

ABSTRACT
Macroautophagy/autophagy is an evolutionarily conserved intracellular process that recycles and degrades intracellular components to sustain homeostasis in response to deficiency of nutrients or growth factors. PAQR3 is a newly discovered tumor suppressor that also regulates autophagy induced by nutrient starvation via AMPK and MTORC1 signaling pathways. In this study, we investigated whether PAQR3 modulates EGFR-mediated autophagy and whether such regulation is associated with the tumor suppressive activity of PAQR3. PAQR3 is able to inhibit the in vitro and in vivo growth of non-small cell lung cancer (NSCLC) cells. PAQR3 potentiates autophagy induced by EGFR inhibitor erlotinib. Knockdown of PAQR3 abrogates erlotinib-mediated reduction of BECN1 interaction with autophagy inhibitory proteins RUBCN/Rubicon and BCL2. PAQR3 blocks the interaction of BECN1 with the activated form of EGFR and inhibits tyrosine phosphorylation of BECN1. Furthermore, inhibition of autophagy by knocking down ATG7 abrogates the tumor suppressive activity of PAQR3 in NSCLC cells. Collectively, these data indicate that PAQR3 suppresses tumor progression of NSCLC cells through modulating EGFR-regulated autophagy.
Abbreviations
AKT: thymoma viral proto-oncogene; ATG5: autophagy related 5; ATG7: autophagy related 7; ATG14: autophagy related 14; BCL2: B cell leukemia/lymphoma 2; BECN1: beclin 1; CCK-8: cell counting kit-8; CQ: chloroquine diphosphate; DMEM: Dulbecco’s modified Eagle’s medium; EdU: 5-ethynyl-2ʹ-deoxyuridine; EGFR: epidermal growth factor receptor; FBS: fetal bovine serum; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; IgG: Immunoglobulin G; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MTOR: mechanistic target of rapamycin kinase; MTORC1: mechanistic target of rapamycin kinase complex 1; MTT: thiazolyl blue tetrazolium bromide; NSCLC: Non-small cell lung cancer; MAP2K/MEK: mitogen-activated protein kinase kinase; MAPK/ERK: mitogen-activated protein kinase; PAQR3: progestin and adipoQ receptor family member 3; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4/VPS15: phosphoinositide-3-kinase regulatory subunit 4; PRKAA/AMPK: protein kinase, AMP-activated alpha catalytic; RUBCN: rubicon autophagy regulator; RPS6: ribosomal protein S6; RAS: Ras proto-oncogene; RAF: Raf proto-oncogene; TKI: tyrosine kinase inhibitor; TUBA4A: tubulin alpha 4a; UVRAG: UV radiation resistance associated.
KEYWORDS: ATG7, autophagy, BECN1, EGFR, lung cancer, tumor suppressor
Introduction
Autophagy is a critical mechanism to maintain cellular homeostasis that allows cells to survive in times of nutrient deprivation via self-digestion of proteins and organelles [1–3]. Autophagy initiation requires a series of autophagy-related (ATG) proteins that function in a sequential order. The BECN1/Beclin1 (Vps30/Atg6 in yeast) complex containing BECN1, PIK3C3/VPS34, ATG14, and PIK3R4/VPS15 functions as a class III PtsIns3K kinase and plays an essential role in autophagy initiation by orchestrating multiple autophagy initiating signals [4–6]. Loss-of-function mutations of autophagy-related genes, such as BECN1, ATG5 and ATG7, results in spontaneous tumorigenesis [7]. Thus, dysregulation of autophagy has been considered as an important mechanism involved in the development of different cancers [8–11].
Lung cancer is the leading cause of cancer death in the world. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer and accounts for 80% of all lung cancers [12,13]. Most NSCLC harbors specific activating mutations in the tyrosine kinase domain of EGFR (epidermal growth factor receptor) [14,15]. EGFR is an oncogenic receptor tyrosine kinase and links extracellular signals to cellular homeostasis. As previously reported, EGFR has a double effect on regulation of autophagy. Active EGFR suppresses autophagy through tyrosine phosphorylation of BECN1 [16], while inactive EGFR interacts with the oncoprotein LAPTM4B (lysosome-associated protein transmembrane 4 beta) to promote autophagy initiation [17]. In NSCLC, suppression of autophagy by active EGFR contributes to tumor progression [16].
PAQR3, a recently characterized tumor suppressor, has an inhibitory function in many types of tumors through negative regulation of RAS-RAF-MAP2K/MEK-MAPK/ERK and PI3K-AKT signaling pathways [18–20]. In addition, PAQR3 was recently discovered to play an important role in modulating autophagy upon nutrient deprivation. PAQR3 controls autophagy by integrating AMPK signaling to improve ATG14-associated class III PI3K activity in response to glucose starvation [21]. PAQR3 also modulates autophagy induced by amino acid starvation by regulating MTORC1 activity [22]. However, it is currently unknown whether PAQR3 has a functional role in growth factor-mediated autophagy. Furthermore, it is unknown whether PAQR3-modulated autophagy is linked to its tumor suppressive activity. In this study, we investigated the potential function of PAQR3 in EGFR-mediated autophagy in NSCLC cells and revealed that PAQR3 inhibits cell growth and tumorigenesis of NSCLC cells via modulation of autophagy.
Results
PAQR3 inhibits the proliferation of NSCLC cells
PAQR3 is a newly discovered tumor suppressor that is deregulated in different types of human cancer including colon cancer, gastric cancer, bladder cancer, liver cancer, osteosarcoma, breast cancer, laryngeal squamous cell carcinoma and prostate cancer [23–30]. However, it is little known whether PAQR3 has a functional role in NSCLC. To investigate whether PAQR3 impacts the growth of human NSCLC cells, we established HCC827 cells with stable overexpression or knockdown of PAQR3. HCC827 is a human NSCLC cell line that bears a EGFRΔ746–750 activating mutation endowing the cells sensitive to inhibition by tyrosine kinase inhibitor (TKI) [31–33]. PAQR3 was either knocked down or overexpressed in HCC827 cells by infection of the cells with lentivirus containing either PAQR3-specific shRNA or PAQR3-overexpressing plasmid (Figure S1). We then investigated the cell proliferation status of these cells. EdU assay, used to analyze newly synthesized DNA, revealed that growth of HCC827 cells was significantly increased by PAQR3 knockdown (Figure 1A) and significantly reduced by PAQR3 overexpression (Figure 1B). Cell counting by CCK-8 assay also demonstrated that the proliferation rate of HCC827 cells was negatively regulated by PAQR3 (Figure 1C,D). MTT assay, used to evaluate the mitochondrial activity, showed that the viability of HCC827 cells was negatively modulated by PAQR3 (Figure S2). Consistently, colony formation of HCC827 cells was significantly elevated by PAQR3 knockdown (Figure 1E) and significantly reduced by PAQR3 overexpression (Figure 1F). We also analyzed a different PAQR3-specific shRNA in HCC827 cells and found that this shRNA also increased the cell proliferation rate of HCC827 cells (Figures S3A-S3C). In addition, we analyzed the effect of PAQR3 on other NSCLC cells. In both H1703 and H460 cells, knockdown of PAQR3 significantly stimulated the proliferation of these cells as analyzed by EdU assay, cell counting and colony formation assay (Figures S1, S4 and S5). On the other hand, overexpression of PAQR3 significantly inhibited the cell proliferation rate of H1703 and H460 cells (Figures S1, S4 and S5). Together, these data indicated that PAQR3 has a negative effect on the growth of human NSCLC cells.
Figure 1.
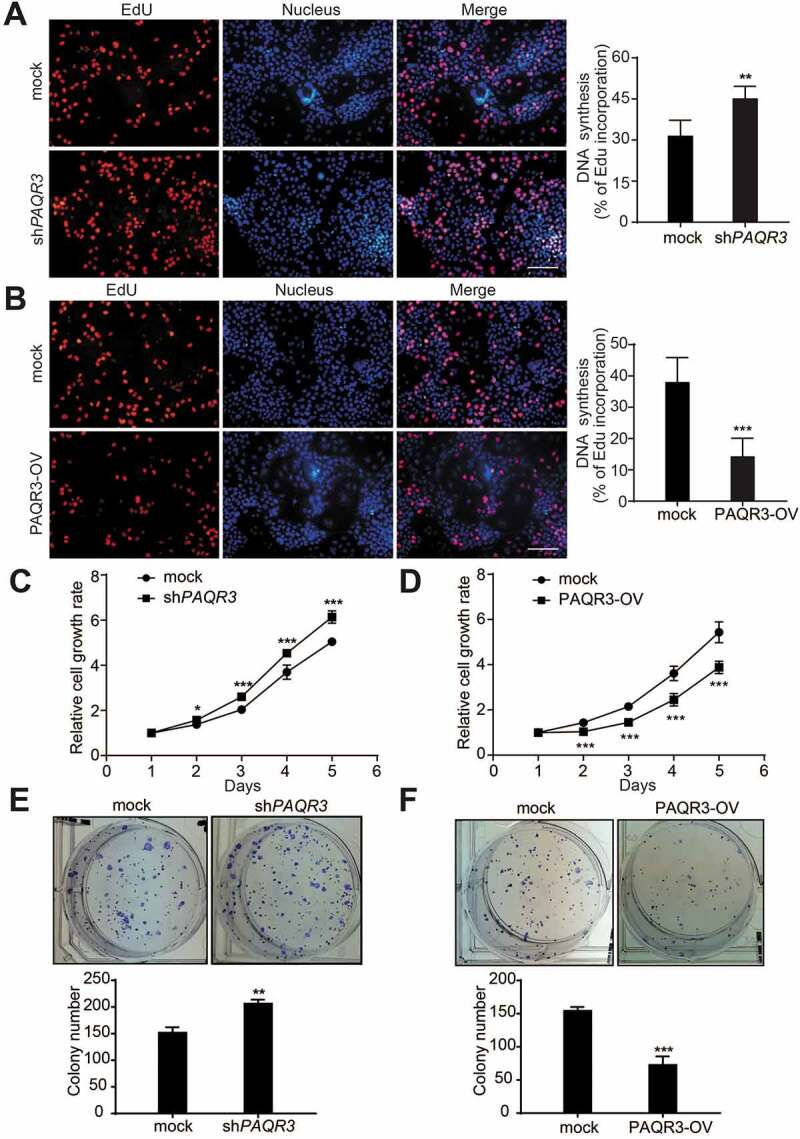
PAQR3 inhibits cell growth and colony formation of HCC827 cells.
In order to further elucidate the tumor suppressive activity of PAQR3 in NSCLC in vivo, we applied a xenograft model to investigate the effects of PAQR3 on tumor growth. HCC827 cells with stable expression of either control vector or PAQR3 knockdown vector were implanted into the nude mice. The growth of the HCC827 cells in the mice was measured by tumor size, tumor weight and tumor volume. Consistent with the in vitro study, tumor size, tumor weight and tumor volume were all increased by PAQR3 knockdown (Figure 2). These observations, therefore, clearly indicated that PAQR3 has a tumor suppressive activity in human NSCLC cells.
Figure 2.
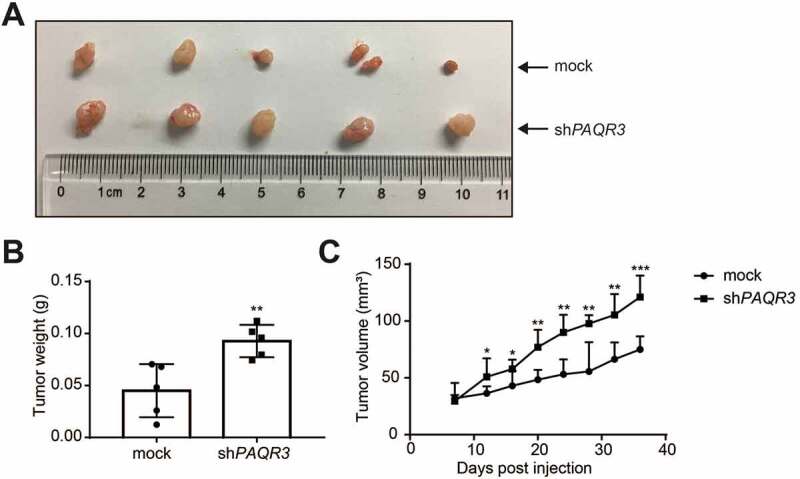
PAQR3 affects the growth of HCC827 cell xenografts in nude mice.
PAQR3 modulates erlotinib-induced autophagy in NSCLC cells
Starvation of nutrients such as glucose and amino acid as well as deficiency of growth factors can induce autophagy. In previous reports, we found that PAQR3 can modulate autophagy via regulating AMPK- and MTORC1-mediated signaling cascades in response to glucose starvation and amino acid starvation respectively [21,22]. Thus, we wondered whether or not PAQR3 could regulate autophagy induced by inhibition of growth factor-mediated signaling. As expected, treatment of HCC827 cells with erlotinib, an EGFR TKI, was able to induce autophagy shown as an increased accumulation of LC3B-II by an autophagy flux assay (Figure 3A). Consistently, knockdown of EGFR was able to abrogate erlotinib-induced autophagy in NSCLC cells (Figure S6). Knockdown of PAQR3 significantly decreased erlotinib-induced accumulation of LC3B-II in HCC827 cells (Figure 3A). A different PAQR3-specific shRNA also blocked erlotinib-induced LC3B-II accumulation in HCC827 cells (Figure S3D). On the other hand, PAQR3 overexpression increased erlotinib-induced LC3B-II accumulation in HCC827 cells (Figure 3A). We also analyzed the effect of PAQR3 on erlotinib-induced autophagy in other NSCLC cells. Knockdown of PAQR3 could abrogate erlotinib-induced LC3B-II accumulation in H1703 and H460 cells (Figure S7). Consistently, overexpression of PAQR3 significantly enhanced erlotinib-induced LC3B-II accumulation in H1703 and H460 cells (Figure S7).
Figure 3.
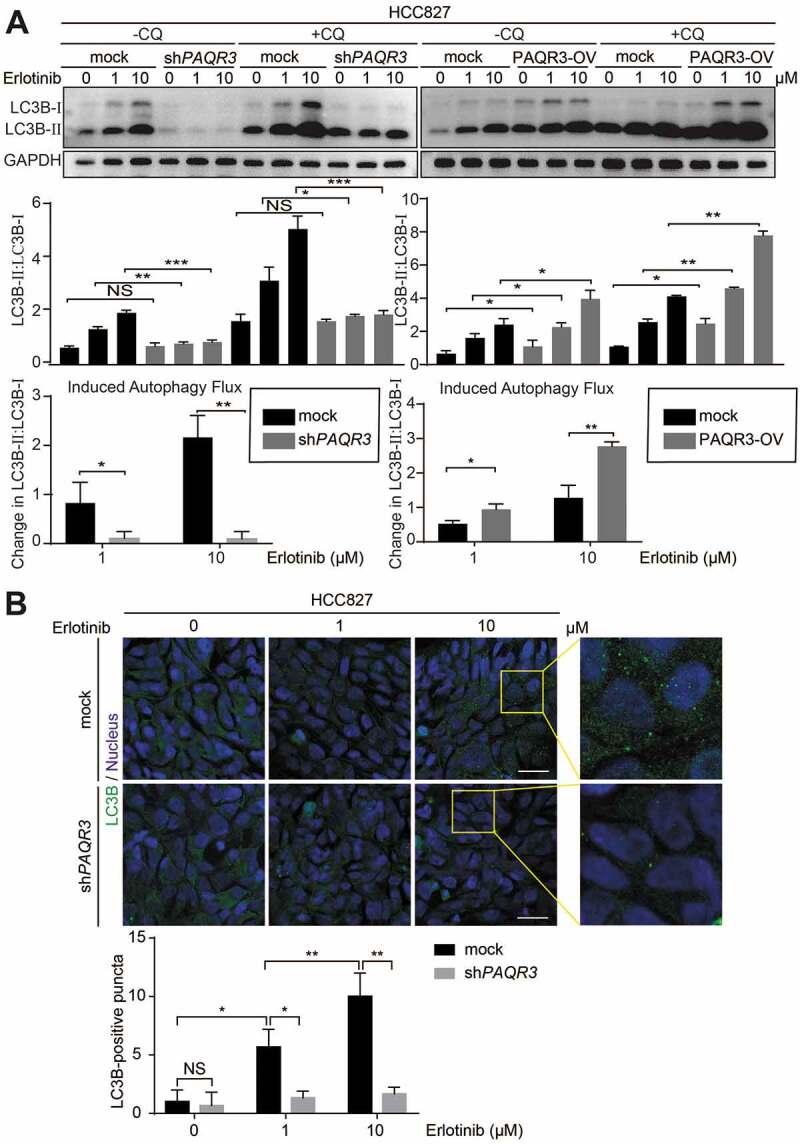
PAQR3 regulates erlotinib-induced autophagy in HCC827 cells.
It is known that the accumulation of LC3B-II is mainly achieved by two mechanisms: promotion of autophagosome formation and inhibition of LC3B-II degradation. To discriminate between these two possibilities, we treated the cells with a lysosomal inhibitor CQ (chloroquine diphosphate salt). We found that CQ-induced LC3B-II accumulation was also reduced by PAQR3 knockdown and increased by PAQR3 overexpression in the three NSCLC cell lines (Figure 3A and Figure S7), indicating that PAQR3 mainly promotes autophagosome formation instead of blocking LC3B-II degradation in these cells (Figure 3A and Figure S7). Consistent with a previous report [21], PAQR3 also positively regulated autophagy flux under the condition of glucose starvation in NSCLC cells (Figure S8).
We also analyzed autophagosome formation by LC3B immunostaining assay. Erlotinib treatment dose-dependently increased the formation of autophagosome shown as LC3B-positive staining in HCC827 cells (Figure 3B). However, erlotinib-induced autophagosome formation was drastically reduced by PAQR3 knockdown (Figure 3B). Collectively, these data suggested that PAQR3 has a positive effect on erlotinib-induced autophagy in NSCLC cells.
PAQR3 disrupts the interaction of EGFR with BECN1 and inhibits BECN1 phosphorylation
As PAQR3 can inhibit MTORC1 activity to regulate autophagy [22], we analyzed whether PAQR3 could also modulate autophagy via altering MTORC1 activity in NSCLC cells. We found that erlotinib treatment reduced MTORC1 activity shown as reduced phosphorylation of MTOR and RPS6 in HCC827, H1703 and H460 cells (Figure S9). However, knockdown or overexpression of PAQR3 could not alter erlotinib-mediated reduction of MTOR and RPS6 phosphorylation in these cells (Figure S9). These data thus indicated that the modulation of erlotinib-induced autophagy by PAQR3 is not through regulation of MTORC1 activity in NSCLC cells. We therefore explored other mechanisms underlying the regulation of PAQR3 on erlotinib-induced autophagy.
Previously, it was discovered that EGFR regulates autophagy in NSCLC cells via interacting with BECN1 complex [16]. EGFR binds BECN1 and phosphorylates multiple tyrosine sites of BECN1, leading to enhanced binding of BECN1 with autophagy-inhibitory proteins while decreasing BECN1-associated PIK3C3/VPS34 kinase activity [16]. In addition, we previously reported that PAQR3 could bind BECN1 and modulate PIK3C3/VPS34 kinase activity [21]. Thus, we hypothesized that PAQR3 might compete with EGFR for binding with BECN1, thereby affecting erlotinib-induced autophagy. To test this hypothesis, we performed a co-immunoprecipitation experiment to analyze the interaction of BECN1 with other proteins. Similar to what was previously reported [16], erlotinib disrupted the interaction of EGFR with BECN1 in HCC827, H1703 and H460 cells (Figure 4A, Figures S10A and S10B). Meanwhile, erlotinib reduced the interaction of BECN1 with autophagy-inhibitory proteins including RUBCN and BCL2 (Figure 4A, Figures S10A and S10B). However, PAQR3 knockdown abrogated the effects of erlotinib on the interactions of BECN1 with EGFR, RUBCN and BCL2 in NSCLC cells (Figure 4A, Figures S10A and S10B).
Figure 4.
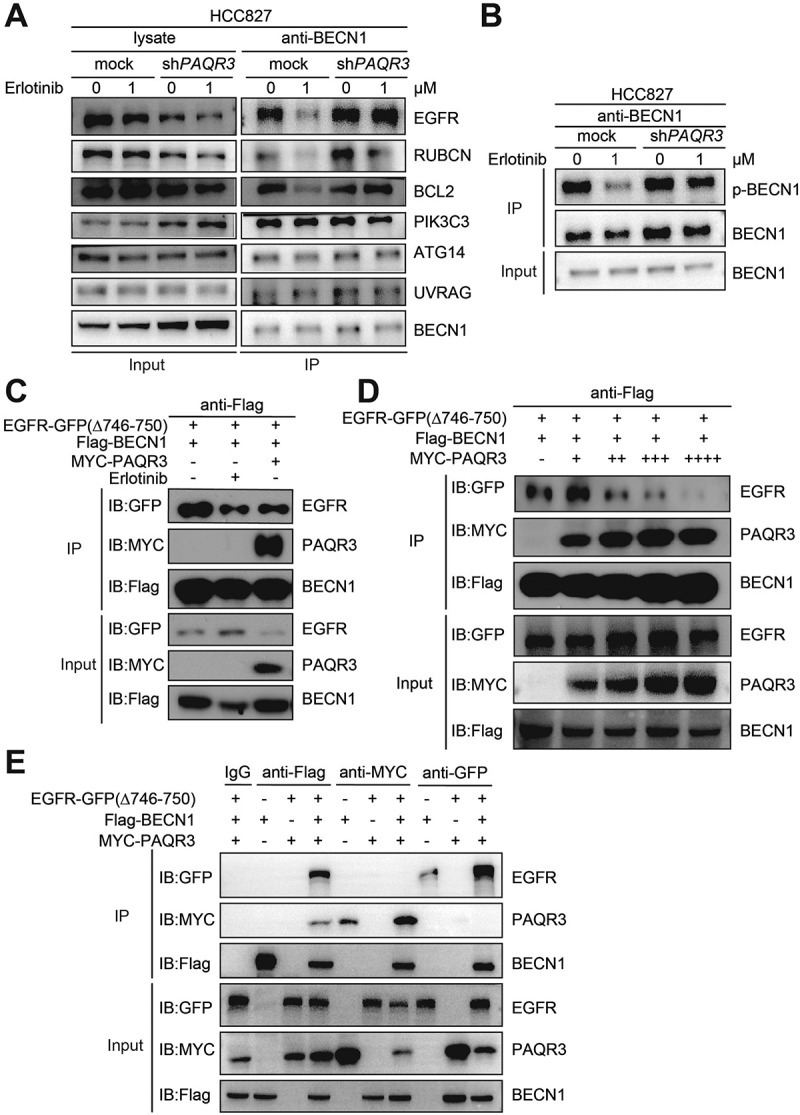
PAQR3 inhibits the interaction of EGFR with BECN1 and phosphorylation of BECN1.
Consistent with the notion that EGFR could phosphorylate BECN1 [16], we found that erlotinib reduced BECN1 phosphorylation in NSCLC cells (Figure 4B, Figures S10C and S10D). Knockdown of PAQR3 abrogated the effect of erlotinib on BECN1 phosphorylation in these cells (Figure 4B, Figures S10C and S10D).
We next analyzed whether PAQR3 could alter the interaction of BECN1 with the activated form of EGFRΔ746–750. HEK293T cells were used to analyze the interaction between ectopically expressed proteins due to their high efficiency of transfection. As expected, erlotinib treatment reduced the interaction of EGFRΔ746-750 with BECN1 by a co-immunoprecipitation assay (Figure 4C). Interestingly, overexpression of PAQR3 disrupted the interaction of EGFRΔ746-750 with BECN1 (Figure 4C). To consolidate this result, we found that overexpression of PAQR3 could dose-dependently reduce the interaction of EGFRΔ746-750 with BECN1 (Figure 4D). We also investigated whether the interactions of BECN1 with EGFR and PAQR3 were mutually exclusive. Immunoprecipitation of BECN1 could pull down both EGFR and PAQR3 (Figure 4E). Immunoprecipitation of PAQR3 could only pull down BECN1 but not EGFR, while immunoprecipitation of EGFR could only pull down BECN1 but not PAQR3 (Figure 4E). Collectively, these data indicated that PAQR3 regulates erlotinib-induced autophagy via blocking the interaction EGFR with BECN1 and phosphorylation of BECN1, leading to alteration of BECN1 interaction with inhibitory proteins involved in the regulation of autophagy.
The suppression of PAQR3 on tumor growth is dependent on autophagy
The relationship between autophagy and cancer development is complex. Some studies have shown that autophagy has a tumor suppressive activity, while others have demonstrated that it promotes tumor growth. Autophagy plays a different role in cancer biology depending on the type and context of the tumor [34]. Previously, it was found that EGFR-mediated suppression of BECN1 and inhibition of autophagy may contribute to tumor progression in NSCLC cells [16]. Considering our findings that PAQR3 modulates erlotinib-induced autophagy in HCC827 cells, we postulated that the tumor suppressive activity of PAQR3 is dependent on its regulation on autophagy. To test this hypothesis, we disrupted autophagy in HCC827 cells and investigated the tumor suppressive activity of PAQR3.
We analyzed a panel of shRNAs to knock down ATG7, a crucial gene required for autophagy, and found two of the shRNAs were effective to silence ATG7 expression in NSCLC cells (Figure 5A and Figure S11A). Consistently, these two ATG7-specific shRNAs were able to abrogate erlotinib-induced autophagy in HCC827 cells, shown as blocking erlotinib-induced LC3B-II accumulation and autophagy flux (Figure 5B). As expected, PAQR3 overexpression augmented the accumulation of LC3B-II upon erlotinib treatment (Figure 5C, left panels), while ATG7 knockdown clearly abrogated the effect of PAQR3 overexpression on LC3B-II accumulation in HCC827 cells (Figure 5C, right panels). Consistently, ATG7 knockdown also abrogated the effect of PAQR3 overexpression on LC3B-II accumulation in two other NSCLC cells (Figures S11B and S11C).
Figure 5.
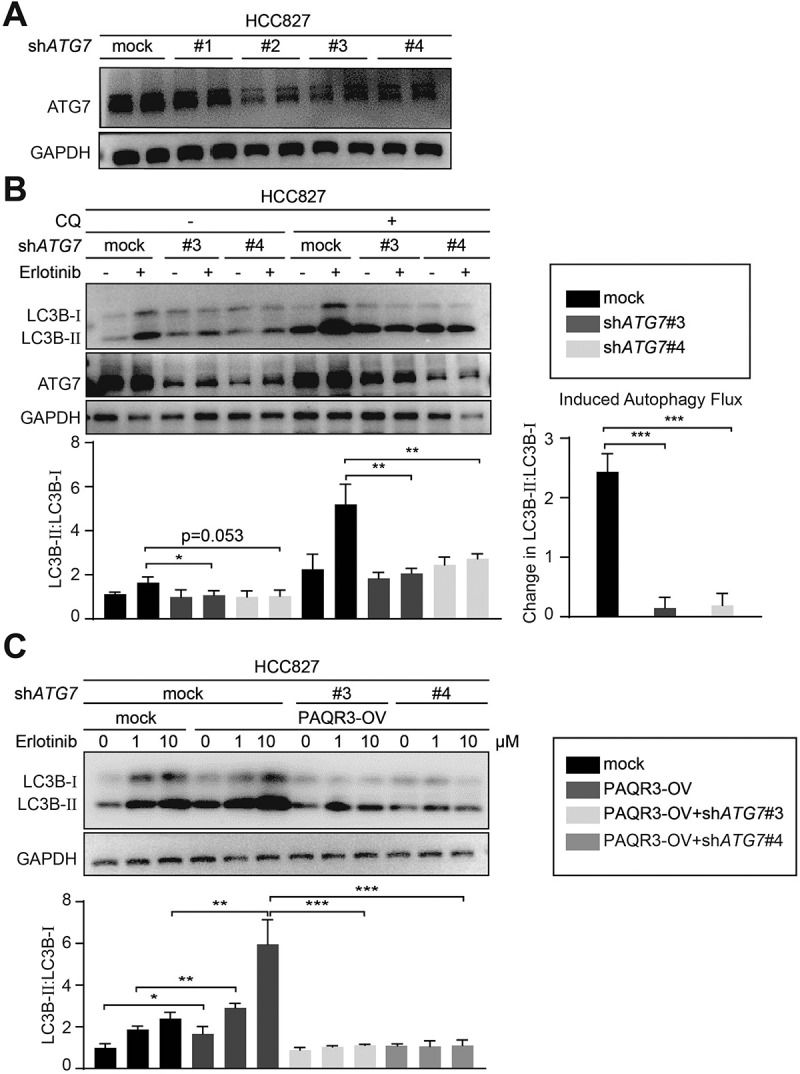
Knockdown of ATG7 abrogates the stimulatory effect of PAQR3 on autophagy.
Next, we analyzed the effect of ATG7 knockdown on the suppressive activity of PAQR3 in lung cancer cell proliferation. EdU assay revealed that the inhibitory effect of PAQR3 on the cell proliferation rate in NSCLC cells was completely abrogated by ATG7 knockdown (Figure 6A and Figure S12). Cell counting also demonstrated that the inhibitory effect of PAQR3 on the cell growth rate of HCC827 cells was abrogated by ATG7 knockdown (Figure 6B). Furthermore, PAQR3 overexpression could no longer inhibit colony formation of HCC827 cells when ATG7 was silenced (Figure 6C). These data, therefore, indicated that the suppressive activity of PAQR3 on NSCLC cell proliferation is dependent on autophagy.
Figure 6.
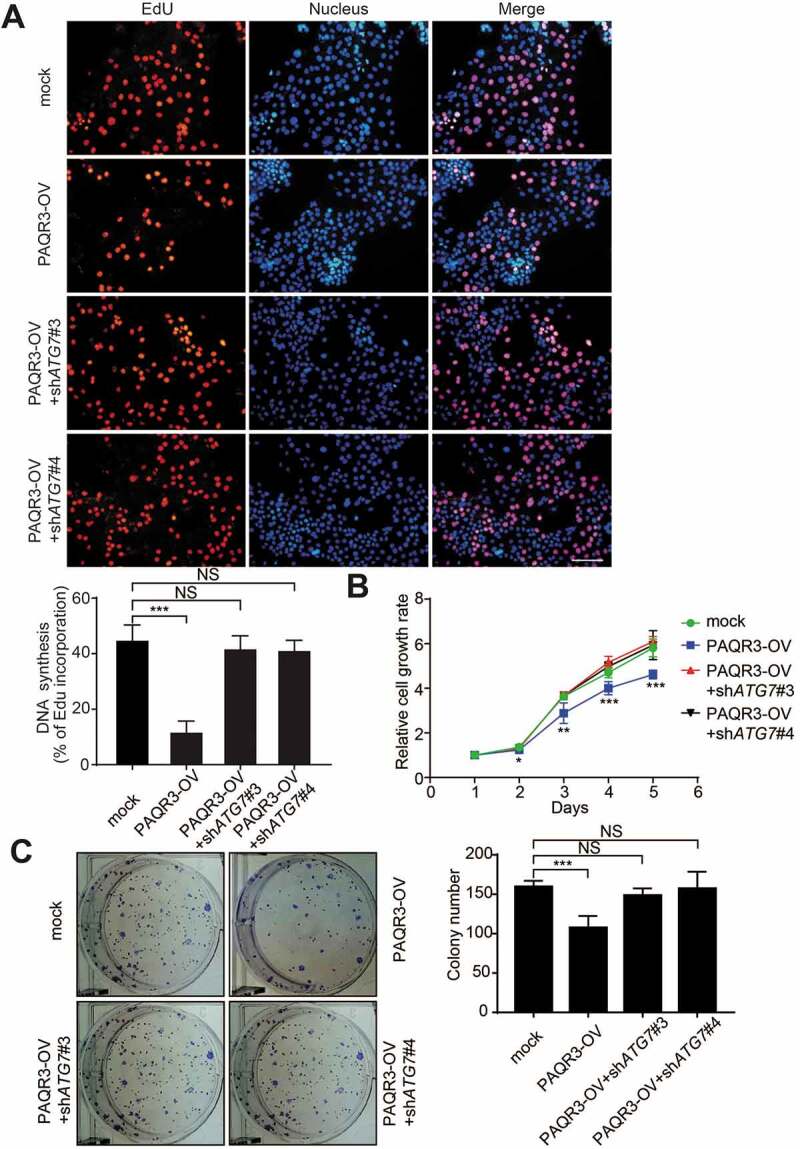
Effect of ATG7 knockdown on HCC827 cell growth in vitro.
We next performed a xenograft model to further test the hypothesis in vivo. HCC827 cells were infected with lentivirus containing control vector, PAQR3 overexpressing plasmid and/or ATG7-specific shRNA. The cells were implanted into the nude mice. As expected, the tumor size, tumor weight and tumor volume were all decreased by PAQR3 overexpression in the mice (Figure 7). However, knocking down ATG7 in PAQR3-overexpression cells abrogated the inhibitory effect of PAQR3 overexpression on tumor growth (Figure 7). Together, these data further demonstrated that the inhibitory effect of PAQR3 on NSCLC tumor growth is dependent on autophagy.
Figure 7.
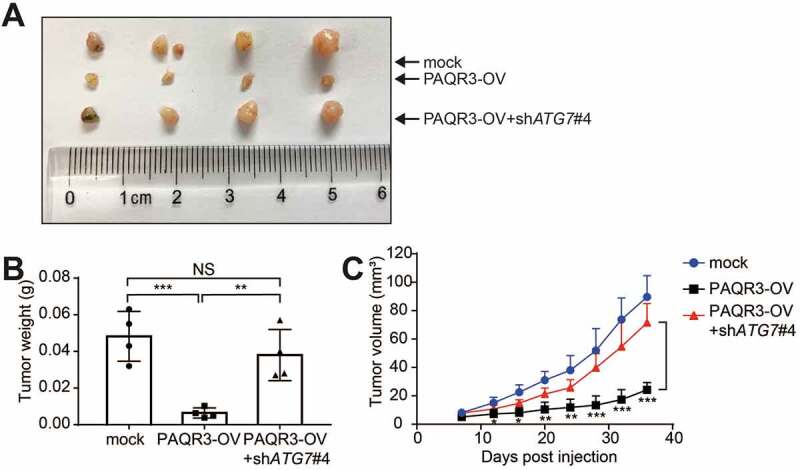
Effect of ATG7 knockdown on HCC827 cell growth in vivo.
Discussion
Our study reveals for the first time that the tumor suppressive activity of PAQR3 is linked to its regulation of autophagy. PAQR3 is dysregulated in different types of human cancer including colon cancer, gastric cancer, bladder cancer, liver cancer, osteosarcoma, breast cancer, laryngeal squamous cell carcinoma, and prostate cancer [23–30]. In this study, we also found that PAQR3 inhibits the growth of NSCLC cells both at the cell and animal levels. These results further indicate that PAQR3 has a tumor suppressive activity in a broad spectrum of human cancers.
Our study reveals for the first time that the tumor suppressive activity of PAQR3 is dependent on autophagy. Knockdown of ATG7 to block autophagy almost completely abrogates the inhibitory effect of PAQR3 on the proliferation of NSCLC cells (Figure 6). In addition, the suppressive activity of PAQR3 on the tumor growth of NSCLC cells is abrogated by ATG7 knockdown (Figure 7). These in vitro and in vivo data suggest that autophagy is crucial to mediate the anti-tumor effect of PAQR3.
Mechanistically, our study demonstrates that PAQR3 modulates TKI-induced autophagy in lung cancer cells by blocking EGFR interaction with BECN1 and inhibiting BECN1 phosphorylation (Figure 4 and Figure S13). It was reported that EGFR regulates autophagy in NSCLC cells via interacting with BECN1 complex [16]. Activated EGFR binds BECN1 and phosphorylates multiple tyrosine sites of BECN1, leading to enhanced binding of BECN1 with autophagy-inhibitory proteins such as RUBCN and BCL2 [16]. Consequently, the BECN1-associated PIK3C3/VPS34 kinase activity is inhibited and autophagy is compromised [16]. We found that erlotinib-mediated dissociation of BECN1 with EGFR, RUBCN and BCL2 was abrogated by PAQR3 knockdown. Erlotinib-mediated reduction of BECN1 phosphorylation was also abrogated by PAQR3 knockdown. BECN1 mutually exclusively interacts with EGFR and PAQR3. As a result, PAQR3 dose-dependently competes off the interaction of EGFR with BECN1, thus blocking the inhibitory effect of activated EGFR on autophagy (Figure S13).
To further consolidate our results that PAQR3 competes off EGFR interaction with BECN1 to regulate autophagy, we analyzed a PAQR3 mutant that lacks a BECN1 binding domain as reported by a previous study [21]. Both EdU assay and cell counting revealed that such a PAQR3 mutant could no longer inhibit the cell proliferation of HCC827 cells (Figure S14). These results, therefore, indicated that the suppressive activity of PAQR3 in NSCLC cells is dependent on its BECN1 interaction.
A previous study revealed that combination AKT inhibitor with gefitinib to inhibit autophagy can synergistically induce cell death in HCC827 cells [35]. In vitro, AKT inhibitor MK-2206 synergizes with erlotinib to inhibit cell proliferation and induce apoptosis of NSCLC cells [36]. In vivo, phase I and phase II clinical studies further demonstrated that AKT inhibitor MK2206 plus erlotinib shows better anti-tumor activity than erlotinib alone in NSCLC patients [36,37]. Intriguingly, PAQR3 can also inhibit AKT activation by inhibiting signaling of G protein βγ-subunit and by inhibiting PI3K via spatial regulation of PIK3CA/p110α subunit [19,20]. We also identified that the N-terminal 6–55 amino acid residues of PAQR3 is sufficient for its interaction with PIK3CA [38]. A synthetic peptide, P6-55, that contains these amino acid residues is able to disrupt the interactions of PIK3CA with PAQR3 and inhibit the oncogenic activities of PI3K, contributing to reduced progression of gastric cancer cells [38]. Therefore, it can be postulated that the inhibition of AKT activation and potentiation of autophagy by PAQR3 as reported here may synergistically execute the tumor suppressive activity of PAQR3 in NSCLC cells. Furthermore, PAQR3 is able to inhibit RAS-RAF-MAP2K/MEK-MAPK/ERK signaling cascade by spatial regulation of Raf kinase [18,39]. In this regard, PAQR3 can suppress tumor growth via three pathways: inhibition of RAS-RAF-MAP2K/MEK-MAPK/ERK pathway, inhibition of PI3K-AKT pathway and stimulation of autophagy. Considering such important and multi-faceted activities of PAQR3, it will be of great importance to explore PAQR3 as a potential therapeutic target in many types of cancers including NSCLC in the future. For example, it will be tempting to test whether the PAQR3-based peptide, that has been shown to block AKT activation [38], is able to enhance the anti-tumor activity of erlotinib or gefitinib in NSCLC cells.
Materials and methods
Antibodies and reagents
The antibodies used in the study were as follows: LC3B (2775, Boston, USA), PIK3C3/VPS34 (4263), ATG14 (5504S), phosphorylated MTOR (Ser2448; 2971), RPS6/S6 ribosomal protein (2317S), phosphorylated RPS6/S6 ribosomal protein (Ser235/236; 2211S), RUBCN/rubicon (8465S) were purchased from Cell Signaling Technology. BECN1 (Abcam, ab207612, Cambridge, UK), UVRAG (MBL International Corporation, M160-3, Tokyo, Japan), ATG7 (ABclonal, A091, Boston, USA), BCL2 (ABclonal, A0208). EGFR (sc-373746, Santa Cruz, CA), pTyr99-HRP (sc-7020), GAPDH (sc-365062), MYC (sc-40) were purchased from Santa Cruz Biotechnology. TUBA4A/tubulin (Sigma-Aldrich, T5168), Flag (Sigma-Aldrich, F3165). MTT (Sigma-Aldrich, M2128) and erlotinib (Selleck Chemicals, S7786, Houston, USA).
Plasmid construction
The MYC-tagged PAQR3 and Flag-tagged BECN1 were described previously [21]. The wild-type (WT) and active mutant EGFRΔ746-750 were cloned by RT-PCR from human cells and confirmed by DNA sequencing.
Cell culture and transfection
HEK293T cells were grown in DMEM (Thermo Fisher Scientific, 11995–065) medium with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, 10100154) and 1% penicillin/streptomycin. The NSCLC cell lines including HCC827, H1703 and H460 cells (Cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences, SCSP-538, SCSP-593 and SCSP-584) were maintained in RPMI-1640 medium (Thermo Fisher Scientific, 11875–093) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and 1% penicillin-streptomycin. Stable knockdown and overexpression of PAQR3 in NSCLC cells were generated by lentivirus infection as previous described. Another short hairpin RNA (shRNA) construct specific for PAQR3 and two shRNA constructs for EGFR were inserted into the hRNU6-MCS-CMV-Puro vector (LncBio Co., shRNA-304, Shanghai, China). The shRNA targeting sequences were 5′-GCTTTGCTCTGTGGGCTATCA-3′ for human PAQR3; 5′-GCAGTGACTTTCTCAGCAACA-3′ and 5′-GCGAAGGGCCTTGCCGCAAAG-3′ for human EGFR. The shRNA constructs for ATG7 were inserted into the RNU6-MCS-Ubiquitin-Cherry-IRES-puromycin vector (Genechem Co., GV298, Shanghai, China). The shRNA targeting sequences were 5′-CAGCTATTGGAACACTGTA-3′ and 5′- CTGCTGAGGAGCTCTCCAT −3′ for human ATG7. Transient transfection in HEK293T cells was performed using polyethylenimine (Sigma-Aldrich, 408727).
Autophagy studies
Autophagy induced by glucose starvation was performed by culturing cells in the no-glucose RPMI-1640 (Thermo Fisher Scientific, 11879–020) for 6 h. Autophagy was also analyzed in cells treated with pharmacological inhibitors of EGFR (Erlotinib). Autophagy was examined by measuring the LC3B-II:LC3B-I ratio by immunoblotting analysis. The levels of LC3B-II and LC3B-I were normalized to GAPDH or TUBA4A/α-tubulin. The cells were also treated with 20 µM chloroquine diphosphate (CQ; Millipore Sigma, C6628) for 4 h to inhibit lysosome function. Autophagy flux was measured by subtracting the LC3B-II:LC3B-I ratio without CQ treatment from the LC3B-II:LC3B-I ratio with CQ treatment. Induced autophagy flux was calculated by comparing the autophagy flux with and without autophagy induction (e.g. erlotinib treatment or glucose starvation).
Co-immunoprecipitation and immunoblotting
For co-immunoprecipitation (Co-IP) assays, the HEK293T cells were transfected with indicated plasmids for 24 h, then lysed with ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 300 mM NaCl, 10% glycerol, 3 mM EDTA, 1 mM MgCl2, 1% Triton X-100 and protease inhibitor cocktail) as described previously [22]. The homogenates were centrifuged for 15 min at 134,000 x g at 4°C. About 10% of the supernatant was harvested as inputs, and the remaining cell lysate was incubated with indicated antibodies overnight at 4°C. Protein A/G plus agarose (Genescript Co., L00209) was added at 4°C for 2 h, and then washed with lysis buffer for 5 times. The resulting proteins were used for western blotting analysis.
Immunofluorescence staining
Confocal microscopy analysis was performed as previously reported [21]. Autophagosomes were stained with antibodies against LC3B. Nuclei were stained with Hoechst 33342 (Molecular Probes, H-3570). Pictures in the same panel were taken under the same excitation conditions in order to precisely examine autophagy under different conditions.
Cell proliferation assays
Cell proliferation rate was detected with EdU (5-ethynyl-2ʹ-deoxyuridine) Cell Proliferation Kit (C0075S) and Cell Counting Kit-8 (CCK-8, C0038) from Beyotime Biotechnology. For EdU assay, NSCLC cells were inoculated into 6-well culture plate and analyzed according to the instruction of the kit. For cell counting assay, NSCLC cells were seeded at a density of 5 × 103 cells/well into a 96-well culture plate. The cells were then incubated with 10 μL of CCK-8 for 2 h at 37°C. The absorbance was measured at 450 nm wavelength with a spectrophotometer. For MTT assay, HCC827 cells were seeded at a density of 5 × 103 cells/well into a 96-well culture plate and analyzed as previously described [30]. For colony formation assay, the cells were inoculated into 6-well culture plate with 500 cells/well and cultured for 10 d. Then the cells were fixed with 4% paraformaldehyde and stained with crystal violet (Sigma, C3886).
Nude mice xenograft model
All of the animal studies were carried out in accordance with the Chinese Academy of Sciences ethics commission with an approval number 2010-AN-8. Nude mice were purchased from SLAC (Shanghai, China) and maintained on a 12 h light/dark cycle at 25°C. The indicated HCC827 cells in the logarithmic phase of growth were trypsinized, centrifuged and rinsed with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) three times. Each five nude mice (4 weeks old, female) were injected with a clonal population of HCC827 cells (5 X 106 cells) in 0.1 ml of RPMI-1640 with 50% Matrigel (Corning Incorporated, 354248) in the upper right shoulders subcutaneous. Xenograft tumor sizes were measured by vernier caliper with two perpendicular diameters every other day and calculated according to the formula: 0.5 x length x width2.
Statistical analyses
Student’s t-test was used for all the statistical analyses. The P value < 0.5 was considered statistically significant.
Supplementary Material
Funding Statement
This study was supported by research grants from National Natural Science Foundation of China [31630036 to Y.C.], Ministry of Science and Technology of China [2016YFA0500103 to Y.C.], Chinese Academy of Sciences [XDA12010102, QYZDJ-SSW-SMC008, ZDRW-ZS-2016-8, and CAS Interdisciplinary Innovation Team to Y.C.].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher‘s website.
References
- [1].Levine B, Klionsky DJ.. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. [DOI] [PubMed] [Google Scholar]
- [2].Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Galluzzi L, Pietrocola F, Levine B, et al. Metabolic Control of Autophagy. Cell. 2014;159:1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. [DOI] [PubMed] [Google Scholar]
- [5].He CC, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Feng YC, He D, Yao ZY, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].White E. The role for autophagy in cancer. J Clin Invest. 2015;125:42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Folkerts H, Hilgendorf S, Vellenga E, et al. The multifaceted role of autophagy in cancer and the microenvironment. Med Res Rev. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell Metab. 2017;25:1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. [DOI] [PubMed] [Google Scholar]
- [13].Roman M, Baraibar I, Lopez I, et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer. 2018;17:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. [DOI] [PubMed] [Google Scholar]
- [15].Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wei Y, Zou Z, Becker N, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154:1269–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tan X, Thapa N, Sun Y, et al. A kinase-independent role for EGF receptor in autophagy initiation. Cell. 2015;160:145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Feng L, Xie XD, Ding QR, et al. Spatial regulation of Raf kinase signaling by RKTG. Proc Natl Acad Sci U S A. 2007;104:14348–14353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jiang YH, Xie XD, Li ZG, et al. Functional Cooperation of RKTG with p53 in Tumorigenesis and Epithelial-Mesenchymal Transition. Cancer Res. 2011;71:2959–2968. [DOI] [PubMed] [Google Scholar]
- [20].Wang X, Wang LD, Zhu L, et al. PAQR3 modulates insulin signaling by shunting phosphoinositide 3-Kinase p110 alpha to the golgi apparatus. Diabetes. 2013;62:444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xu DQ, Wang Z, Wang CY, et al. PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. Embo J. 2016;35:496–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang L, Pan Y, Huang MQ, et al. PAQR3 augments amino acid deprivation-induced autophagy by inhibiting mTORC1 signaling. Cell Signal. 2017;33:98–106. [DOI] [PubMed] [Google Scholar]
- [23].Wang X, Li XB, Fan FJ, et al. PAQR3 plays a suppressive role in the tumorigenesis of colorectal cancers. Carcinogenesis. 2012;33:2228–2235. [DOI] [PubMed] [Google Scholar]
- [24].Ling ZQ, Guo W, Lu XX, et al. A Golgi-specific protein PAQR3 is closely associated with the progression, metastasis and prognosis of human gastric cancers. Ann Oncol. 2014;25:1363–1372. [DOI] [PubMed] [Google Scholar]
- [25].Xiu YC, Liu Z, Xia SY, et al. MicroRNA-137 upregulation increases bladder cancer cell proliferation and invasion by targeting PAQR3. Plos One. 2014;9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [26].Wu HG, Zhang WJ, Ding Q, et al. Identification of PAQR3 as a new candidate tumor suppressor in hepatocellular carcinoma. Oncol Rep. 2014;32:2687–2695. [DOI] [PubMed] [Google Scholar]
- [27].Ma ZQ, Wang YL, Piao TK, et al. The tumor suppressor role of PAQR3 in osteosarcoma. Tumor Biol. 2015;36:3319–3324. [DOI] [PubMed] [Google Scholar]
- [28].Li ZH, Ling ZQ, Guo WW, et al. PAQR3 expression is downregulated in human breast cancers and correlated with HER2 expression. Oncotarget. 2015;6:12357–12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu Q, Zhuang K, Li HJ. PAQR3 plays a suppressive role in laryngeal squamous cell carcinoma. Tumor Biol. 2016;37:561–565. [DOI] [PubMed] [Google Scholar]
- [30].Huang WQ, Guo WW, You X, et al. PAQR3 suppresses the proliferation, migration and tumorigenicity of human prostate cancer cells. Oncotarget. 2017;8:53948–53958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ciardello F, Tortora G. EGFR antagonists in cancer treatment (vol 358, pg 1160, 2008). New Engl J Med. 2009;360:1579. [DOI] [PubMed] [Google Scholar]
- [32].Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New Engl J Med. 2004;350:2129–2139. [DOI] [PubMed] [Google Scholar]
- [33].Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wilde L, Tanson K, Curry J, et al. Autophagy in cancer: a complex relationship. Biochem J. 2018;475:1939–1954. [DOI] [PubMed] [Google Scholar]
- [35].Bokobza SM, Jiang Y, Weber AM, et al. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget. 2014;5:4765–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Molife LR, Yan L, Vitfell-Rasmussen J, et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol. 2014;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lara PN Jr., Longmate J, Mack PC, et al. Phase II study of the AKT Inhibitor MK-2206 plus erlotinib in patients with advanced non-small cell lung cancer who previously progressed on erlotinib. Clin Cancer Res. 2015;21:4321–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guo W, You X, Wang X, et al. A synthetic peptide hijacks the catalytic subunit of class I PI3K to suppress the growth of cancer cells. Cancer Lett. 2017;405:1–9. [DOI] [PubMed] [Google Scholar]
- [39].Fan F, Feng L, He J, et al. RKTG sequesters B-Raf to the Golgi apparatus and inhibits the proliferation and tumorigenicity of human malignant melanoma cells. Carcinogenesis. 2008;29:1157–1163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.