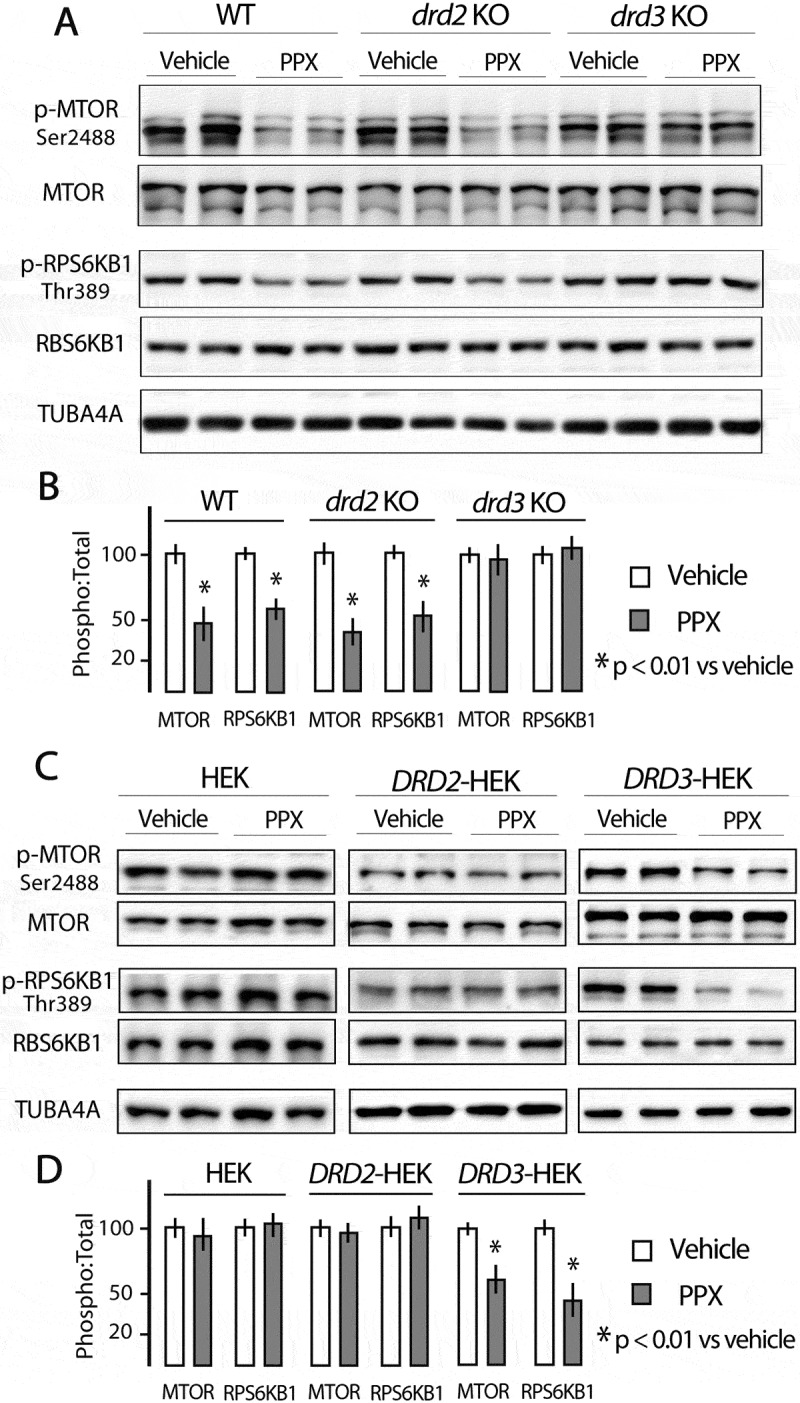
Prolonged PPX treatment promotes DRD3-dependent MTORC1 inhibition. Western blot for total and phosphorylated forms of MTOR at Ser2488 (p-MTOR) and its effector RPS6KB1/p70S6K (ribosomal protein S6 kinase) at Thr389 (p-RPS6KB1) in the striatum of WT, drd2 KO and drd3 KO mice (A and B), and in untransfected and DRD2- and DRD3-transfected HEK cells (C and D). The expression of the phosphorylated form of MTOR (p-MTOR) and RPS6KB1 (p-RPS6KB1) is significantly lower in the striatum of WT and drd2 KO mice and in DRD3-transfected HEK cells after PPX treatment, but not in the striatum of drd3 KO mice, and in untransfected and DRD2-transfected cells.
As parts of the same pathway, it is to be expected that MTOR inhibition may be a consequence of AKT dephosphorylation. Consistent with previous studies by Salles et al. [28], acute administration of PPX in mice led to an increase of phosphorylation of both Thr308-AKT and Ser2488-MTOR (Figure S1A and 3). However, in contrast to MTOR, after 6 d of PPX treatment, Thr308-AKT phosphorylation returned to its basal levels but was not dephosphorylated compared to untreated mice (Figure S3). Therefore, these data suggest that the mechanism responsible for MTORC1 inhibition is AKT-independent.
One of the major regulators of MTORC1 is the key energy sensor AMP-activated protein kinase (AMPK). AMPK is a heterotrimeric protein complex formed by a catalytic α subunit and two regulatory, β and γ, subunits [41]. Its activation involves phosphorylation of Thr172 within the α subunit PRKAA [42]. This occurs when the AMP:ATP ratio increases, but also in response to multiple upstream signals, including G protein-coupled receptor activation [43]. AMPK suppresses MTOR kinase activity through direct phosphorylation of RPTOR/raptor (regulatory associated protein to MTOR) on two conserved serine residues, Ser722 and Ser792 [44–46]. Some reports indicate that DA receptors can modulate AMPK, and subsequently MTORC1 through raptor phosphorylation [47,48]. Accordingly, the possible involvement of AMPK on MTOR inhibition by PPX was explored in DRD3- and DRD2-HEK cells. As shown in Figure 4(a,b), PPX induced an increase in Thr172-PRKAA and Ser792-RPTOR phosphorylation in DRD3- (p < 0.01 vs. vehicle) but not in DRD2-HEK cells. Interestingly, the amount of RPTOR, and particularly its Ser792 phosphorylated form, recovered from immunoprecipitated MTOR also increased after PPX treatment in DRD3- but not in DRD2-cells Figure 4(c). Furthermore, the increase of MTOR-RPTOR interaction induced in DRD3-HEK cells was reversed by co-treatment with the selective DRD3 antagonist NGB2904 Figure 4(c). In sum, these findings indicate that MTORC1 inhibition induced by prolonged PPX treatment is mediated by the selective effects of DRD3 on AMPK activation and RPTOR-MTOR interaction.
Figure 4.
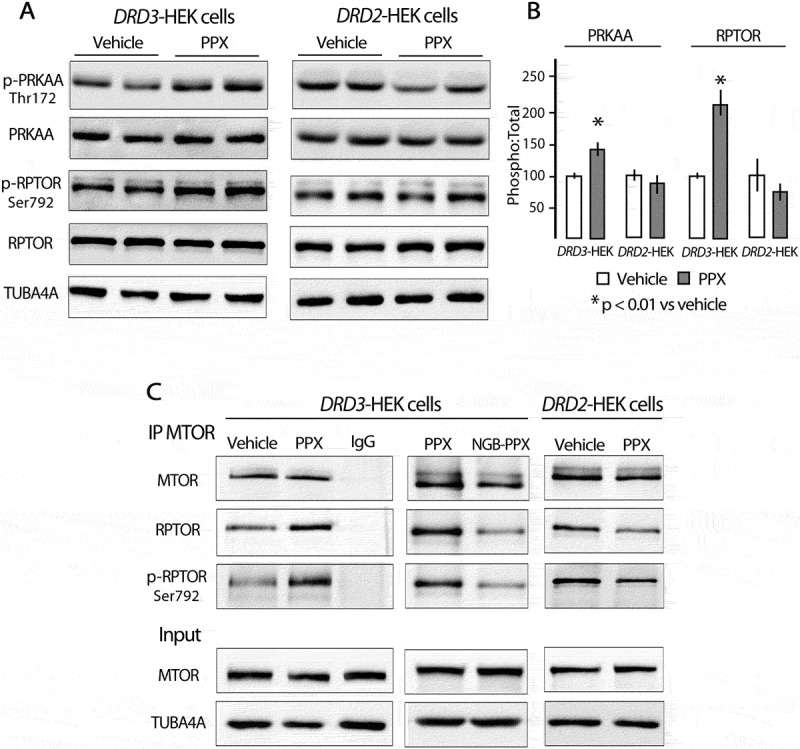
PPX promotes phosphorylation of PRKAA (Thr172) and RPTOR (Ser792), and strengthens RPTOR-MTOR interaction through DRD3 but not DRD2. (A and B) Western blot for total and phosphorylated forms of PRKAA at Thr172 (p-PRKAA) and RPTOR at Ser792 (p-RPTOR). (C) Immunoprecipitation for MTOR (IP MTOR) followed by western blot for RPTOR and p-RPTOR in DRD3- and DRD2-HEK cells. PRKAA and RPTOR phosphorylation, and the interaction RPTOR (p-RPTOR)-MTOR only increased in DRD3-HEK cells, and this effect was blocked by co-treatment with NGB2904.
Cellular response to autophagy and MTORC1 inhibition induced by pramipexole
It should be noted that although autophagy usually acts as a protector, under particular conditions, it can also lead to apoptosis or other forms of cell death [10]. In addition, MTORC1 is a central player in many cellular processes [40], and the loss of its kinase activity can interfere with the synthesis of essential proteins and promote apoptosis and oxidative stress-induced cell death [49]. In view of our findings, cell viability, activity (phosphorylation status) of MTORC1 downstream targets engaged in the control of mRNA translation, and protein synthesis were investigated in DRD3-HEK cells and mice treated with PPX and direct allosteric (rapamycin/CCI-779) or catalytic (torin1) MTORC1 inhibitors. The study of cell viability in DRD3-HEK cells included the colorimetric analysis of MTT tetrazolium reduction and clonogenic cell survival assays. Given the high sensitivity of neuronal membranes to lipid peroxidation in response to different conditions [50], MDA (malonildialdehyde) levels were also studied in mouse striata. As shown in Figure 5(a), cell viability was significantly reduced in DRD3-HEK cells after treatment with rapamycin or torin1 (0.01–10 μM, 72 h), but not after PPX treatment. Furthermore, the ability of cells to proliferate was substantially affected in cells treated with rapamycin but not in those treated with PPX (Figure S4). Further, striatal levels of MDA were significantly increased in mice treated with CCI-779 (20 mg/kg/48 h i.p. x4) but not in those treated with PPX (0.15 mg/kg/d, 6 d; Figure 5(b)). These findings indicate that, in contrast to direct MTOR inhibitors, MTOR inhibition and autophagy induced by PPX do not affect cell viability.
Figure 5.
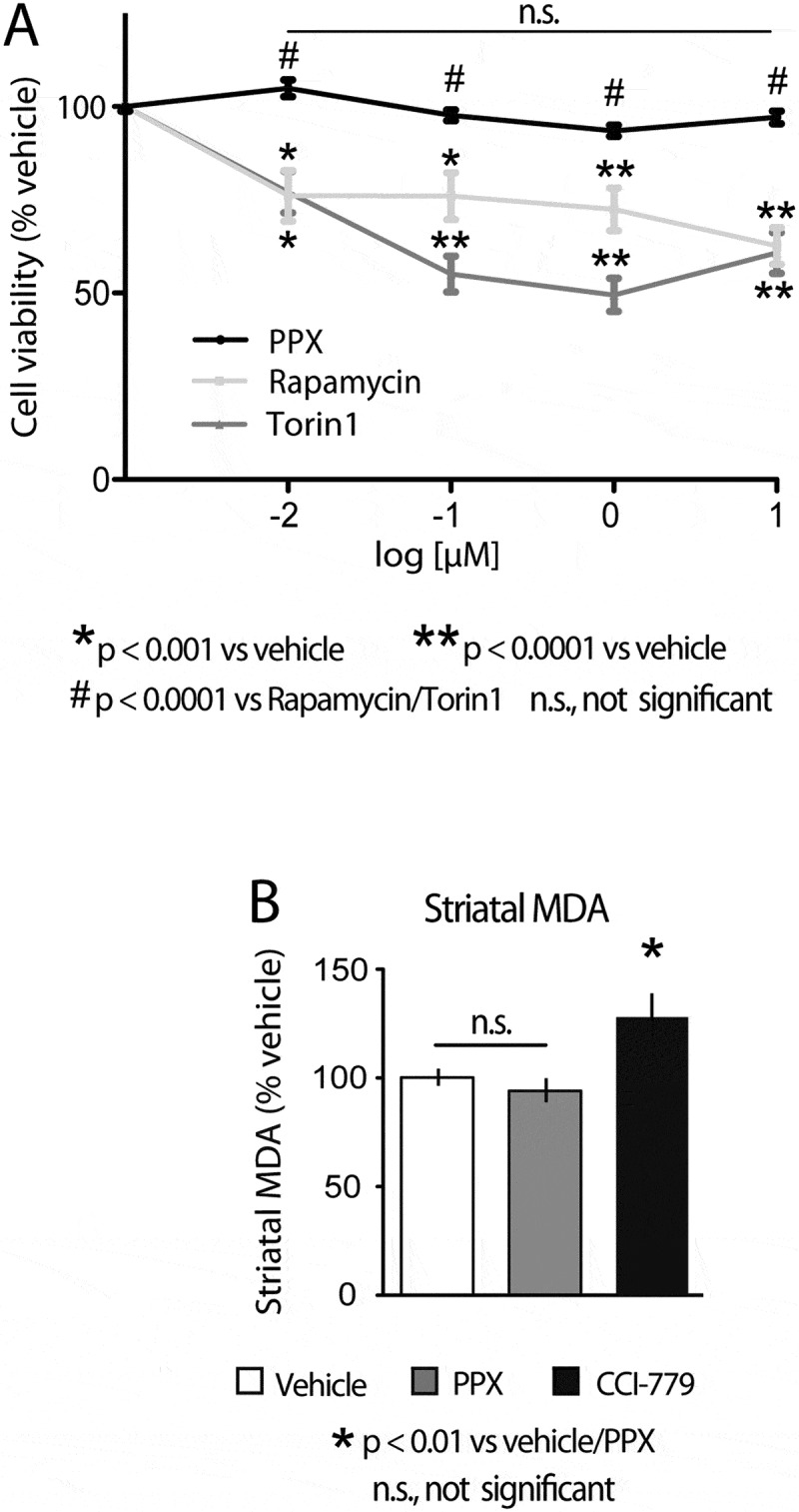
Cell viability is affected by direct MTOR inhibitors but not by DRD3-induced MTOR inhibition. (A) MTT assay in DRD3-HEK cells treated with rapamycin, torin 1 and PPX (0.01–10 µM, 72 h). Cell viability was significantly reduced in DRD3-HEK cells treated with rapamycin and torin 1 but not in cells treated with PPX. (B) Quantitative analysis of malonildialdehyde (MDA) in the striatum of WT mice treated with PPX (0.15 mg/kg/d/6 d) or CCI-779 (20 mg/kg/48 h i.p. x4). Striatal MDA levels were significantly increased in mice treated with CCI-779 but not in those treated with PPX.
Active MTORC1 promotes protein synthesis via phosphorylation of two main mRNA translation regulators, RPS6KB1 and EIF4EBP1/4E-BP1 (eukaryotic translation initiation factor 4E binding protein 1) [36,39,51]. RPS6KB1 is the main kinase of RPS6, phosphorylating four serine residues, Ser235, Ser236, Ser240 and Ser244, in its C-terminal domain [52]. Previous studies indicate that phosphorylation at Ser235 and Ser236 directly promote RPS6 interaction with the 5ʹ end of mRNA and polysome assembly, two steps required for translation initiation [53]. Interestingly, RPS6 phosphorylation at Ser235 and Ser236 was preserved in WT mice and DRD3-HEK treated with PPX despite RPS6KB1 dephosphorylation Figure 6(a,b). It should be noted that these serine residues may also be phosphorylated by the MTOR-independent MAPK1/ERK2-MAPK3/ERK1-downstream kinase RPS6KA/p90S6K/RSK (ribosomal protein S6 kinase A [53]). The fact that PPX treatment promotes acute MAPK1/3 activation in WT mice Figure 6(a) and in DRD3- and DRD2-HEK (Figure 6(b,c) and S1C), prompted us to explore whether activation of the MAPK1/3-RPS6KA pathway is maintained after prolonged PPX treatment as a compensatory response directed to preserve RPS6 activity. As shown in Figure 6(a,d), the initial increase in Thr202/Tyr204-MAPK1/3 phosphorylation in mouse striata was paralleled by an increase in Ser380-RPS6KA phosphorylation that was maintained after 6 d of PPX treatment (p < 0.01 vs. vehicle). Similarly, MAPK1/3 and RPS6KA phosphorylation was also increased after 3 h of PPX treatment in both DRD3- and DRD2-HEK cells (p < 0.01 vs. vehicle; Figure 6(b,c,e,f)). The fact that MAPK1/3-RPS6KA signaling is also activated in DRD2-HEK cells indicates that this effect is independent of AMPK activation and MTORC1 inhibition. The question is whether MAPK1/3-RPS6KA upregulation can contribute to maintain RPS6 activity in DRD3-HEK cells where RPS6KB1 is inhibited. To address this issue, DRD3-HEK cells were treated with PPX (10 μM, 3 h), the MEK inhibitor (indirect MAPK1/3 inhibitor) PD98059 (25 µM, 1 h; Figure S5), or both (PD98059-PPX). We found that similarly to what happens in PPX treated cells, RPS6 phosphorylation was not affected in cells treated with PD98059 Figure 6(g,h), confirming previous reports supporting that RPS6KB1 is sufficient to maintain RPS6 activity [51,52]. However, when cells treated with PPX also received PD98059 (PD98059-PPX; Figure 6(g)), RPS6 phosphorylation was significantly reduced (p < 0.01 vs. PPX). Accordingly, the decrease of RPS6KB1 activity induced by PPX can be compensated by the parallel MAPK1/3-RPS6KA upregulation induced through both DRD2 and DRD3.
Figure 6.
The MAPK1/3-RPS6KA pathway is activated through DRD3 and DRD2 maintaining RPS6 phosphoryation in PPX treated cells. (A-F) Western blot for total and phosphorylated forms of MAPK1/3 at Thr202/Tyr204 (p-MAPK1/3), RPS6KA at Ser380 (p-RPS6KA), and RPS6 at Ser235/236 (p-RPS6) in mice (A and D), DRD3- (B and E) and DRD2-HEK cells (C and F). MAPK1/3-RPS6KA signaling is acutely activated by PPX in WT mice and in DRD3- and DRD2-HEK cells, and the activity is maintained after prolonged treatment. (G and H) Western blot for RPS6 and its phosphorylated form at Ser235/236 in DRD3-HEK cells treated with either PPX, the MAPK1/3 inhibitor PD98059 or both (PD98059-PPX). RPS6 phosphorylation was impaired in PPX cells only when MAPK1/3 was inhibited.
EIF4EBP1 is a translational repressor that in its active hypophosphorylated form binds to the translation factor EIF4E, impeding its coupling to the 5ʹ end of mRNA. In contrast, inactive phosphorylated EIF4EBP1 releases EIF4E, allowing it to recruit the 5ʹ end of mRNA to initiate translation [51]. Four of the seven known phosphorylation sites in EIF4EBP1, Thr37, Thr46, Thr70 and Ser65, are important in EIF4EBP1 release, with phosphorylation at Thr37 and Thr46 acting as the priming event, and phosphorylation at Ser65 being the last [51,54]. Here we show that EIF4EBP1 phosphorylation at Thr37, Thr46 and Ser65 was preserved in DRD3-HEK cells Figure 7(a) and mouse striata Figure 7(e) after PPX treatment. However, as previously reported [55], a marked dephosphorylation of RPS6KB1 and RPS6 with partial dephosphorylation (Ser65) of EIF4EBP1 was found in rapamycin-treated cells (20 nM, 3 h; Figure 7(a)). Phosphorylation of RPS6KB1, RPS6 and EIF4EBP1 was also abolished by torin1 (250 nM, 3 h; Figure 7(a)). Likewise, MTOR, RPS6KB1, RPS6 and EIF4EBP1 were dephosphorylated in mice treated with the rapamycin analog CCI-779 (20 mg/kg/48 h i.p. x4). It should be mentioned that different degrees of EIF4EBP1 phosphorylation may be revealed by the presence of 2–3 bands in western-blot. Following Brunn et al. [56], these bands are designated as α, β and γ, with α corresponding to the hypophosphorylated form, and γ to the most phosphorylated form. In our study, bands β and γ were detected in mice Figure 7(e). Considered together, the labeling intensity of both bands decreased in pThr37/46-EIF4EBP1 but not in pSer65-EIF4EBP1 of CCI-779 treated mice Figure 7(e,g). However, when bands were analyzed individually, a molecular mass shift from γ to β was evident in both total- and pSer65-EIF4EBP1 forms Figure 7(e,g), indicating EIF4EBP1 dephosphorylation. Furthermore, in contrast to PPX, RPS6KA phosphorylation was not upregulated by rapamycin/CCI-779 and torin1.
Figure 7.
MTORC1 downstream targets involved in RNA translation are affected by rapamycin/CCI-779 and torin 1 but not by PPX. Western blot for MTOR, RPS6KB1, RPS6, RPS6KA and EIF4EBP1 and their phosphorylated forms at Ser2488, Thr389, Ser235/236, Ser380, and Thr37/46 and Ser65, respectively, in DRD3-HEK cells (A-D) and the striatum of WT mice (E-G) treated with PPX, rapamycin/CCI-779 and torin 1. In spite of MTOR and RPS6KB1 dephosphorylation, RPS6 and EIF4EBP1 phosphorylations are maintained, and RPS6KA is over-phosphorylated in DRD3-HEK cells and mice treated with PPX. However, RPS6 and EIF4EBP1 are dephosphorylated and RPS6KA phosphorylation is unchanged in rapamycin- and torin1-treated DRD3-HEKcells and CCI-779 treated mice. Dephosphorylation of EIF4EBP1 in its total and pSer65 forms is reflected by a molecular mass shift from the most phosphorylated band (γ) to the partially phosphorylated band (β; E and Gi).
To determine whether differences between the effects of PPX and direct MTOR inhibitors on MTORC1 downstream targets correlate with the impact on protein synthesis, the rate of protein synthesis was analyzed in DRD3-HEK cells using western blot for puromycin (SUnSET assay) and fluorescent detection of OPP (O-propargyl-puromycin) into nascent proteins. As shown in Figure 8(a,b), puromycin labeling was virtually blocked by the protein synthesis inhibitor cycloheximide (lane 5), indicating that puromycin exclusively labels nascent proteins. The incorporation of puromycin was also significantly reduced in torin 1 (p < 0.001 vs. vehicle) and rapamycin (p < 0.05 vs. vehicle) treated cells but not in PPX treated cells. Likewise, fluorescent OPP staining was severely affected in torin1 (p < 0.001 vs. vehicle) and rapamycin (p < 0.001 vs. vehicle) treated cells, but not in those treated with PPX Figure 8(c,d). Together, these data indicate that PPX inhibits MTORC1 and activates autophagy without affecting the translation machinery.
Figure 8.
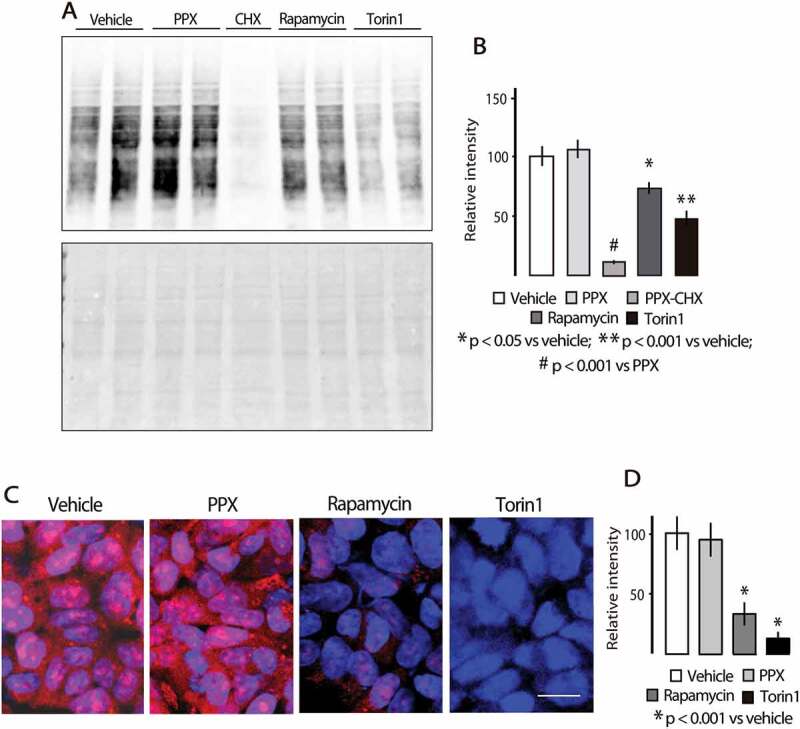
Protein synthesis is affected by rapamycin and torin1 but not by PPX. (A and B) Surface Sensing of Translation (SUnSET) western blot assay (top) and Ponceau S staining for detection of protein bands (bottom). (C and D) Fluorescent OPP Protein synthesis assay. The incorporation of puromycin in nascent proteins is not affected in PPX-treated DRD3-HEK cells, while it is inhibited in rapamycin- and torin1-treated cells. Protein extract of lane 5 in A comes from cells treated with the protein synthesis inhibitor cycloheximide (CHX, 10 µM, 3 h). Bar in C: 10 µm.
Discussion
Several studies in the last decades indicate that DRD2-DRD3 agonists have neuroprotective effects on dopaminergic neurons, and that these effects can be mediated by both dopaminergic and dopamine-independent actions [15,21]. Recently, neuroprotection of DRD2-DRD3 agonists has been associated with their ability to induce autophagy and reduce protein aggregation [12,27,57]. However, relevant aspects of this effect are still unknown. The present study shows that prolonged treatment with the DRD2-DRD3 agonist PPX induces autophagy through a DRD3-dependent DRD2-independent mechanism and MTORC1 inhibition without affecting MTORC1 downstream targets engaged in translation and protein synthesis.
The selective DRD3-dependence of the effect was demonstrated by different autophagy markers and autophagosome dynamics monitoring in drd2 KO and drd3 KO mice, striatal cell cultures and DRD2- and DRD3-transfected HEK cells. Considered separately, the decline of LC3-II induced by PPX in all the experimental models could be due to either an increase in autophagy flux or inhibition of autophagosome synthesis. However, the blockade of autophagosome-lysosome fusion in striatal cells and HEK transfected cells confirmed the increase in autophagy flux (see also [31,34]), and consequently, that autophagy is selectively activated through DRD3. Since PPX has a higher affinity for DRD3 than for DRD2 [58], one could propose that DRD3 selectivity is a result of the PPX dose used here was not being high enough to activate DRD2. However, MAPK1/3 signaling was activated through both receptors with the same dose of PPX in HEK-transfected cells. Hence, reasons other than receptor affinity must underlie the DRD3 selectivity of this effect.
Although displaying high amino acid sequence homology [59] and sharing several signaling pathways [59,60], DRD2 and DRD3 show differences in their action and regulation mechanisms. For example, with respect to the specific subunit of the G protein family of GPCR (G protein-coupled receptor) involved in adenylyl cyclase inhibition, experiments in HEK cells indicate that DRD2 uses the G subunit and DRD3 the βγ subunit [61]. Furthermore, whereas desensitization of DRD2 is strongly associated with GRK (G protein-coupled receptor kinase)-mediated receptor phosphorylation, interaction with ARRB (β-arrestin) and internalization, DRD3 undergoes PRKC (protein kinase C)-mediated phosphorylation, internalization and degradation, or translocation into membrane hydrophobic domains where it becomes less accessible to ligands [62,63]. Interestingly, studies in mouse striata and dopaminergic neurons have shown that the AKT-MTORC1 kinase pathway is transiently activated by DRD3 but not by DRD2 stimulation [28,64,65]. In the present study, the AKT-MTOR pathway was also activated a few minutes after PPX treatment. However, when treatment was prolonged, while MTOR phosphorylation decreased below basal levels, indicating inhibition of its kinase activity, AKT phosphorylation returned to basal levels, suggesting that the pathway is desensitized but not inhibited. Therefore, MTOR signaling can be activated or inhibited depending on the time of DRD3 activation, and although MTOR activation seems to be AKT-dependent, its inhibition is AKT-independent. In this respect, previous reports indicate that MTORC1 is negatively regulated by AMPK through phosphorylation of Ser792-RPTOR and reinforcement of its association with MTOR [44–46]. Our results revealed that PPX activates AMPK, phosphorylates RPTOR at Ser792, and enhances MTOR-RPTOR interaction in DRD3- but not in DRD2-HEK cells. Although other ways of AMPK activation of autophagy cannot be ruled out, acting either through TSC2 (TSC complex subunit 2) on MTORC1 or directly on ULK1 [36,43,45], the DRD3 dependence of AMPK activation explains the inhibition of MTORC1 and the subsequent enhancement of autophagy. These findings differ from those of Wang et al. [12], according to whom autophagy may be activated through both DRD2 and DRD3 and the mechanism involves intracellular Ca2+ mobilization as well as PLC (phospholipase C) and IP3 signaling but not MTORC1 inhibition. It should be mentioned that the PPX doses and treatment times used in the above study were substantially higher than those used here. So, it is possible that differences in treatment conditions determine selectivity and underlying mechanisms.
MTORC1 inhibitors are clinically used as immunosuppressant medication in anti-cancer treatment [66], and in the prevention of transplant rejection [67] and artificial coronary stent re-endothelialization [68]. Numerous experimental studies have also demonstrated the beneficial effects of MTORC1 inhibitors as autophagy inducers, promoting the clearance of aggregate-prone proteins, reducing neuronal loss and improving cognitive and motor functions in different animal models of neurodegenerative diseases [7,69]. However, their use in the treatment of neurodegenerative diseases is considered unwise. MTOR is a master regulator in the synthesis of proteins critical for cellular survival and specific brain functions such as the maintenance of neuronal circuits, synaptic plasticity, and axonal regeneration in response to different brain injuries [70,71]. Since neurodegenerative diseases require chronic treatment, prolonged administration of MTOR inhibitors may have serious consequences for cell homeostasis and brain functioning [72]. Consequently, efforts have been directed toward the search for autophagy inducers acting through MTOR-independent mechanisms [1,73].
The finding of MTOR (Ser2488), RPS6KB1 (Thr389) and ULK1 (Ser757) dephosphorylation suggests that RPS6 and the transcription repressor EIF4EBP1, primary targets of MTORC1 kinase activity in cap-dependent transcription [55,74], are also dephosphorylated, which would interfere with the synthesis of essential proteins. However, unlike observations in cells and mice treated with direct MTOR inhibitors, phosphorylation of RPS6 and EIF4EBP1 as well as protein synthesis and diverse markers of cellular viability were preserved in PPX-treated cells and mice. The fact that the MAPK1/3-RPS6KA pathway is also upregulated through both DRD2 and DRD3, and that RPS6 phosphorylation in PPX treated DRD3-cells only declines when this pathway is inhibited, indicates that MAPK1/3 activation exerts a compensatory effect that helps preserve the activity of RPS6 (Figure 9). Cross-talk between MAPK1/3 and MTOR could also contribute to maintaining EIF4EBP1 phosphorylation [75,76]. Besides direct phosphorylation of RPS6, RPS6KB1 can also activate MTORC1 through inhibition of the suppressor function of TSC [76], resulting in increased MTOR and RPS6KB1 signaling. However, despite RPS6KA activation MTOR and RPS6KB1 were inhibited, indicating that these mechanisms are not involved in the response to PPX. Growing evidence shows that EIF4EBP1 can also be phosphorylated by MTOR-independent kinases such as CDK1, GSK3B/GSK3β, MAPK14/p38MAPK, PIM2 and LRRK2 [77,78]. Thus, EIF4EBP1 phosphorylation is now regarded as a point of convergence of MTOR and MTOR-independent kinases that cooperate to protect the availability of EIF4E for the translation machinery [51]. However, the efficacy of these alternative pathways seems to be limited or absent in response to rapamycin or torin 1, and further studies will determine whether they are operative in other forms of MTOR inhibition, including DRD3-induced autophagy. In any case, the facts that EIF4EBP1 remains inactive, as reflected by the maintained phosphorylation of its three residues analyzed (Thr37, Thr46 and Ser65), and that the activity RPS6 is also preserved through the MAPK1/3-RPS6KA pathway, indicate that MTORC1 inhibition through G protein-coupled DRD3 and AMPK activation is restricted to the autophagy axis (Figure 9).
Figure 9.
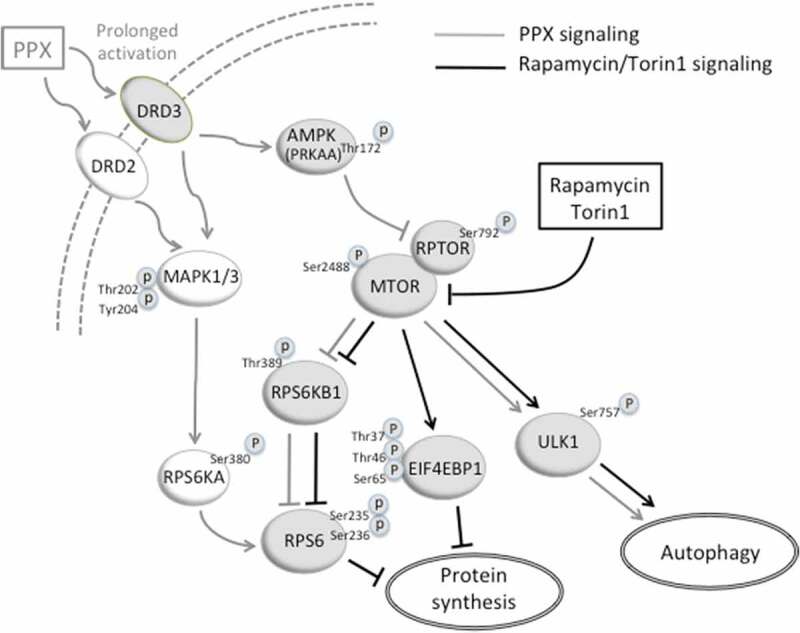
A diagram showing the signaling pathways that activate autophagy through direct MTOR inhibition (black) and prolonged DRD3 activation (gray). DRD3 activates AMPK that phosphorylates RPTOR and reinforces RPTOR (p-RPTOR)-MTOR interaction. Consequently, MTORC1 and its downstream target RPS6KB1 are inhibited (dephosphorylated), and ULK1 is activated (dephosphorylated), initiating the autophagy cascade. Rapamycin and torin1 act directly on MTOR. The translation repressor EIF4EBP1 is activated (dephosphorylated) by rapamycin (partially) and torin1 (totally), but not by prolonged DRD3 stimulation. On the other hand, DRD3 and DRD2 activate the MAPK1/3-RPS6KA pathway that maintains the RPS6 activated (phosphorylated) despite RPS6KB1 inhibition. Consequently, autophagy is promoted by both rapamycin/torin1 and DRD3, and protein synthesis is arrested by rapamycin/torin1 but not by DRD3. Arrows and blocked lines indicate activation or inhibition of the downstream target, respectively. Phosphorylation residues responsible for kinase activation or inhibition are indicated.
The fact that autophagy may be activated through DRD3 but not through DRD2 has important clinical implications, directing our interest to specific neuronal populations. Although DRD2 and DRD3 are coexpressed, and probably form heterodimers in many midbrain dopaminergic and striatal neurons [20,79,80], their distribution differs throughout the brain. DRD2s are widely expressed in the dopaminergic midbrain, ventral and dorsal striatum and cerebral cortex. However, DRD3s are mostly expressed in the ventral striatum, with a lower expression in the dopaminergic midbrain and the dorsal striatum, and very low in the cerebral cortex [20,81]. Based on this expression pattern, we would expect the effect to be restricted to the ventral striatum. However, PPX has been shown to reverse rotenone-induced SNCA/α-synuclein accumulations in PC12 cells and in midbrain dopaminergic neurons [12], where DRD3 levels are significantly lower than in the ventral striatum. On the other hand, in R6/1 mice, a genetic model of Huntington disease in which DRD2 expression virtually disappears but DRD3 is preserved [57], PPX induces autophagy and reduces the levels of soluble mutated huntingtin in both the ventral and dorsal striatum [57]. Confirming their DRD3 dependence, the effects were inhibited by the DRD3 selective antagonist NGB2904, and not detected in the somatosensory cortex where DRD3 is virtually absent. Therefore, we can say that in midbrain dopaminergic neurons and medium spiny neurons of the dorsal striatal, the main target of degeneration in Parkinson disease and Huntington disease, respectively, the levels of DRD3 are high enough to induce autophagy. Bearing in mind that DRD3 are expressed in neuronal populations involved in different neurodegenerative and psychiatric diseases, this autophagy activation mechanism can contribute to the clearance of misfolded proteins and to enhancing the therapeutic efficacy of antidepressant medication.
Most neurotransmitters in the brain signal through GPCRs and most of their effects are mediated by MTOR. For example, as described for DRD3 [64], acute stimulation of cholinergic, serotoninergic, adrenergic, opioid and cannabinoid receptors activate MTORC1 promoting protein synthesis required in synaptic plasticity coupled to memory and neuroprotection [82,83]. So, it is possible that their prolonged activation might also lead to MTOR inhibition and autophagy. Contrasting with direct inhibition of MTORC1 that affects all its targets, MTORC1 inhibition elicited by sustained inhibition of DRD3 restricts to autophagy, abrogating the risk of protein synthesis disruption. Therefore, both activation and inhibition of MTOR following acute and prolonged administration of DRD3, respectively, may have beneficial effects on the brain, particularly under pathological conditions. Although GPCR can induce autophagy through a variety of mechanisms [84], the results described here indicate that ligands promoting activation of both AMPK and MAPK1/3 pathways may be a promising strategy for inducing autophagy in selective neuronal populations since they permit MTORC1 inhibition without disruption of protein synthesis.
Material and methods
Animals and treatment
The experiments were carried out on 22–25 g (4 to 6 months old) male C57BL/6J mice, drd2 KO, and drd3 KO mice and their wild-type (WT) littermates. The drd2 KO mice were generated by homologous recombination as previously described. drd2 KO mice (B6.129S2-Drd2tm1Low/J) and their WT littermates were obtained by mating heterozygous mice [85]. Mice drd3 KO were obtained from the Jackson Laboratory (B6.129S4-Drd3Tm1Dac/J) [86]. Homozygous drd3 KO mice were generated on a pure genetic background (C57BL/6J). Genotype was determined by polymerase chain reaction. Experimental protocols were approved by the Ethical committee of the University of La Laguna (Reference # CEIBA2013-0083), and are in accordance with the ARRIVE guidelines and the European Communities Council Directive of 22 September 2010 (2010/63/EU) regarding the care and use of animals for scientific purposes. Animals were housed in groups of 3–4 per cage, in conditions of constant temperature (21–22°C), a 12 h light/dark cycle, and given free access to food and water. They received a daily dose of pramipexole dihydrochloride (PPX; Sigma-Aldrich, A1237) or its vehicle (100 µL 0.9% sterile saline i.p.) for 6 d, and were sacrificed on day seven. Each experimental group consisted of at least 5 mice.
Striatal primary cell cultures
Cell cultures were generated following protocols previously described by Kowalski and Giraud [87]. Embryos from pregnant mice (E14.5 gestation stage) were dissected and collected in ice-cold dissection solution consisting of Hanks’ balanced salt solution (HBSS; Thermo Fisher Scientific, 14,170–088), 1% penicillin-streptomycin (Biowest, L0018-100) and 1% D-glucose (Sigma-Aldrich, G8270), enriched with 0.1% ascorbic acid (Sigma-Aldrich, A5060). Striata were dissected from embryonic brains and placed in Petri dishes containing ice-cold dissection solution. Cells were dissociated by incubation in 0.1% trypsin (Sigma-Aldrich, L0930-500) and 0.05% DNase (Sigma-Aldrich, 04716728001) in HBSS for 5 min at 37ºC, and thereafter by repeated pipetting in DNase ⁄ HBSS. After centrifugation at 4°C for 5 min at 200 g, they were resuspended in Neurobasal medium (Thermo Fisher Scientific, 21,203–049) containing 2% B27 serum-free supplement (Thermo Fisher Scientific, 08-0085SA), 0.25 mM L–glutamine (Thermo Fisher Scientific, 25,030–081), 0.1% ascorbic acid and 10% fetal bovine serum (FBS; Biowest, S1810-500), and plated onto 35-mm Petri dishes or coverslips coated with poly-L-lysine (10 μg/mL; Sigma-Aldrich, P4707). The number of viable cells was estimated by trypan blue-exclusion assay (Sigma-Aldrich, T8154), seeded at a density of 1.5 × 105 cells ⁄ cm2, and maintained under control conditions (Neurobasal medium containing 2% B27 serum-free supplement, 0.25 mM L–glutamine, 0.1% ascorbic acid and 10% FBS, at 37°C in a humidified 5% CO2 incubator) for 7 d. The medium was removed every 2 d until treatment. On day 7, the cells were treated with PPX. In some cases, the autophagy inhibitor chloroquine diphosphate (CQ, 20 μM; Sigma-Aldrich, C6628) or CQ and the selective DRD3 antagonist NGB2904 (1 μM; Sigma-Aldrich, 189,061-11-8) or the DRD2 antagonist L741,626 (1 μM; Sigma-Aldrich, L135) were added to the medium 30 min before PPX. Each experiment was performed in triplicate.
Ectopic DRD2 and DRD3 expression
HEK293 cells were cultured in Dulbecco´s Modified Eagle´s Medium (DMEM; Biowest, L104) supplemented with 10% FBS and 1% penicillin-streptomycin. Cultures were maintained in a humidified incubator set at 37ºC and 5% CO2. Semiconfluent cells were transfected with pCEP4-EGFP-DRD3- or pcDNA3.1+/Hygro-GFP-DRD2- using Lipofectamine 2000 (Thermo Fisher Scientific, 11,668–027) at a ratio of 2:1 according to the manufacturer´s protocol. The plasmids were a gift from Dr. Jean-Michel Arrang (Addegene, 24,098 and 24,099 [88]). Stable cell lines expressing EGFP-DRD3- and GFP-DRD2- were obtained by growth in selective medium containing 200 μg/ml hygromycin B (Thermo Fisher Scientific, 1068–7010). After 8–10 d of selection in G418 (Sigma-Aldrich, A1780), individual clones were expanded in multi-well plates and examined for expression by western-blot and immunofluorescence for GFP. Several positive clones were identified and used for subsequent experiments. Subconfluent cell cultures were grown and treated with PPX as indicated in the text and figure legends. In some cases, the autophagy inhibitor chloroquine diphosphate (CQ, 20 μM, 30 min before PPX), or the MEK/MAPK1/3 inhibitor PD98059 (25 μM, 1 h; Cell Signaling Technology, 99,005) were added to the medium. Each experiment was performed in triplicate.
Western blot
Mouse striata were dissected in ice from freshly obtained mouse brains using a brain blocker. Protein extracts were obtained in 200–300 µL M-PER (Thermo Fisher Scientific, 78,501). Striatal cells were harvested using trypsin/EDTA (Biowest, L0930) and HEK293 cells in ice-cold phosphate-buffered saline (PBS; 137 mM NaCl [Merck, 106,404], 2.7 mM KCl [PanReac AppliChem, 131,494.1210], 10 mM Na2HPO4 [PanReac AppliChem, A2943], 1.8 mM KH2PO4 [Merck, 529,568], pH 7.4). The cell suspensions were centrifuged for 5 min at 500 g, the pellets were washed in PBS twice and resuspended in M-PER. After sonication (3 bursts of 5 s in ice), lysates were centrifuged at 17,000 g for 5 min and the supernatants were collected. Proteins were quantified using the bicinchoninic acid (Sigma Aldrich, B9643) method and bovine serum albumin (Sigma-Aldrich, 1,076,192) as standard. Protein samples were diluted in Laemmli’s loading buffer (62.5 mM Tris-HCl [Sigma-Aldrich, T5941], 20% glycerol [Sigma-Aldrich, G5516], 2% sodium dodecyl sulfate [SDS; Sigma-Aldrich, L3771], 1.7% β-mercaptoethanol [Sigma-Aldrich, M6250] and 0.05% bromophenol blue [Sigma-Aldrich, B0126], pH 6.8), denatured (90ºC, 5 min), separated by electrophoresis in 10% (15% in the case of LC3 western blot analysis) SDS-polyacrylamide gel, and transferred to nitrocellulose (Schleicher & Schuell, 1,620,115) or PVDF (Bio-Rad, 162–0175; in the case of LC3 western blot analysis) membranes. Blots were blocked for 1 h at room temperature (RT) with 5% nonfat dry milk (5% BSA in the case of phosphorylated forms) in TBST (250 mM NaCl, 50mM Tris, pH 7.4, and 0.05% Tween20 [Sigma-Aldrich, P1379]), and incubated overnight at 4ºC in blocking solution with one of the following antibodies: rabbit polyclonal anti-LC3 (1:1000; Novus Biologicals, NB100-2220), guinea pig polyclonal anti-SQSTM1 protein (1:1000; Progen, GP62-C), mouse monoclonal anti-BECN1/Beclin 1 (1:1500; Santa Cruz Biotechnology, SC48341), rabbit polyclonal anti-MAPK1/ERK2-MAPK3/ERK1 (1:7000; R&D systems, AF1576), mouse monoclonal anti-GFP (1:4000; Roche, 11,814,460,001), rabbit polyclonal anti-phosphoThr202/Tyr204-MAPK1/ERK2-MAPK3/ERK1 (1:5000; Cell Signaling Technology, 9101); rabbit polyclonal anti AKT (1:4000; Cell Signaling Technology, 9272S), rabbit monoclonal anti-phosphoThr308-AKT (1:4000; Cell Signaling Technology, 2965S); rabbit monoclonal anti-MTOR (1:4000; Cell Signaling Technology, 2983), rabbit polyclonal anti-phosphoSer2448-MTOR (1:2000; Millipore, 09213), rabbit polyclonal anti-PRKAA/AMPKα (1:2000; Cell Signaling Technology, 2532), rabbit monoclonal anti-phosphoThr172-PRKAA/AMPKα (1: 2000; Cell Signaling Technology, 2535), rabbit monoclonal anti-RPTOR/Raptor (1:2000; Cell Signaling Technology, 2280); rabbit monoclonal anti-phosphoSer792-RPTOR/Raptor (1:2000; Cell Signaling Technology, 2083), rabbit monoclonal anti-ULK1 (1:2500; Cell Signaling Technology, 8054), rabbit polyclonal anti-phosphoSer757-ULK1 (1:2500; Cell Signaling Technology, 6888); rabbit polyclonal anti-RPS6KB1/p70S6 kinase (1:2000; Cell Signaling Technology, 9202), mouse monoclonal anti-phosphoThr389-RPS6KB1/p70S6 kinase (1:2000; Cell Signaling Technology, 9206), rabbit monoclonal anti-RPS6KA/p90S6 kinase (1:2000; Cell Signaling Technology, 9355), rabbit monoclonal anti-phosphoSer380-RPS6KA/p90S6 kinase (1:2000; Cell Signaling Technology, 11,989); rabbit monoclonal anti-RPS6 (1:3000; Cell Signaling Technology, 2217), rabbit polyclonal anti-phosphoSer235/236-RPS6 (1:5000; Cell Signaling Technology, 2211), rabbit monoclonal anti-EIF4EBP1/4E-BP1 (1:3000; Cell Signaling Technology, 9644), rabbit polyclonal anti-phosphoSer65-EIF4EBP1/4E-BP1 (1:3000; Cell Signaling Technology, 9451), rabbit monoclonal anti-phosphoThr37/46-EIF4BP1/4E-BP1 (1:3,500; Cell Signaling Technology, 2855), mouse anti-ACTB/β-Actin (1:15,000; Sigma-Aldrich, A5441) and anti-TUBA4A/α-Tubulin (1:30,000; Sigma-Aldrich, T6074). After several rinses in TBST-5% milk, the membranes were incubated for 1 h in horseradish peroxidase conjugated anti-mouse-IgG (1:50,000; Jackson-ImmunoResearch Laboratories, 115-035-146), anti-guinea pig-IgG (1:50,000; Jackson-ImmunoResearch Laboratories, 106-035-003) or anti-rabbit-IgG (1:50,000; Jackson-ImmunoResearch Laboratories, 111-035-144). Immunoreactive bands were visualized using enhanced chemiluminiscence (Immun-Star; Bio-Rad, 170–5061) and a Chemi-Doc gel documentation system (Bio-Rad, Hercules, CA). Different protein quantities, antibody dilutions and exposure times were tested to establish the working range of each antibody. The labeling densities were compared with those of ACTB or TUBA4A by using a densitometry software (Bio-Rad). A rectangle of uniform size and shape was placed over each band, and the density values were calculated by subtracting the background at approximately 2 mm above each band. Data are presented as percentages of their respective controls (100%).
Co-immunoprecipitation
Co-immunoprecipitation was performed in DRD2- and DRD3-HEK cells. They were harvested in ice-cold PBS, centrifuged (1,000 g, 5 min, 4ºC) and resuspended in IP buffer (10 mM KH2PO4 pH 7.2, 10 mM MgCl2, 0.3% CHAPS [3-{(3-cholamidopropyl) dimethylammonio}-1 propanesulfonate]; [Thermo Fisher Scientific, 28,300], 1 mM EDTA [Ethylenediaminetetracetic cid; Sigma-Aldrich, EDS], 5 mM EGTA [Ethylene glycol-bis {2-aminoethylether} N,N,N’,N’-tetracitic acid; Sigma-Aldrich, E4378], and protease-phosphatase inhibitors [Thermo Fisher Scientific, A32959]). After 30 min of gentle shaking at 4ºC, the samples were centrifuged again (14,000 g, 5 min), the pellets were discarded, and the protein concentration was quantified in the supernatants. Aliquots of 1.5 mg proteins were pre-cleared using Protein A/G Plus-Agarose beads (Santa Cruz Biotechnology, SC2003) and rabbit IgGs (Santa Cruz Biotechnology, SC3888) for 1 h by gentle rocking. After centrifugation, the pre-cleared supernatants were incubated with 3 μl rabbit polyclonal anti-MTOR (Cell Signaling Technology) overnight at 4ºC in continuous shaking. Samples were incubated with new beads for 3 h. Immunocomplexes were precipitated by gentle centrifugation. After extensive washing in IP buffer, they were resuspended in 40 μl Laemmli’s buffer, heated for 5 min at 95ºC, and centrifuged to collect bead-free supernatants. Samples were separated by electrophoresis in 10% SDS-polyacrylamide gel and transferred to nitrocellulose. MTOR immunoprecipitates were blotted for MTOR, RPTOR and phosphoSer792-RPTOR.
Autophagy flux assessment
Autophagy activity was evaluated in striatal neurons and DRD2- and DRD3-transfected cells by immunoblot analysis of LC3-II with and without the presence of the autophagy inhibitor chloroquine. Chloroquine prevents lysosomal acidification and autophagosome-lysosome fusion, leading to autophagosome accumulation, and consequently, to an increase of LC3-II levels. Cells were treated with chloroquine 20 µM for 30–60 min before adding 10 µM PPX for 2 h. Autophagy flux was also assessed using the Cyto-ID® fluorescence assay (Enzo Life Sciences, 51,031–0050). The probe is based on a green cationic amphiphilic tracer with special affinity for autophagic vesicles and minimal staining of lysosomes and endosomes [89]. Following the manufacturer’s instructions, DRD2- and DRD3-transfected HEK cells were grown in coverslips to 70%-80% confluence and treated as follows: control; chloroquine 20µM, 1 h (CQ), and PPX 10 µM 3 h + CQ. Thereafter cells were washed twice in assay buffer and incubated in dual detection reagent (Cyto-ID® green dye + Hoechst 33,342 nuclear stain) for 30 min at 37ºC in the dark. After two washes in assay buffer, cells were fixed in 4% formaldehyde (Panreac, 141,451.1211) for 20 min, washed three times in PBS and analyzed by confocal microscopy using appropriate filters. The number and size of autophagic vesicles was quantified by using the ImageJ (NIH) standard program. Twelve 30 µm x 30 µm fields of DRD2-HEK and DRD3-HEK cell cultures were randomly selected in each experimental condition (vehicle, CQ and CQ + PPX) from three different experiments. Images were acquired at 60X (1024 x 1024 pixels) with microscopic and computer parameters remaining constant throughout the analysis.
Protein synthesis assay
Protein synthesis was investigated in DRD3-HEK cells using the Surface Sensing of Translation (SUnSET) western blot assay and its fluorescent variant (Click-iT Plus OPP protein synthesis assay; Thermo Fisher Scientific, C10457). This method is based on the incorporation of puromycin or an alkyne analog of puromycin, O-propargyl-puromycin, into nascent protein during protein synthesis [90]. For the western blot analysis, cells were treated (PPX 10 µM, rapamycin 20 nM or torin1 250 nM; 3 h) and thereafter labeled with a short pulse of puromycin dihydrochloride (10 µg/ml, 30 min; Santa Cruz Biotechnology, SC108071). After two washes in PBS they were harvested and processed as described in the western blot section using a mouse monoclonal anti-puromycin antibody (1: 10,000; Sigma-Aldrich, MABE343).
For fluorescence analysis, cells were incubated for 30 min in 20 µM Click-iT OPP in culture medium at 37ºC. Thereafter, they were washed in PBS, fixed in 4% formaldehyde in PBS for 20 min, and permeabilized in 0.5% Triton X-100 (Sigma-Aldrich, X100) for 15 min. After two washes in PBS, cells were incubated in the reaction cocktail containing 0.25% Alexa Fluor 594 picolyl azide (Thermo Fisher Scientific, C10457) for 30 min in the dark at room temperature, washed three times in PBS and analyzed by confocal microscopy. The intensity of OPP labeling was quantified by using the ImageJ standard program. Twelve 220-µm x 220-µm fields of 70-80% confluent DRD3-HEK cell cultures were randomly selected from three different experiments in each experimental condition. Images were acquired at 60X (1024 x 1024 pixels) and labeling was taken from a 3-µm x 3-µm cytosolic area of randomly selected cell profiles. At least 10 cells were analyzed per field. The intensity of OPP labeling is expressed as a percentage of the average fluorescence (arbitrary fluorescence units, range 0–255) in vehicle treated cells (100%). Microscopic and computer parameters were kept constant throughout the sample analysis.
Study of cell viability
The viability of DRD3-HEK cells was assessed using MTT (3[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) and clonogenic cell survival assays. The MTT assay reflects cell viability based on the colorimetric quantification of NAD(P)H-dependent oxidoreductase activity. Cells were seeded in 24-well dishes at a density of 20,000 cells/well (in 400 μl medium) and grown for 48 h. After treatment (0.01–10 μM, PPX/Rapamycin/Torin1, 72 h), 50 μl of MTT reagent (0.5% MTT [Sigma-Aldrich, M2128] in PBS, 0.1% final concentration) was added to each well and incubated at 37°C for 4 h. Thereafter, 500 μl of solubilization buffer (20% sodium dodecyl sulfate (SDS) in 0.01 M HCl) was added and incubated overnight at 37°C. The optical density of the samples was then measured at 570 nm with reference at 630 nm. MTT reduction was expressed as a percentage of the untreated DRD3-HEK cells. For the clonogenic cell survival assay, cells were seeded in 6-well dishes at a density of 120 cells/well and grown in the presence of PPX or rapamycin (0.1–10 μM). After 2 weeks, visible single-cell clones were counted by two independent observers.
The levels of MDA, a lipid peroxidation marker, were also analyzed in mouse striata. After treatment samples were homogenized in ice-cold saline solution. Aliquots of 0.2 ml homogenates were used for MDA determination. The analytical measures of MDA were referred to as TBARS (thiobarbituric acid-reactive substance) containing 0.11 M TBA (2-thiobarbituric acid; Sigma-Aldrich, T-5500) and following the method described by Kikugawa et al [91]. The pink complex of samples was extracted in 10.9 M n-butanol (Sigma-Aldrich, 33,065). Each one was placed in a 96 well plate and read at 535 nm in a microplate spectrophotometer reader (Spectra MAX-190; Molecular Devices, Sunnyvale, CA). The detection limit of this assay was 0.079 nmol/ml; the intra- and inter-assay coefficients of variation were 1.82% and 4.01%, respectively. The sample concentration of MDA was expressed in nmol/mg protein. To avoid possible interferences of compounds that react or absorb at 532 nm, each sample was compared with our blank tube (sample without thiobarbituric acid reagent) and the absorbance was subtracted from each sample tube. Furthermore, in this assay, the use of n-butanol as extracting agent of TBARS complex prevents many of these interferences [92].
Statistics
Data were plotted using Graph Pad Prism software (San Diego, CA). Unpaired t-test or Mann-Whitney U test were performed for parametric or non-parametric analysis, respectively. ANOVA or the Kruskall-Wallis test, followed by the Tukey’s or Dunn’s multiple comparison tests, were used when more than two groups were analyzed. A level of p < 0.05 was considered as critical for assigning statistical significance.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Philippe Marin at the University of Monpellier, France, for his feedback and helpful comments in the preparation of this manuscript.
Funding Statement
This work was supported by the Spanish Ministerio de Economía y Competitividad [BFU2016-77363-R] and Gobierno Autónomo de Canarias [2018-00000034] to T.G.H. D.L.R. was supported by the “Programa Agustín de Betancourt” (Cabildo Insular de Tenerife). F.F.R. and A.F.C. were supported by the Programme “Ayudas para contratos predoctorales para la formación de doctores”, Spanish Ministerio de Economía y Competitividad [BES-2014-067781 and BES-2017‐079923, respectively]; Consejería de Educación, Universidades y Sostenibilidad, Gobierno de Canarias [2018-00000034]; Ministerio de Economía, Industria y Competitividad, Gobierno de España [BFU2016-77363-R]; Ministerio de Economía, Industria y Competitividad, Gobierno de España [BES-2014-067781]; Ministerio de Economía, Industria y Competitividad, Gobierno de España [BES-2017‐079923]; Cabildo de Tenerife (ES) [Programa Agustín de Betancourt].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Bioch Soc Trans. 2013;41:1103–1130. [DOI] [PubMed] [Google Scholar]
- [2].Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:2937–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. [DOI] [PubMed] [Google Scholar]
- [5].Merenlender-Wagner A, Malishkevich A, Shemer Z, et al. Autophagy has a key role in the pathophysiology of schizophrenia. Mol Psychiatry. 2015;20:126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jia J, Le W. Molecular network of neuronal autophagy in the pathophysiology and treatment of depression. Neurosci Bull. 2015;31:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. [DOI] [PubMed] [Google Scholar]
- [8].Rose C, Menzies FM, Renna M, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet. 2010;19:2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Metcalf DJ, García-Arencibia M, Hochfeld WE, et al. Autophagy and misfolded proteins in neurodegeneration. Exp Neurol. 2012;238:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mariño G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang L, Yu J, Pan H, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci USA. 2007;104:19023–19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang JD, Cao YL, Li Q, et al. A pivotal role of FOS-mediated BECN1/Beclin1 upregulation in dopamine D2 and D3 receptor agonist-induced autophagy activation. Autophagy. 2015;11:2057–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wei C, Gao J, Li M, et al. Dopamine D2 receptors contribute to cardioprotection of ischemic post-conditioning via activating autophagy in isolated rat hearts. Int J Cardiol. 2016;203:837–839. [DOI] [PubMed] [Google Scholar]
- [14].Yan H, Li WL, Xu JJ, et al. D2 dopamine receptor antagonist raclopride induces non-canonical autophagy in cardiac myocytes. J Cell Biochem. 2013;114:103–110. [DOI] [PubMed] [Google Scholar]
- [15].Ling ZD, Robie HC, Tong CW, et al . Both the antioxidant and D3 agonist actions of pramipexole mediate its neuroprotective actions in mesencephalic cultures. J Pharmacol Exp Ther. 1999;289:202–210. [PubMed] [Google Scholar]
- [16].Li M, Yang Z, Vollmer LL, et al. AMDE-1 is a dual function chemical for autophagy activation and inhibition. PLoS One. 2015;10:e0122083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Y, McGreal S, Zhao J, et al. A cell-based quantitative high-throughput image screening identified novel autophagy modulators. Pharmacol Res. 2016;110:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Meador-Woodruff JH, Damask SP, Wang J, et al. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology. 1996;15:17–29. [DOI] [PubMed] [Google Scholar]
- [19].Diaz J, Pilon C, Le Foll B, et al. Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J Neurosci. 2000;20:8677–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Araki KY, Sims JR, Bhide PG. Dopamine receptor mRNA and protein expression in the mouse corpus striatum and cerebral cortex during pre- and postnatal development. Brain Res. 2007;1156:31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Joyce JN, Millan MJ. Dopamine D3 receptor agonists for protection and repair in Parkinson’s disease. Curr Opin Pharmacol. 2007;7:100–105. [DOI] [PubMed] [Google Scholar]
- [22].Rao NP, Remington G. Targeting the dopamine receptor in schizophrenia: investigational drugs in Phase III trials. Expert Opin Pharmacother. 2014;15:373–383. [DOI] [PubMed] [Google Scholar]
- [23].Moritz AE, Benjamin Free R, Sibley DR. Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds. Cell Signal. 2018;41:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Antonini A, Barone P, Ceravolo R, et al. Role of pramipexole in the management of Parkinson’s disease. CNS Drugs. 2010;23:829–841. [DOI] [PubMed] [Google Scholar]
- [25].Zarate CA Jr, Payne JL, Singh J, et al. Pramipexole for bipolar II depression: a placebo-controlled proof of concept study. Biol Psychiatry. 2004;56:54–60. [DOI] [PubMed] [Google Scholar]
- [26].Fawcett J, Rush AJ, Vukelich J, et al. Clinical experience with high-dosage pramipexole in patients with treatment-resistant depressive episodes in unipolar and bipolar depression. Am J Psychiatry. 2016;173:107–111. [DOI] [PubMed] [Google Scholar]
- [27].Li C, Guo Y, Xie W, et al. Neuroprotection of pramipexole in UPS impairment induced animal model of Parkinson’s disease. Neurochem Res. 2010;35:1546–1556. [DOI] [PubMed] [Google Scholar]
- [28].Salles MJ, Hervé D, Rivet JM, et al. Transient and rapid activation of Akt/GSK-3β and mTORC1 signaling by D3 dopamine receptor stimulation in dorsal striatum and nucleus accumbens. J Neurochem. 2013;125:532–544. [DOI] [PubMed] [Google Scholar]
- [29].Mannoury La Cour C, MJ S, Pasteau V, et al. Signaling pathways leading to phosphorylation of Akt and GSK-3β by activation of cloned human and rat cerebral D2 and D3 receptors. Mol Pharmacol. 2011;79:91–105. [DOI] [PubMed] [Google Scholar]
- [30].Zapata A, Kivell B, Han Y, et al. Regulation of dopamine transporter function and cell surface expression by D3 dopamine receptors. J Biol Chem. 2007;282:35842–35854. [DOI] [PubMed] [Google Scholar]
- [31].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. [DOI] [PubMed] [Google Scholar]
- [33].Mizushima N, Yoshimori T, Levine N. Methods in mammalian autophagy research. Cell. 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Streeter A, Menzies FM, Rubinsztein DC. LC3-II Tagging and western blotting for monitoring autophagic activity in mammalian cells. Methods Mol Biol. 2016;1303:161–170. [DOI] [PubMed] [Google Scholar]
- [35].Bjorkoy G, Lamark T, Pankiv S, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. [DOI] [PubMed] [Google Scholar]
- [36].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biol. 2011;13:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Menon MB, Beclin DS. 1 phosphorylation - at the center of autophagy regulation. Front Cell Dev Biol. 2018;6:137. e Collection 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pearson RB, Dennis PB, Han JW, et al . The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. Embo J. 1995;14:5279–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69:1821–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes. 2008;32(Suppl 4):S7–S12. [DOI] [PubMed] [Google Scholar]
- [42].Hawley SA, Davison M, Woods A, et al. Characterization of the AMP-activated protein kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. [DOI] [PubMed] [Google Scholar]
- [43].Tamargo-Gomez I, Mariño G. AMPK: regulation of metabolic dynamics in the context of autophagy. Int J Mol Sci. 2018;19:pii: E3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;331:456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee JW, Park S, Takahashi Y, et al. The association of AMPK with ULK regulates autophagy. PLoS One. 2010;5:e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. [DOI] [PubMed] [Google Scholar]
- [47].Sutton LP, Caron MG. Essential role of D1R in the regulation of mTOR complex 1 signaling induced by cocaine. Neuropharmacology. 2015;99:610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xu S, Kang UG. Region-specific activation of the AMPK system by cocaine: the role of D1 and D2 receptors. Pharmacol Biochem Behav. 2016;146-147:28–38. [DOI] [PubMed] [Google Scholar]
- [49].Maiese K. targeting molecules to medicine with mTOR, autophagy and neurodegenerative disorders. Br J Clin Pharmacol. 2016;82:1245–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013;62:157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle. 2016;15:781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Meyuhas O. Ribosomal protein S6 phosphorylation: four decades of research. Int Rev Cell Mol Biol. 2015;320:41–73. [DOI] [PubMed] [Google Scholar]
- [53].Roux PP, Shahbazian D, Vu H, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ayuso MI, Hernández-Jiménez M, Martín ME, et al. New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress. J Biol Chem. 2010;285:34355–34363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Thoreen CC, Kang SA, Chang JW, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Brunn GJ, Hudson CC, Sekulic A, et al. Phosphorylation of the translational repressor PHA-l by the mammalian target of rapamycin. Science. 1997;277:99–101. [DOI] [PubMed] [Google Scholar]
- [57].Luis-Ravelo D, Estévez-Silva H, Barroso-Chinea P, et al. Pramipexole reduces soluble mutant huntingtin and protects striatal neurons through dopamine D3 receptors in a genetic model of Huntington’s disease. Exp Neurol. 2018;299:137–147. [DOI] [PubMed] [Google Scholar]
- [58].Piercey MF, Hoffmann WE, Smith MW, et al. Inhibition of dopamine neuron firing by pramipexole, a dopamine D3 receptor-preferring agonist: comparison to other dopamine receptor agonists. Eur J Pharmacol. 1996;312:35–44. [DOI] [PubMed] [Google Scholar]
- [59].Sokoloff P, Giros B, Martres MP, et al. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. [DOI] [PubMed] [Google Scholar]
- [60].Beaulieu JM, Gainetdinov RR. The physiology, signalling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. [DOI] [PubMed] [Google Scholar]
- [61].Jin M, Min C, Zheng M, et al. Multiple signalling routes involved in the regulation of adenylyl cyclase and extracellular regulated kinase by dopamine D(2) and D(3) receptors. Pharmacol Res. 2013;67:31–41. [DOI] [PubMed] [Google Scholar]
- [62].Cho EY, Cho DI, Park JH, et al. Roles of protein kinase C and actin-binding protein 280 in the regulation of intracellular trafficking of dopamine D3 receptor. Mol Endocrinol. 2007;21:2242–2254. [DOI] [PubMed] [Google Scholar]
- [63].Min C, Zheng M, Zhang X, et al. Novel roles of β-arrestins in the regulation of pharmacological sequestration to predict agonist-induced desensitization of dopamine D3 receptors. Br J Pharmacol. 2013;170:1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Collo G, Bono F, Cavalleri L, et al. Nicotine-induced structural plasticity in mesencephalic dopaminergic neurons is mediated by dopamine D3 receptors and Akt-mTORC1 signaling. Mol Pharmacol. 2013;83:1176–11189. [DOI] [PubMed] [Google Scholar]
- [65].Cavalleri L, Merlo Pich E, Millan MJ, et al. Ketamine enhances structural plasticity in mouse mesencephalic and human iPSC-derived dopaminergic neurons via AMPAR-driven BDNF and mTOR signaling. Mol Psychiatry. 2018;23:812–823. [DOI] [PubMed] [Google Scholar]
- [66].Francipane MG, Lagasse E. Therapeutic potential of mTOR inhibitors for targeting cancer stem cells. Br J Clin Pharmacol. 2016;82:1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Baroja-Mazo A, Revilla-Nuin B, Ramirez P, et al. Immunosuppressive potency of mechanistic target of rapamycin inhibitors in solid-organ transplantation. World J Transplant. 2016;6:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Habbib A, Finn AV. Antiproliferative drugs for restenosis prevention. Interv Cardiol Clin. 2016;5:321–329. [DOI] [PubMed] [Google Scholar]
- [69].Caccamo A, Majumder S, Richardson A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84:275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bockaert J, Marin P. mTOR in brain physiology and pathologies. Physiol Rev. 2015;95:1157–1187. [DOI] [PubMed] [Google Scholar]
- [72].Ben-Sahra I, Manning BD . mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017;45:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mishra P, Dauphinee AN, Ward C, et al. Discovery of pan autophagy inhibitors through a high-throughput screen highlights macroautophagy as an evolutionarily conserved process across 3 eukaryotic kingdoms. Autophagy. 2017;13:1556–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chauvin C, Koka V, Nouschi A, et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene. 2014;33:474–483. [DOI] [PubMed] [Google Scholar]
- [75].Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Roux PP, Ballif BA, Anjum R, et al. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA. 2004;101:13489–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Greenberg VL, Zimmer SG. Paclitaxel induces the phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 through Cdk-1dependent mechanism. Oncogene. 2005;24:4851–4860. [DOI] [PubMed] [Google Scholar]
- [78].Shin S, Wolgamott L, Tcherkezian J, et al. Glycogen synthase kinase-3β positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene. 2014;33:1690–1699. [DOI] [PubMed] [Google Scholar]
- [79].Mengod G, Villaró MT, Landwehrmeyer GB, et al. Visualization of dopamine D1, D2 and D3 receptor mRNAs in human and rat brain. Neurochem Int. 1992;20:33S–43S. [DOI] [PubMed] [Google Scholar]
- [80].Maggio R, Scarselli M, Capannolo M, et al. Novel dimensions of D3 receptor function: focus on heterodimerisation, transactivation and allosteric modulation. Eur Neuropsychopharmacol. 2015;25:1470–1479. [DOI] [PubMed] [Google Scholar]
- [81].Murray AM, Ryoo HL, Gurevich E, et al. Localization of dopamine D3 to mesolimbic and D2 receptors to mesostriatal regions of human forebrain. Proc Natl Acad Sci USA. 1994;91:11271–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Giovannini MG, Lana D, Pepeu G. The integrated role of Ach, ERK and mTOR in the mechanisms of hippocampal inhibitory avoidance memory. Neurobiol Learn Mem. 2015;119:18–33. [DOI] [PubMed] [Google Scholar]
- [83].Karl T, Garner B, Cheng D. The therapeutic potential of the phytocannabinoid cannabidiol for Alzheimer’s disease. Behav Pharmacol. 2017;28:142–160. [DOI] [PubMed] [Google Scholar]
- [84].Boland B, Yu WH, Corti O, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018;17:660–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Granado N, Ares-Santos S, Oliva I, et al. Dopamine D2-receptor knockout mice are protected against dopaminergic neurotoxicity induced by methamphetamine or MDMA. Neurobiol Dis. 2011;42:391–403. [DOI] [PubMed] [Google Scholar]
- [86].Accili D, Fishburn CS, Drago J, et al. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci USA. 1996;93:1945–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kowalski C, Giraud P. Dopamine decreases striatal enkephalin turnover and proenkephalin messenger RNA abundance via D2 receptor activation in primary striatal cell cultures. Neuroscience. 1993;53:665–672. [DOI] [PubMed] [Google Scholar]
- [88].Jeanneteau F, Diaz J, Sokoloff P, et al. Interactions of GIPC with dopamine D2, D3 but not D4 receptors define a novel mode of regulation of G protein-coupled receptors. Mol Biol Cell. 2004;15:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Guo S, Liang Y, Murphy SF, et al. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy. 2015;11:560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Liu J, Xu Y, Stoleru D, et al. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Natl Acad Sci USA. 2012;109:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kikugawa K, Kojima T, Yamaki S, et al. Interpretation of the thiobarbituric acid reactivity of rat liver and brain homogenates in the presence of ferric ion and ethylediaminotetraacetic acid. Anal Biochem. 1992;202:249–255. [DOI] [PubMed] [Google Scholar]
- [92].Valenzuela A. The biological significance of malondialdehyde determination in the assessment of tissue oxidative stress. Life Sci. 1991;48:301–309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.