

ABSTRACT
We recently identified the mitochondrial peptidase, neurolysin (NLN), as a top hit in an acute myeloid leukemia (AML) viability screen. Using chemical and genetic approaches, we demonstrated that loss of NLN disrupted respiratory chain supercomplex assembly and impaired oxidative metabolism in AML. Moreover, inhibition of NLN in vitro and in vivo reduced the growth of AML cells.
KEYWORDS: Acute myeloid leukemia, metabolism, respiratory chain supercomplexes, neurolysin, LETM1
Acute myeloid leukemia (AML) is an aggressive hematological malignancy that is defined by the uncontrolled proliferation of immature blast cells. The rapid proliferation of these immature blasts, called myeloblasts, impairs normal hematopoiesis, resulting in infection, anemia, and hemorrhage. Despite recent advances and new therapies for this disease, the prognosis for most patients with AML remains poor. Patients with relapsed and refractory disease have a particularly dismal outcome. Likewise, patients who are unfit for chemotherapy have a median survival of only 6–9 months.1
Recently, we and others demonstrated that AML cells and stem cells have unique mitochondrial properties that can be therapeutically targeted.2,3 Among these properties is overexpression of mitochondrial proteases and dysregulated mitochondrial protein quality control pathways.4,5 In a genetic screen to identify members of the mitochondrial proteome that are essential for AML viability, we identified the mitochondrial peptidase, neurolysin (NLN), as a top hit.4 NLN’s mitochondrial function is not well characterized and its role in AML has not been previously studied.
NLN is a zinc metallopeptidase that was originally identified in the nervous system where it was shown to cleave vasoactive peptides such as bradykinin and neurotensin.6 However, this function appears redundant as NLN knockout mice do not have abnormalities with blood pressure regulation. Instead, NLN knockout mice are viable with mild metabolic defects, such as fewer oxidative muscle fibers and poor performance in endurance exercise tests.7 These results suggest that loss of NLN impairs metabolism.
We began our study by investigating NLN’s expression in AML using a publicly available dataset of 536 AML and 73 normal control bone marrow samples. NLN mRNA was overexpressed in a subgroup of AML patient samples and was equally expressed in bulk and progenitor populations. To confirm the results of our shRNA screen and to investigate NLN’s importance in AML growth, we knocked down NLN using shRNA in OCI-AML2, NB4, TEX, and MV4-11 leukemia cell lines. NLN knockdown reduced the growth of all tested cell lines and also reduced the colony-forming potential of OCI-AML2 and NB4 cells, suggesting that NLN knockdown targets both bulk and progenitor AML cells. In line with this finding, knockdown of NLN reduced the engraftment of TEX cells into mouse marrow.5
NLN’s mitochondrial function has not been well defined. To understand NLN’s role in the mitochondria, we identified its mitochondrial interactors via proximity-dependent biotin labeling coupled with mass spectrometry (BioID-MS). NLN interacted with several proteins involved in respiratory electron transport and assembly. Based on these findings, we investigated NLN’s role in oxidative metabolism and found that NLN knockdown reduced oxygen consumption rates (OCR) in leukemic cells. Moreover, NLN knockdown impaired the formation of large quaternary structures called respiratory chain supercomplexes (RCS).5 RCS consist of complexes I, III, and IV and increase the efficiency of electron transport.8
RCS have not been previously studied in leukemia. We found that RCS were increased in a subset of AML patient samples, compared to normal hematopoietic cells. Moreover, RCS expression correlated with NLN levels. To understand how NLN affects RCS, we returned to our BioID-MS results and found that one of NLN’s top interactors was LETM1 (leucine zipper-EF-hand containing transmembrane protein 1).5 LETM1 has previously been shown to regulate RCS assembly and forms two complexes, the minor and major.9 NLN knockdown reduced the formation of both the LETM1 minor and major complexes. Of note, knockdown of LETM1 also reduced AML growth and oxidative metabolism.5
We assessed the effects of pharmacologically inhibiting NLN in AML using the previously reported NLN inhibitor, R2 (3-[(2 S)-1-[(3 R)-3-(2-Chlorophenyl)-2-(2-fluorophenyl)pyrazolidin-1-yl]-1-oxopropan-2-yl]-1-(adamantan-2-yl)urea).10 Treatment of AML cells in vitro with R2 impaired growth of AML cells and primary AML culture models. Moreover, R2 treatment reduced RCS levels, LETM1 complex formation, and oxidative metabolism. In vivo, R2 reduced the leukemic burden in mice engrafted with primary AML patient samples and also reduced engraftment in secondary transplantations. Importantly, R2 treatment did not affect the engraftment of normal human cord blood cells. Collectively, these data suggest that pharmacological inhibition of NLN selectively targets bulk and progenitor AML cells without affecting normal hematopoietic cells.5 Although an effective tool compound, additional work will be required to develop more potent NLN inhibitors and identify potential leads for therapeutic candidates. Moreover, R2 is predicted to cross the blood-brain barrier.10 NLN inhibitors that do not cross the blood brain barrier may have a superior therapeutic index.
AML cells and stem cells depend on oxidative metabolism for their survival. NLN promotes the assembly of RCS, which drives efficient mitochondrial energy production. We showed that genetic and chemical inhibition of NLN targeted AML cells by disrupting RCS formation and OCR (Figure 1). However important biological questions remain unanswered and will form the basis for future studies. Future work will explore the precise mechanism by which NLN mediates LETM1 complex and RCS formation and whether this is related to its peptidase activity. In addition to uncovering important new biology, determining whether the effects of NLN on LETM1 and RCS formation are dependent or independent of its peptidase function will help guide the development of NLN inhibitors. Moreover, our findings do not exclude the possibility that NLN regulates RCS directly and future mechanistic studies will aim to address this. Finally, little is known about the composition of the LETM1 minor and major complexes. Characterizing these complexes will provide new insight into how LETM1 promotes RCS and potentially new ways to target RCS formation. In summary, our work demonstrates the importance of RCS in AML and supports inhibition of NLN as a novel therapeutic approach.
Figure 1.
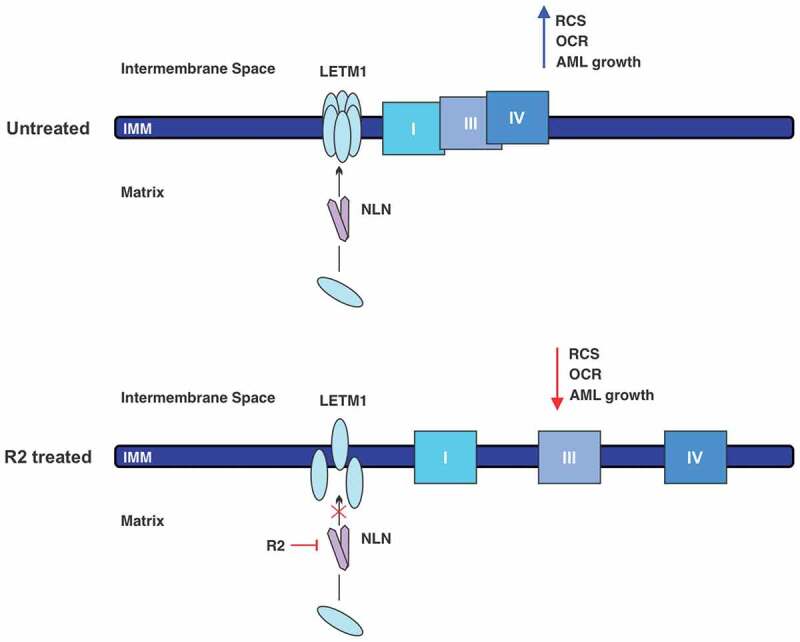
Chemical inhibition of NLN disrupts RCS and growth in AML cells. (Top) NLN (neurolysin) promotes the formation of LETM1 (leucine zipper-EF-hand containing transmembrane protein 1) complexes through an unknown mechanism. LETM1 complexes stabilize RCS (respiratory chain supercomplexes) and promote efficient oxidative metabolism and AML (acute myeloid leukemia) growth. (Bottom) Inhibition of NLN by R2 disrupts the assembly of LETM1 complexes and destabilizes RCS. Consequently, OCR (oxygen consumption rate) is impaired and AML growth is reduced.
Acknowledgments
We thank Jill Flewelling (Princess Margaret Cancer Center) for administrative assistance. This work was supported by the Canadian Institutes of Health Research, the Ontario Institute of Cancer Research with funding provided by the Ontario Ministry of Research and Innovation, the Princess Margaret Cancer Centre Foundation, the Ministry of Long Term Health and Planning in the Province of Ontario. A.D.S. holds the Barbara Baker Chair in Leukemia and Related Diseases.
Funding Statement
This work was supported by the Ontario Institute for Cancer Research; Canadian Institutes of Health Research (CA); Canadian Institutes of Health Research (CA); Princess Margaret Cancer Foundation.
Disclosure of potential conflicts of interest
A.D.S. has received honorariums or consulting fees from Novartis, Jazz, Otsuka, and Takeda Pharmaceuticals and research support from Medivir AB and Takeda. A.D.S. owns stock in Abbvie Pharmaceuticals and is named on a patent application for the use of DNT cells for the treatment of leukemia.
References
- 1.Short NJ, Rytting ME, Cortes JE.. Acute myeloid leukaemia. Lancet. 2018;392(10147):1–3. doi: 10.1016/S0140-6736(18)31041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, D’Alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(1859–1866):1859–1866. doi: 10.1038/s41591-018-0233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20(674–688):674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2015;27(864–876):864–876. doi: 10.1016/j.ccell.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirali S, Botham A, Voisin V, Xu C, St-Germain J, Sharon D, Hoff FW, Qiu Y, Hurren R, Gronda M, et al. The mitochondrial peptidase neurolysin, regulates respiratory chain supercomplex formation and is necessary for AML viability. Sci Transl Med 2020;12. [DOI] [PubMed] [Google Scholar]
- 6.Checler F, Barelli H, Dauch P, Dive V, Vincent B, Vincent JP. Neurolysin: purification and assays. Methods Enzymol. 1995;248:593–614. [DOI] [PubMed] [Google Scholar]
- 7.Cavalcanti DM, Castro LM, Rosa Neto JC, Seelaender M, Neves RX, Oliveira V, Forti FL, Iwai LK, Gozzo FC, Todiras M, et al. Neurolysin knockout mice generation and initial phenotype characterization. J Biol Chem. 2014;289(15426–15440):15426–15440. doi: 10.1074/jbc.M113.539148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greggio C, Jha P, Kulkarni SS, Lagarrigue S, Broskey NT, Boutant M, Wang X, Conde Alonso S, Ofori E, Auwerx J, et al. Enhanced respiratory chain supercomplex formation in response to exercise in human skeletal muscle. Cell Metab. 2017;25(301–311):301–311. doi: 10.1016/j.cmet.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Tamai S, Iida H, Yokota S, Sayano T, Kiguchiya S, Ishihara N, Hayashi J-I, Mihara K, Oka T. Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J Cell Sci. 2008;121(2588–2600):2588–2600. doi: 10.1242/jcs.026625. [DOI] [PubMed] [Google Scholar]
- 10.Hines CS, Ray K, Schmidt JJ, Xiong F, Feenstra RW, Pras-Raves M, de Moes JP, Lange JHM, Melikishvili M, Fried MG, et al. Allosteric inhibition of the neuropeptidase neurolysin. J Biol Chem. 2014;289(35605–35619):35605–35619. doi: 10.1074/jbc.M114.620930. [DOI] [PMC free article] [PubMed] [Google Scholar]