

ABSTRACT
Mitophagy is a critical process that safeguards mitochondrial quality control in order to maintain proper cellular homeostasis. Although the mitochondrial-anchored receptor Atg32-mediated cargo-recognition system has been well characterized to be essential for this process, the signaling pathway modulating its expression as a contribution of governing the mitophagy process remains largely unknown. Here, bioinformatics analyses of epigenetic or transcriptional regulators modulating gene expression allow us to identify the Paf1 complex (the polymerase-associated factor 1 complex, Paf1C) as a transcriptional repressor of ATG genes. We show that Paf1C suppresses glucose starvation-induced autophagy, but does not affect nitrogen starvation- or rapamycin-induced autophagy. Moreover, we show that Paf1C specifically regulates mitophagy through modulating ATG32 expression. Deletion of the genes encoding two core subunits of Paf1C, Paf1 and Ctr9, increases ATG32 and ATG11 expression and facilitates mitophagy activity. Although Paf1C is required for many histone modifications and gene activation, we show that Paf1C regulates mitophagy independent of its positive regulatory role in other processes. More importantly, we also demonstrate the mitophagic role of PAF1C in mammals. Overall, we conclude that Paf1C maintains mitophagy at a low level through binding the promoter of the ATG32 gene in glucose-rich conditions. Dissociation of Paf1C from ATG32 leads to the increased expression of this gene, and mitophagy induction upon glucose starvation. Thus, we uncover a new role of Paf1C in modulating the mitophagy process at the transcriptional level.
Abbreviations
AMPK: AMP-activated protein kinase; ATP5F1A: ATP synthase F1 subunit alpha; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CCCP: chlorophenylhydrazone; DFP: chelator deferiprone; GFP: green fluorescent protein; H2B-Ub1: H2B monoubiquitination; HSPD1/HSP60: heat shock protein family D (Hsp60) member 1; KD: kinase dead; OPTN, optineurin; Paf1: polymerase-associated factor 1; PINK1: PTEN induced kinase 1; PRKN/Parkin: parkin RBR E3 ubiquitin protein ligase; RT-qPCR: real-time quantitative PCR; SD-N: synthetic dropout without nitrogen base; TIMM23: translocase of inner mitochondrial membrane 23; TOMM20: translocase of outer mitochondrial membrane 20; WT: wild-type; YPD: yeast extract peptone dextrose; YPL: yeast extract peptone lactate
KEYWORDS: Atg32, glucose starvation, mitophagy, Paf1 complex, transcription
Introduction
Mitochondria are the powerhouses of the cell, where the majority of cellular energy is usually generated. Mitochondria are also dynamic organelles that function in many other cellular processes, such as cell death, calcium homeostasis and autophagy [1,2]. Degradation of excess or damaged mitochondria is one of the major concerns that eukaryotes have to face in maintaining cellular metabolism and adjusting cellular activities to environmental changes [3]. Improper accumulation of damaged molecules in mitochondria would cause aging and many human diseases, such as cancer and neurodegenerative disease [4,5]. Thus, a conserved protection system called autophagy is prevalently utilized to maintain the proper homeostasis of various intracellular constituents in the cell. During macroautophagy/autophagy phagophores, transient double-membrane compartments, sequester cytoplasm and then mature into double-membrane autophagosomes that subsequently transport their cargo to vacuoles in yeast or lysosomes in mammals for degradation and recycling [6,7].
Besides nonselective bulk autophagy, selective autophagy has been appreciated in the past years as a specific degradation pathway for removal of useless or damaged components and organelles through particular cargo-receptor recognition under certain nutrient or environmental cues [8,9]. For example, in budding yeast, selective degradation of the endoplasmic reticulum, termed reticulophagy, is mediated by two receptor proteins, Atg39 and Atg40, during nutrient starvation [10]; selective degradation of mitochondria, termed mitophagy, is mediated by the outer mitochondria membrane receptor Atg32 under respiratory conditions [11,12]; and the selective removal of peroxisomes by autophagy, pexophagy, occurs when cells are shifted from oleic acid to glucose [13]. Pexophagy, reticulophagy and mitophagy require that receptors interact with Atg8 and Atg11, proteins critical for recognition of the cargo [9,10,14,15]. Mitophagy in mammals is similar, but is mechanistically more complicated than in yeast. It has been reported that several receptors, including SQSTM1/p62 (sequestsome 1), OPTN (optimeurin), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), BNIP3L (BCL2 interacting protein 3 like) and FUNDC1 (FUN14 domain containing 1), bind to MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta, one of the yeast Atg8-family homologs) and mediate mitophagy in a PINK1 (PTEN induced kinase 1)-PRKN/Parkin (parkin RBR E3 ubiquitin protein ligase)-dependent or -independent manner (reviewed in [8,16]). In addition, BCL2L13 (BCL2 like 13) has been suggested as the yeast Atg32 homolog to mediate mitophagy [17]. Recently, the inner mitochondrial membrane protein PHB2 (prohibitin 2) was identified to promote mitophagy in a proteasome- and PINK1-PRKN-dependent manner [18]. These molecular studies of mitophagy have led to an understanding of the receptor-mediated autophagy machinery; yet, regulatory mechanisms that act at multiple levels to govern their functions remain largely unknown. Particularly, the transcriptional regulation of the ATG genes is still obscure.
So far, 42 ATG genes have been identified in fungi and many have homologs in more complex eukaryotes. The expression of many, if not all, of the ATG genes as well as their encoding proteins are substantially increased in response to autophagy induction [19]. This prompt upregulation upon such an induction is critical for optimal autophagy efficiency and energy usage. At present, several studies using yeast illustrate that a few transcriptional regulators are involved in regulation of the expression of ATG genes. For instance, Pho23 represses ATG9 expression under nutrient-rich conditions, which controls the frequency of autophagosome formation [20]. More recently, the histone demethylase Rph1 was discovered as a master transcriptional repressor to control the expression of a subset of ATG genes [21]. Yet, overall, very few transcription factors have been identified as regulating the expression of the ATG genes, and whether the induction of ATG genes contributes to autophagy activity is still poorly understood.
To identify additional transcriptional regulators that function in autophagy, we took advantage of a previously published microarray database that has summarized the global expression patterns from 165 yeast strains bearing individual deletions of epigenetic or transcriptional regulators. Analysis of the expression changes of ATG genes in any deletion strains compared to the wild type led to the finding of Paf1C as a transcriptional repressor of ATG32 to specifically regulate mitophagy.
Results
Deletion of the genes encoding Paf1C subunits leads to upregulated expression of ATG genes
To identify epigenetic or transcriptional regulators of autophagy, we analyzed the expression levels of 36 ATG genes from the 165 deletion mutants in the dataset, compared to the wild-type (WT) strain [22]. We set a fold-change threshold of 1.4 as the selection bottom line (log2 value above 0.5 or below −0.5) in order to generate a list of relevant regulatory components. Intriguingly, 4 subunits of the Paf1 complex appeared on the list (Table S1).
The evolutionarily conserved Polymerase-Associated Factor 1 complex, Paf1C, comprises 5 conserved subunits, Paf1, Ctr9, Cdc73, Rtf1 and Leo1, throughout eukaryotes. Paf1C is well known for its essential roles in promoting RNA polymerase II transcription elongation and transcription-coupled histone modifications [23,24]. Paf1C also functions in controlling gene expression and governing chromatin structure [23]. Notably, Paf1 and Ctr9 have been reported as the core components of Paf1C essential for complex integrity and functional regulation [25,26]. Thus, we particularly paid attention to those 2 proteins and observed similar gene expression patterns in paf1Δ and ctr9Δ mutants relative to each other when compared to WT cells (Figure 1A). Among them, approximately 75% of the 506 upregulated genes overlapped in both paf1Δ and ctr9Δ mutants (Figure 1B). Gene Ontology analyses of biological processes demonstrate that the 506 upregulated genes are associated with multiple cellular events, including autophagy (Figure 1C).
Figure 1.
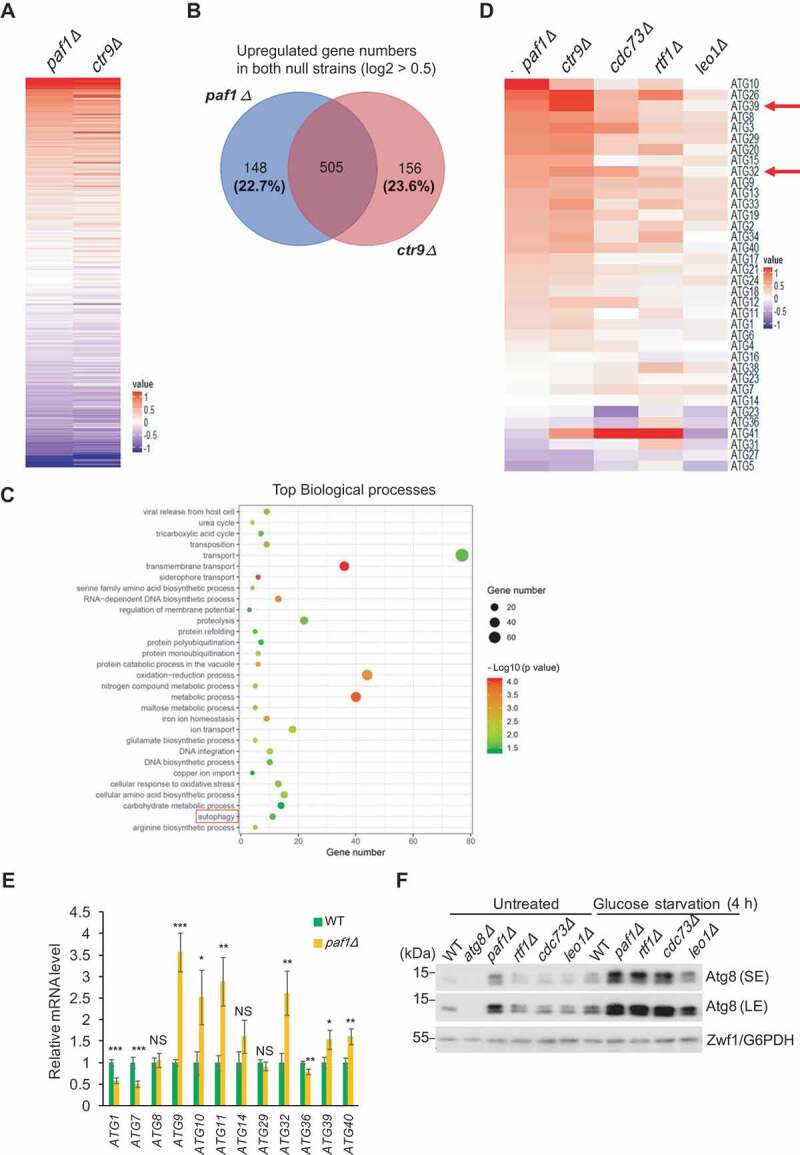
Paf1C regulates ATG gene expression. (A) Heat map of the altered expression patterns in paf1Δ and ctr9Δ strains from an average of 2 biological repeats. The color scales indicate the log2 ratio of relative expression levels. (B) Venn diagram showing the number of overlapping genes among those that are upregulated (1.4-fold increase, log2 > 0.5) in both paf1Δ and ctr9Δ strains. (C) Gene Ontology analyses of biological processes show the significant upregulated genes in (B). The red box points out the autophagy pathway. (D) Heat map of ATG gene expression patterns in the Paf1C-defective strains. The color scales indicate the log2 ratio of relative expression levels. (E) Relative mRNA levels of the indicated ATG genes in WT and paf1Δ cells were quantified by RT-qPCR. Error bars indicate the SD of 3 independent experiments. (F) Cell lysates from the indicated cells were loaded onto an SDS-PAGE gel containing 6 M urea, and Atg8 protein levels were examined by immunoblotting with anti-Atg8 serum. The lower bands indicate phosphatidylethanolamine-conjugated forms of Atg8 (Atg8–PE). Zwf1/G6PDH was used as a loading control. SE, short exposure; LE, long exposure.
When the altered expression profiles of the ATG genes were summarized among Paf1C subunit gene-deleted mutants, we noticed that the majority of genes were upregulated (Figure 1D). Gene expression changes upon PAF1 deletion were confirmed for several genes using real-time quantitative PCR (RT-qPCR) analyses, including ATG9, ATG11, ATG32 and ATG40 (Figure 1E), supporting the hypothesis that in general Paf1C might regulate ATG gene expression and, accordingly, autophagy.
To test whether Paf1C affects ATG gene expression upon different starvation inductions, we compared expression changes of ATG genes in WT and paf1Δ strains under various conditions. Under nitrogen-starvation and rapamycin-treatment conditions, the majority of ATG genes showed no difference between these two strains (Figure S1A, S1B). Intriguingly, ATG11 and ATG32 still exhibited a significant increase in the paf1Δ strain under glucose-starvation conditions, although the divergence of gene expression of other ATG genes became subtle (Figure S1C). These data suggested that Paf1C may specifically regulate glucose starvation-induced autophagy. Indeed, after deleting the genes encoding subunits of Paf1C, both Atg8 and phosphatidylethanolamine (PE)-conjugated Atg8 proteins were detected at higher levels by immunoblotting even in cells cultured in nutrient-rich medium (Figure 1F). Upon glucose starvation, the enrichment of Atg8 proteins significantly increased in Paf1C null mutants compared to WT cells, further suggesting the potential role of Paf1C in autophagy regulation.
Paf1C suppresses glucose starvation-induced autophagy, but does not affect nitrogen starvation- or rapamycin-induced autophagy
To further investigate whether Paf1C regulates autophagy specifically through glucose starvation induction, WT cells and various Paf1 subunit gene-null mutants expressing endogenous promoter-driven Atg8 with an N-terminal GFP (green fluorescent protein) tag (GFP-Atg8) were cultured in different starvation conditions, and GFP-Atg8 processing assays were performed [27,28]. In brief, GFP-Atg8 initially lines both sides of the phagophore. The population on the concave surface becomes trapped within the mature autophagosome and is delivered to the vacuole, where proteolytic degradation releases the relatively stable GFP moiety. After 1.5 h or 3 h of glucose starvation, autophagy was significantly induced to a higher extent in either of the Paf1C subunit gene-null mutants, as indicated by the levels of free GFP, compared to the WT cells (Figure 2A and Figure S2A). In contrast, regardless of short or longer induction by nitrogen starvation or by rapamycin treatment, free GFP moieties were generated to a similar extent among the WT and Paf1C subunit gene-null mutants (Figure 2B, Cand Figure S2B). Altogether, these data suggest that Paf1C plays an important role in glucose starvation-induced autophagy, but has essentially no effect on nitrogen starvation- or rapamycin-induced autophagy.
Figure 2.
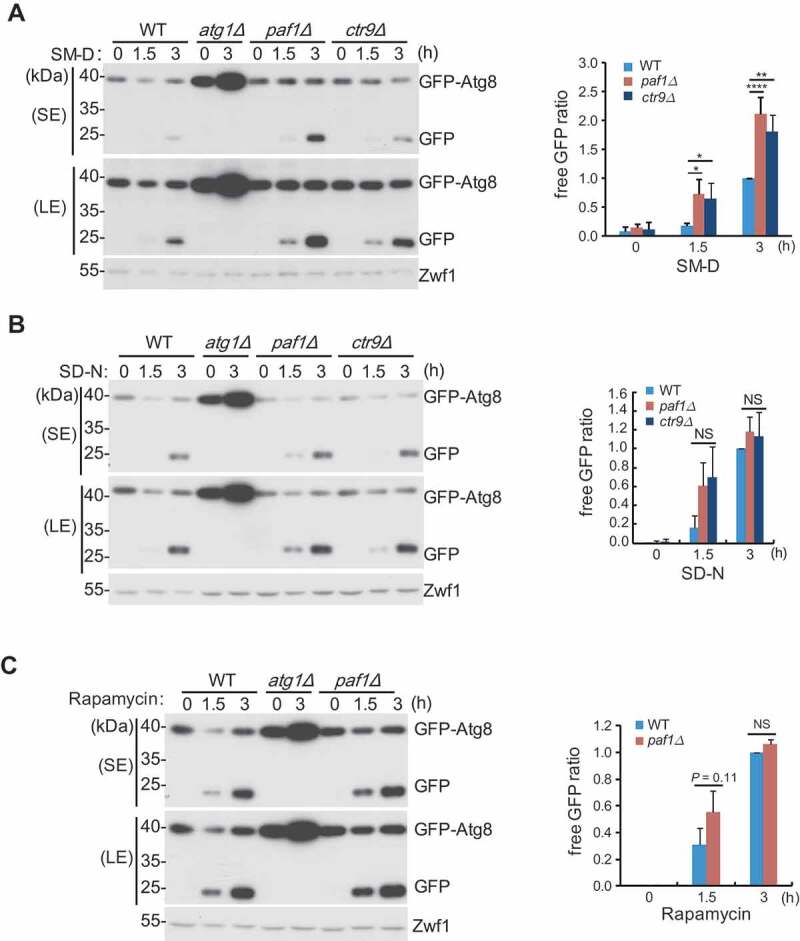
GFP-Atg8 processing assays were performed using the indicated yeast cells expressing the endogenous promoter-driven GFP-Atg8 construct. Cells were cultured in different starvation conditions: (A) SM-D, glucose starvation; (B) SD-N, nitrogen starvation; (C) rapamycin treatment. Cells were collected at the indicated time points after each treatment. GFP-Atg8 degradation was measured by immunoblotting with anti-GFP antibody (left panel). Relative free GFP levels against the 3-h time point in the WT (set as 1) were statistically quantified (right panel). Zwf1 was used as a loading control.
Autophagy is principally a catabolic response to nutrient or energy deprivation [29], which may be induced through different signaling pathways [30–32]. For example, nitrogen starvation and rapamycin stimuli trigger autophagy through inhibition of an MTOR (mechanistic target of rapamycin kinase)-associated cascade, whereas glucose starvation induces autophagy through AMP-activated protein kinase (AMPK) in mammals or Snf1 in yeast. AMPK activates autophagy through a cascade of signaling transduction, including phosphorylation of AMPK itself and subsequent phosphorylation of ULK1 (unc-51 autophagy activating kinase 1, a homolog of Atg1) and so on [29]. Because deletion of PAF1 does not affect ATG gene expression and GFP-Atg8 processing under nitrogen-starvation conditions, we speculate that Paf1C is not likely to be involved in activation of MTOR (mechanistic target of rapamycin kinase)-mediated autophagy. Thus, we decided to explore whether Paf1C participates in Snf1 activation-mediated autophagy. To do this, Snf1 activity and Atg1 phosphorylation were examined under both rich-medium and glucose-starvation condition. We found that, the phosphorylation of Snf1 was enhanced in Paf1C-defective cells grown in yeast extract peptone dextrose (YPD) medium, suggesting pre-activation of Snf1 in those mutant strains (Figure 3A,B). However, the difference of Snf1 phosphorylation states in those strains was negligible upon glucose-starvation induction, indicating full activation of Snf1 under such a circumstance. Consistently, phosphorylation of Atg1, a direct substrate of Snf1, was visible in Paf1C-defective cells by comparing the mobility shift of Atg1 proteins with that in WT cells (Figure 3C, lane 2 vs. lane 3, 4). The WT cells expressing a kinase-dead (KD) mutant of HA-Atg1 served as a control. Upon glucose starvation for 1 h, Atg1 was hyperphosphorylated in PAF1-mutant strains (Figure 3C, lane 6–8), which is in accord with activation of Snf1 (Figure 3B). The pre-activation of Snf1 in Paf1C-defective cells does not result from mitochondrial damage, as the basal respiration and maximum respiratory capacities were largely unaffected based on measuring the oxygen consumption rates (OCR) (Figure 3D,E). Although we observed a reduction of cristae in paf1Δ cells by transmission electron microscopy, the respiratory capacity of mitochondria remains normal (Figure S3A). Altogether, we found that Paf1C only affects glucose starvation-induced autophagy by activation of a Snf1-mediated pathway. How deletion of Paf1C stimulates Snf1 activation needs further investigation.
Figure 3.
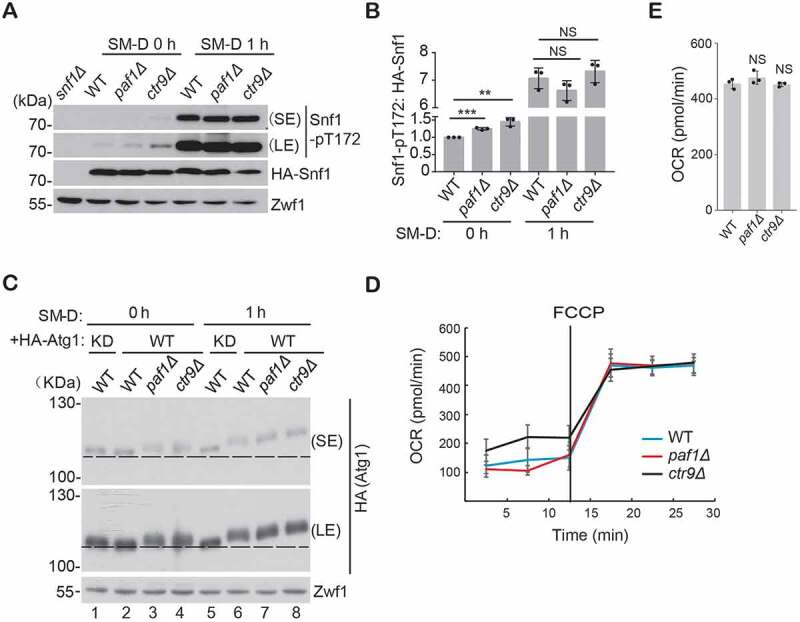
Deficiency of Paf1C promotes Snf1 activation. (A-B) The indicated cells expressing HA-Snf1 were cultured in glucose-starvation conditions for different times, and the phosphorylated status of Snf1 (Thr172) was examined and statistically quantified. Zwf1 was used as a loading control. (C) Yeast cells expressing either WT or KD HA-Atg1 were cultured as in (A). Cell lysates were run on a 6% SDS-PAGE gel under 80V constant voltage at 4ºC for a prolonged time. The phosphorylation of Atg1 was examined by immunoblotting. (D) Basal respiration and maximum respiratory capacity were measured by the OCR. (E) The maximum respiratory capacities of each strain at 30 min were statistically quantified.
Paf1C does not regulate ATG39- and ATG40-mediated reticulophagy
The mRNA levels of ATG39 and ATG40 in the paf1Δ strain were increased (Figure 1E), and Atg39 and Atg40 are required for reticulophagy [10]. Accordingly, we hypothesized that Paf1C might regulate Atg39- and Atg40-mediated reticulophagy. Thus, we generated either pep4Δ or pep4Δ paf1Δ strains with expression of Atg39 or Atg40, both bearing an integrated C-terminal 3xHA-tag (Atg39-HA or Atg40-HA). PEP4 was disrupted to block autophagic degradation of these proteins [10]. As expected, the protein levels of Atg39 or Atg40 were gradually accumulated over time upon rapamycin treatment in the pep4Δ strains (Figure 4A–D). However, deletion of PAF1 did not further increase the levels of Atg39 and Atg40, and even decreased Atg39 levels after prolonged treatment. In addition, the ER membrane protein Sec63 fused with GFP was used to monitor reticulophagy. Degradation of Sec63-GFP produced vacuolar protease-resistant GFP moieties, which were detected by immunoblotting analysis. Deletion of PAF1 did not further increase levels of free GFP or accelerate the kinetics of Sec63-GFP degradation regardless of nitrogen- or glucose-starvation condition (Figure 4E–G). Based on these observations, we conclude that Paf1C is unlikely a regulator of ATG39- and ATG40-mediated reticulophagy; the upregulation of ATG39 and ATG40 gene expression in Paf1C-defective cells probably results from indirect effects.
Figure 4.
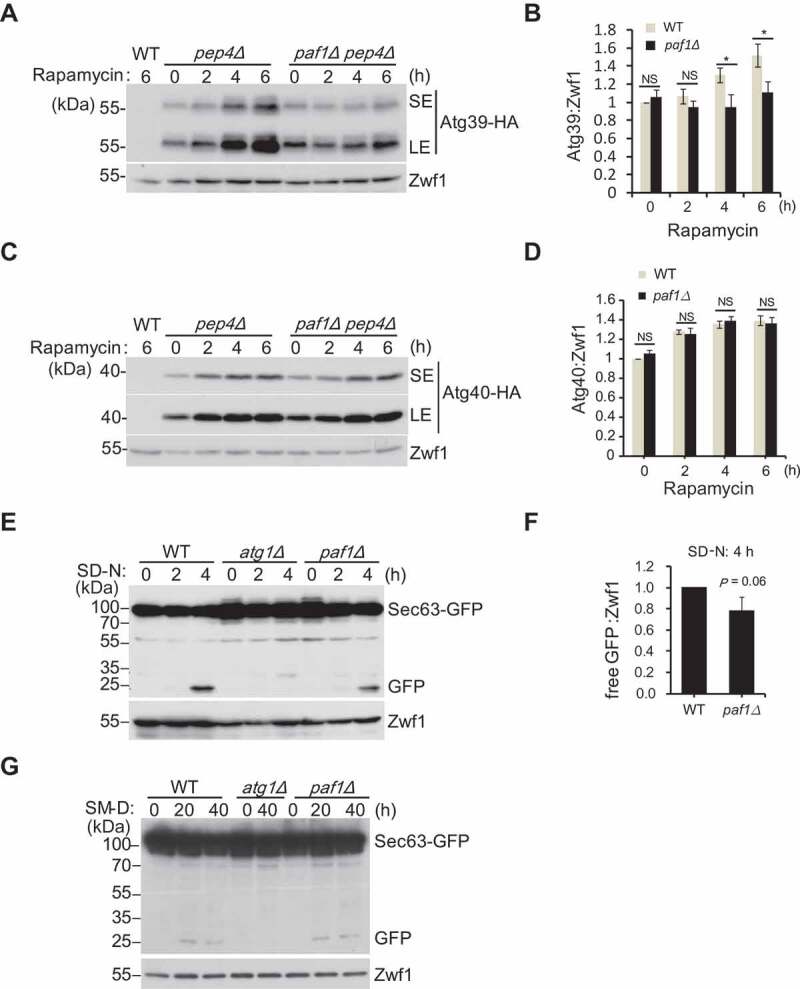
Paf1 does not regulate ATG39- and ATG40-mediated reticulophagy. (A-D) The indicated cells expressing integrated C-terminal 3xHA tagged Atg39 (A) or Atg40 (B) were treated with rapamycin for different times, and cell extracts were immunoblotted with the indicated antibodies. Relative protein levels were statistically quantified (C, D) (E-G) The indicated cells expressing integrated C-terminal GFP-tagged Sec63 were grown in nitrogen starvation condition (E and F) or glucose starvation (G) for different times, and Sec63-GFP degradation was analyzed by immunoblotting. Relative free GFP levels were quantified (F).
Paf1C is a negative regulator of mitophagy
It has been demonstrated that Atg32 acts as a mitophagy receptor to regulate selective degradation of mitochondria through interacting with Atg8 and Atg11 during respiratory growth conditions [9,14]. Since we have determined that Paf1 transcriptionally regulates ATG11 and ATG32, we wondered whether Paf1C could regulate mitophagy. It was reported that mitophagy can be induced when yeast cells are cultured in medium containing a non-fermentable carbon source (such as glycerol or lactate) for a prolonged period [33]. Thus, GFP-Atg8 processing assays were performed in synthetic medium with glycerol as the sole carbon source (SMG). Consistent with previous results done in glucose-starvation conditions, free GFP moieties increased to higher levels in Paf1C-defective cells compared with that in WT cells, suggesting mitophagy occurred at a higher level (Figure 5A,B).
Figure 5.
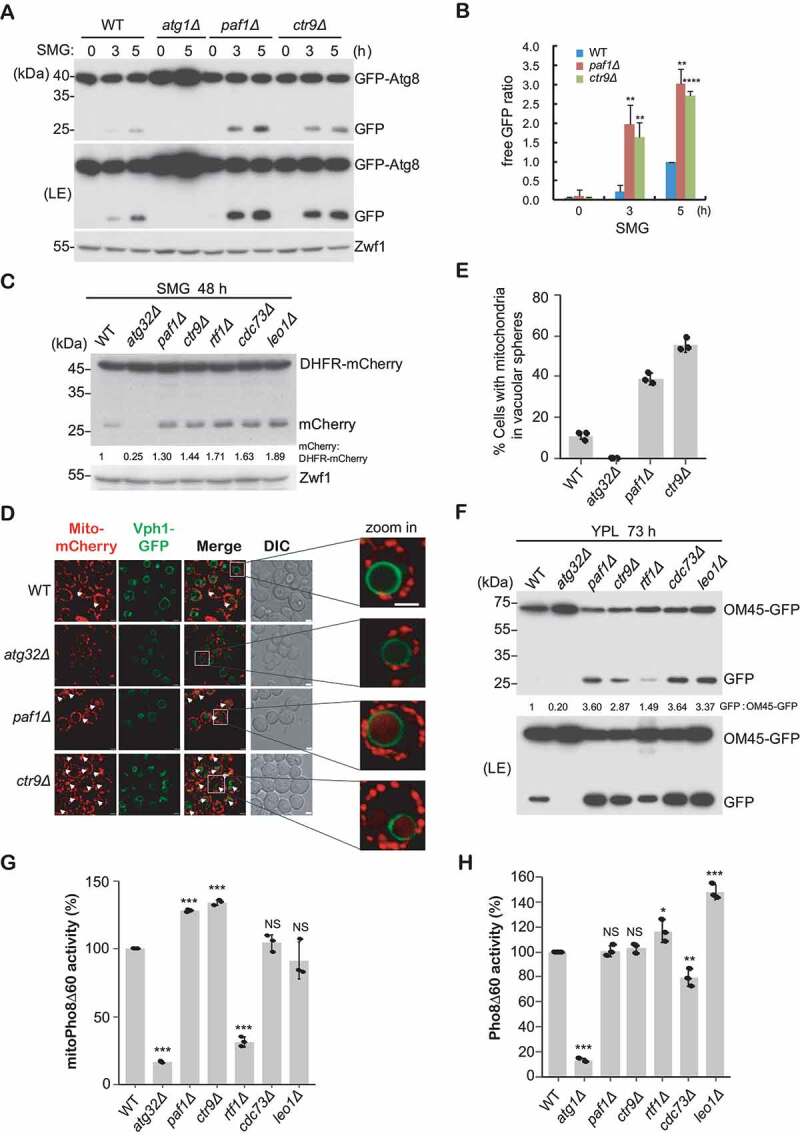
Paf1C negatively regulates mitophagy. (A-B) GFP-Atg8 processing assays were performed as described in Figure 2, except that cells were cultured in SMG medium. Relative free GFP levels against the 5 h time point in the WT strain (set as 1) were statistically quantified (B). (C) The indicated yeast cells bearing integrated mito-DHFR-mCherry were cultured in SMG medium for 48 h. Free mCherry signals were detected by immunoblotting with anti-mCherry antibody. (D-E) The indicated cells with integrated Vph1-GFP and mito-DHFR-mCherry were grown in SMG medium for 36 h. Cell mitophagy patterns were observed using fluorescence microscopy. Arrows highlight vacuolar-localized mitochondria. Representative images were enlarged and shown on the right (D). DIC, differential interference contrast. Scale bar: 2 μm. Cells (n = 100) showing mitochondria (red) surrounded by vacuoles (green) were counted, and cell populations were statistically quantified (E). (F) OM45-GFP-processing assays were performed using the indicated cells expressing integrated OM45-GFP that were cultured in YPL for 73 h. Free GFP indicates the degradation of mitochondria. (G-H) MitoPho8Δ60 activity (G, mitophagy measurement) or Pho8Δ60 activity (H, bulk autophagy measurement) were examined and statistically quantified in the indicated cells. The WT activity was set to 100%, and other strains were normalized to the WT.
Alternatively, cells bearing integrated mito-DHFR (dihydrofolate reductase)-mCherry (that served as a mitochondria-localized marker) were grown in SMG medium for 48 h; the generation of free mCherry produced by vacuolar proteases and accumulated in the vacuole was monitored by western blot analysis. The levels of free mCherry were elevated in Paf1C gene null mutants compared to WT cells (Figure 5C). No signal was detected in the atg32Δ mutant, which served as a negative control. This difference was also observed when these cells were grown in the same conditions for a longer time period (Figure S3B). Transport of mitochondria to the vacuole in yeast cells can also be examined using fluorescence microscopy. Thus, a GFP tag was integrated into the 3ʹ end of the VPH1 gene locus in the strains that express mito-DHFR-mCherry; Vph1 is a component of the vacuolar-type H+-ATPase, and thus marks the vacuole limiting membrane. Taking advantage of this double-color-labeled tool, we could monitor the mitochondrial localization upon mitophagy induction. After 36-h incubation in SMG medium, approximately 10% of the population of WT cells exhibited Atg32-dependent formation of spherical mCherry fluorescence patterns localized within the Vph1-GFP-labeled vacuole (Figure 5D,E). Deletion of PAF1 or CTR9 significantly increased the cell populations displaying intravacuolar mCherry puncta to 38% or 55%, respectively. Furthermore, if we continued culturing cells in SMG medium up to 51 h, cell populations with mCherry fluorescence localized in the vacuole were indistinguishable in those strains, suggesting that the WT strain was ultimately able to catch up with the mutant strains, and achieved full activation of mitophagy (Figure S3C,D).
Moreover, strains expressing the mitochondrial outer membrane protein OM45 fused with GFP (OM45-GFP) were used as an alternative method to monitor mitophagy, in which this chimera is delivered to the vacuole when cells are cultured to the post-logarithmic phase in medium with lactate as the sole carbon source (YPL, yeast extract peptone lactate), or when cells are shifted from YPL to SD-N (synthetic dropout without nitrogen base) [12]. The amount of free GFP processed from OM45-GFP was measured by immunoblotting assays (Figure 5F). Consistently, the levels of free GFP were much more abundant in Paf1C gene null strains than in WT cells, and the relative ratios of free GFP were also higher in those null strains. Note that the enhanced mitophagy activity in paf1Δ and rtf1Δ mutants seen relative to the wild type upon shifting to SD-N for 4 h was not seen after a prolonged induction (Figure S3E), implying that abundant Atg32 levels in WT cells ultimately reached that in mutant strains, thereby minimizing the difference of mitophagy activity between WT and mutants. This assumption was confirmed by showing similar ATG32 levels in these strains after a prolonged mitophagic induction (Figure S3F).
To confirm the specificity of Paf1C in autophagy, we used quantitative Pho8∆60 phosphatase assays to measure selective mitophagy activity or nonspecific bulk autophagy activity [34]. In brief, deletion of 60 N-terminal amino acids of the vacuolar zymogen Pho8 prevents its normal transport to the vacuole through the secretory pathway; delivery and proteolytic activation can occur through autophagy. The major difference between mitophagy and bulk autophagy relies on fusion of Pho8∆60 to the mitochondrial inner membrane protein Cox4 to generate mitoPho8Δ60, which is specifically targeted to mitochondria [34,35]. As expected, compared to the WT strain, paf1Δ and ctr9Δ mutants showed a dramatic increase in mitoPho8Δ60-dependent phosphatase activity following a shift from YPL to SD-N for 4 h (Figure 5G). In contrast, the mitoPho8Δ60 activity in the rtf1Δ strain significantly dropped down, and this phenotype was validated using several colonies bearing different knockout markers. We speculate that Rtf1 might have unknown cellular functions independent of Paf1C. Conversely, the nonspecific Pho8Δ60 assay showed an indistinguishable activity between WT and paf1Δ or ctr9Δ mutants (Figure 5H), suggesting a specific role of Paf1C in suppression of selective mitophagy.
Paf1C negatively regulates ATG32 expression
Next, we wanted to elucidate the molecular mechanism of Paf1C in suppressing mitophagy. It has been reported that a reduction of H2B monoubiquitination (H2B-Ub1) level is associated with the activation of autophagy under starvation conditions in mammals [36]. Because Paf1C is required for the conserved monoubiquitination of histone H2B and H2B-Ub1-dependent methylation of H3K4 and H3K79, we hypothesized that Paf1C might affect mitophagy through directly regulating H2B-Ub1 and histone H3 methylation [37–39]. To test this, the BRE1 gene encoding an E3 ligase responsible for catalyzing H2B-Ub1 was deleted in cells expressing OM45-GFP, and the mitophagy activity was examined by monitoring the free GFP production in glucose-starvation conditions as described above. We found that the bre1Δ mutant did not display higher mitophagy activity compared to the WT after a prolonged incubation in YPL medium (Figure S4A). This result suggests that Paf1C likely suppresses mitophagy independent of H2B-Ub1 regulation.
Alternatively, Paf1C may directly suppress ATG32 gene transcription as a mechanism of regulating mitophagy. Indeed, the transcriptional levels of ATG11 and ATG32 genes were remarkably increased when most of the genes coding Paf1C subunits were deleted, compared to the WT (Figure 6A). Moreover, the protein levels of Atg32 and Atg11 were also increased in paf1Δ and ctr9Δ strains (Figure 6B,C). Intriguingly, we did not observe enhanced levels of ATG11 and ATG32 in cdc73Δ strain (Figure 6A). To exclude the possibility that Paf1 and Ctr9 affect ATG gene expression independent of Paf1C, we investigate the effect of Cdc73 under mitophagy-inducing conditions. As expected, deletion of CDC73 significantly increased both transcriptional levels (Figure S4B) and protein levels (Figure S4C) of Atg32 and Atg11 when cells grown in SMG medium, reinforcing its importance in regulating ATG32 expression and glucose starvation-induced autophagy. Of note, when cells with integrated mito-DHFR-mCherry overexpressed a construct containing ATG32 or ATG11 with a 3xHA tag driven by the ADH1 promoter (ADH1p-ATG32-3HA or ADH1p-ATG11-3HA)) in SMG medium for 48 h, the level of free mCherry was elevated compared to the control cells (Figure S4D-G). Accelerated mitophagy by increasing Atg32 expression in paf1∆ cells is specific, as bulk autophagy (Figure S4H) or reticulophagy (Figure 4G) could not be activated even under prolonged glucose-starvation conditions.
Figure 6.
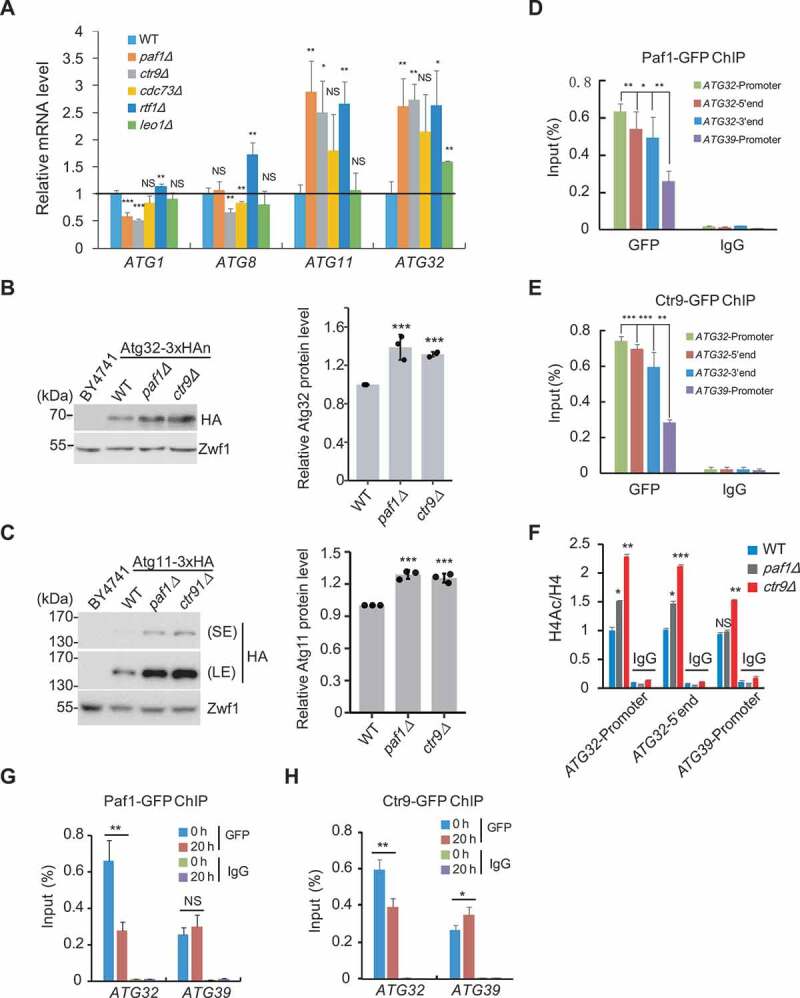
Paf1C negatively regulates ATG32 expression. (A) The indicated cells were grown in YPD to mid-log phase, and mRNA levels of 4 ATG genes were examined. (B) Atg32 protein levels in the indicated cells bearing integrated internally 3xHA-tagged Atg32-3xHAn were immunoblotted and quantified. (C) Atg11 protein levels in cells bearing integrated C-terminal tagged Atg11-3xHA were examined and quantified. (D-E) Yeast cells bearing integrated Paf1-GFP or Ctr9-GFP were subjected to ChIP assays using anti-GFP antibody; IgG served as a negative control. The enrichment of Paf1 or Ctr9 at the indicated regions of the ATG32 gene was examined by ChIP-qPCR. The ChIP data were normalized to input levels. Data represent mean ± SD compared with the promoter region of the ATG39 gene. (F) ChIP assays were performed using anti-pan acetyl and anti-H4 antibodies. Relative enrichment of acetyl histone H4 at the indicated regions of the ATG32 or ATG39 gene normalized to H4 was examined and quantified by ChIP-qPCR. (G-H) ChIP assays were performed using the same strains in (D-E) that were cultured in YPD to mid-log phase, then shifted to SMG medium. The enrichment of Paf1 or Ctr9 at the promoter regions of the ATG32 or ATG39 genes at the indicated time points were quantified by ChIP-qPCR.
Based on these data, we speculate that Paf1C may repress ATG32 transcription though binding the promoter of this gene, similar to the mechanism reported previously for Paf1C function [40]. To test this, chromatin immunoprecipitation (ChIP) assays were performed using Paf1- or Ctr9-expressing cells bearing GFP-tags at the endogenous chromosomal loci to examine the enrichment of Paf1C at the ATG32 promoter. Both Paf1 and Ctr9 subunits accumulated at the ATG32 gene locus, including the promoter region, and the 5ʹ and 3ʹ ends of the gene body, but much less accumulated at the promoter of the ATG39 gene, indicating that Paf1C specifically regulates ATG32 (Figure 6D,E). Moreover, we found that deletion of either PAF1 or CTR9 resulted in higher levels of histone acetylation at the ATG32 gene locus, whereas there was relatively little effect at the ATG39 locus (Figure 6F). This observation suggested that association of Paf1 and Ctr9 at the ATG32 gene locus was accompanied by lower levels of histone acetylation, supporting the idea that Paf1C suppresses ATG32 transcription. Importantly, the relatively stronger binding of Paf1 and Ctr9 with ATG32 in YPD was markedly decreased once cells were shifted to SMGlyCA medium to induce mitophagy, suggesting that the dissociation of Paf1C from the ATG32 gene locus was essential for activation of mitophagy (Figure 6G,H); in contrast, there was very little change in binding at the ATG39 locus. Altogether, our data demonstrated that Paf1C negatively regulates mitophagy through suppression of ATG32 expression.
Paf1C regulates mitophagy in mammalian cells
Because Paf1C is a highly conserved complex from yeast to mammals, we wanted to know whether regulation of mitophagy by Paf1C that we uncovered here would occur in more complex eukaryotes. Because the PINK1-PRKN/Parkin pathway is the most extensively characterized mechanism in mammalian cells, we first examined mitophagic effect of PAF1C in HeLa cells stably expressing GFP-PRKN. Cells with siPAF1 and siCTR9 or HA-PAF1 overexpression were treated with carbonyl cyanide 3-chlorophenylhydrazone (CCCP), a commonly used stimulus, to induce mitophagy [41]. The relative mRNA levels of individual genes were examined by RT-qPCR. Different from the effect of Paf1C on multiple ATG gene expression in yeast, knockdown of PAF1 and CTR9 only increased the MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) level, but not other ATG genes (Figure S5A). Moreover, a reduction of the PAF1 and CTR9 level was associated with an increase of OPTN mRNA level, whereas it had no effect on other mitochondrial receptors (Figure S5A,B). Intriguingly, overexpression of HA-PAF1 did not statistically affect the mRNA level of OPTN, which might be due to rapid activation of mitophagy upon CCCP treatment so that overexpression of PAF1 no longer enhanced OPTN expression (Figure S5C).
To further validate that PAF1C regulates mitophagy in mammalian cells, several mitochondrial proteins were examined in cells with either knockdown or overexpression of PAF1. CCCP treatment resulted in a significant reduction in levels of TOMM20 (translocase of outer mitochondrial membrane 20), TIMM23 (translocase of inner mitochondrial membrane 23), ATP5F1A (ATP synthase F1 subunit alpha) and HSPD1/HSP60 (heat shock protein family D [Hsp60] member 1), and these protein levels were further diminished after knockdown of PAF1C subunits (Figure S5D, 7A). Conversely, HeLa cells overexpressing PAF1 showed an accumulation of these mitochondrial proteins compared to control cells, indicating suppression of mitophagy in this condition (Figure 7B). The fate of the ubiquitin-binding autophagy receptor OPTN was consistent with that of other mitochondrial proteins after PAF1 levels were altered, suggesting a global degradation of damaged mitochondria upon induced mitophagy (Figure S5E,F). Unexpectedly, siPAF1 and siCTR9 further decreased the OPTN protein level, whereas overexpression of PAF1 in turn increased its level, which is opposite to the change in patterns of the OPTN mRNA level (Figure S5B,E). Noticeably, CALCOCO2, which was reported to function redundantly with OPTN [16], was not affected by PAF1C (Figure S5G,H). Moreover, immunofluorescent staining showed that knockdown of PAF1 and CTR9 led to a substantial reduction of the mitochondria chaperone protein HSPD1/HSP60 (Figure 7C). We speculate that the opposite effects might result from a feed-back regulation, in which higher levels of OPTN stimulates mitophagy, and accelerated mitophagy promotes more protein degradation, and then further triggers more OPTN transcription. The greater reduction of OPTN protein in PAF1C-defective cells actually comes from proteasome-mediated protein degradation [41], as addition of MG132 to these cells in the presence of CCCP remarkably restored OPTN protein levels comparable to that in control cells (Figure S5G,H, lane 5 vs. lane 6). The effect of PAF1C on mitophagy also did not rely on expression of PINK1-PRKN, as knockdown of PAF1 and CTR9 or overexpression of PAF1 did not significantly increase PINK1 mRNA levels (Figure S5A) or change GFP-PRKN protein levels (Figure S5G,H). More importantly, PAF1C does not perturb translocation of PRKN/Parkin (Figure S5I). Altogether, these data support the idea that PAF1C is a repressor of mitophagy, at least as it occurs in the PINK1-PRKN-mediated mitophagy pathway (Figure 7C).
Figure 7.
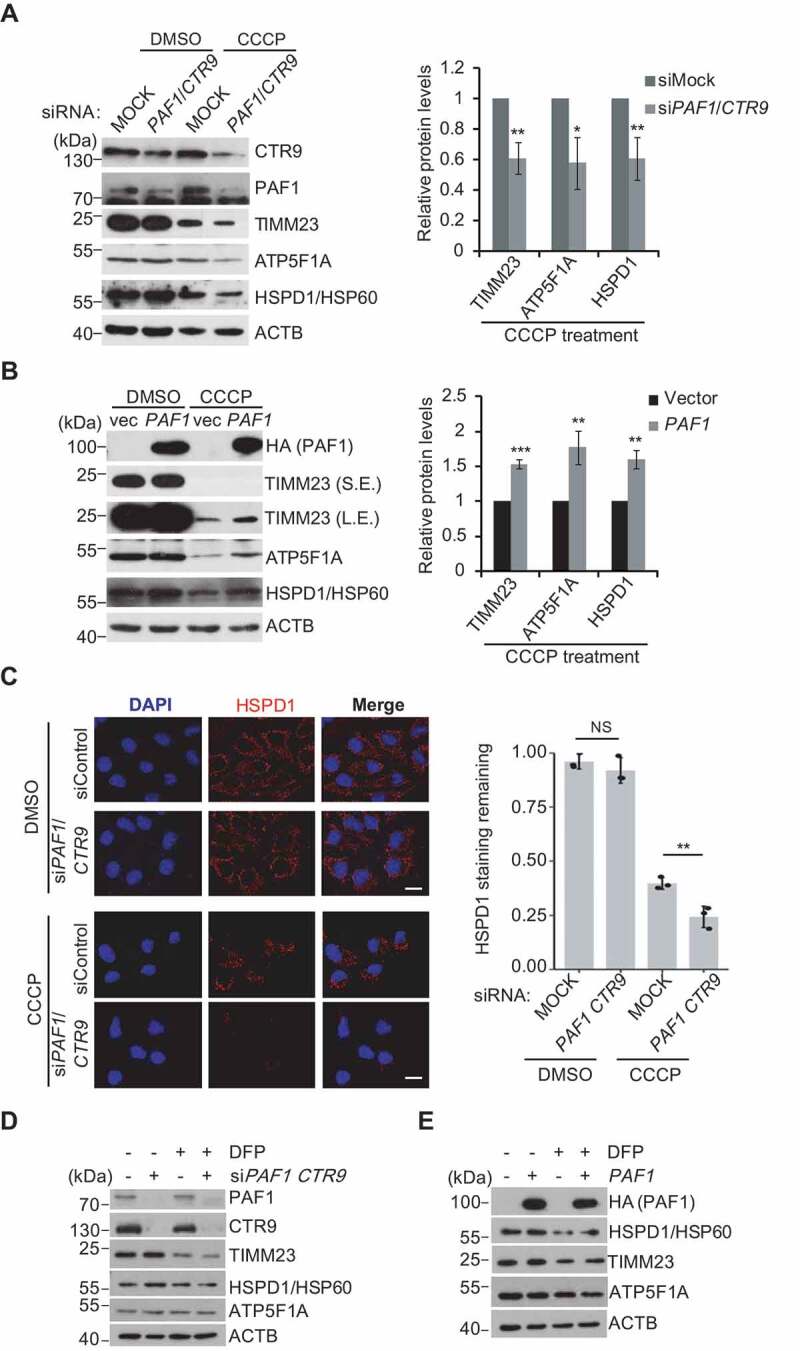
PAF1C affects mitophagy in mammalian cells. (A-B) HeLa cells expressing GFP-PRKN were transfected with siPAF1 and siCTR9 or HA-PAF1, and treated with CCCP (10 μM) for 24 h. Cell lysates were immunoblotted using the indicated antibodies. The relative protein levels were quantified. (C) Cells treated as indicated in (A) with the addition of z-VAD (OME)-FMK were immunostained and analyzed by confocal microscopy. DNA was stained with DAPI. Representative images are shown (left panel) and HSPD1/HSP60 signal intensities were quantified with each field containing at least 50 cells (right panel). Scale bar: 20 μm. (D-E) HeLa cells with siPAF1 and siCTR9 or expressing HA-PAF1 were treated with DFP (1 mM, MedChemExpress, HY-B0568) for 24 h, and cell lysates were immunoblotted using the indicated antibodies.
Besides PINK1-PRKN-dependent activation of mitophagy that is mediated by ubiquitin binding receptors, OTPN and CALCOCO2, PINK1-PRKN-independent activation of mitophagy relies on other mitochondria-anchored receptors, which can be stimulated by treating with the iron chelator deferiprone (DFP) [42]. To test whether PAF1C affects mitophagy in a PINK1-PRKN-independent manner, degradation of mitochondrial proteins were examined in cells with either knockdown or overexpression of PAF1 in the presence of DFP. DFP treatment resulted in a moderate reduction of the levels of TIMM23 and HSPD1/HSP60, and no change of ATP5F1A (Figure 7D,E). Levels of those mitochondria proteins remained constant after knockdown or overexpression of PAF1, suggesting that PAF1 may not be involved in this type of mitophagy. In agreement with this result, we did not see any changes of OTPN, CALCOCO2 and PRKN (Figure S5J,K). Overall, our results indicate that PAF1C regulates mitophagy through a PINK1-PRKN-dependent pathway in mammalian cells, and Paf1C/PAF1C conservatively regulates mitophagy from yeast to mammals.
Discussion
Prompt upregulation of ATG gene expression upon autophagy induction is critical for optimal autophagic efficiency. In this study, bioinformatics analyses of epigenetic or transcriptional regulators modulating gene expression allowed us to identify the Paf1 complex as a transcriptional repressor of autophagy, specifically functioning in glucose starvation-induced mitophagy. Furthermore, we demonstrate that Paf1C negatively regulates mitophagy by repression of ATG32 gene expression. Therefore, we uncovered a new role of Paf1C in transcriptionally modulating the mitophagy process.
Numerous fundamental studies have indicated that the highly conserved Paf1C is important for facilitating transcriptional elongation [23]. During elongation, Paf1C promotes histone modifications associated with active transcription [23]. However, genome microarray analyses show that Paf1C also represses expression of many genes [43]. For example, Paf1C represses ARG1 gene by negatively affecting Gcn4 occupancy at the promoter [44]. In addition, Paf1C represses SER3 transcription by promoting noncoding transcription initiated from its promoter region, primarily dependent on the Paf1 and Ctr9 subunits of this complex in yeast [40]. Consistent with this, our results demonstrated a role of Paf1C in gene repression, which mainly relies on Paf1 and Ctr9. Because structural analyses have indicated that Paf1 and Ctr9 form a core particle essential for complex integrity and functional regulation, it is possible that these two subunits execute more essential roles than other subunits of Paf1C [25,26].
The mitochondrial-anchored receptor Atg32 is critical for linking targeted mitochondria to the autophagic machinery. Atg32 interacts with Atg11 that is necessary for recruitment of mitochondria to the PAS for sequestration [8]. It has been reported that Ume6 forms a complex with Sin3 and Rpd3, and this complex can suppress ATG32 gene expression [45]. Although disruption of Sin3 or Rpd3 causes a high level of ATG32 expression, it does not simultaneously lead to mitophagy induction even under conditions of nitrogen starvation, implying the presence of another regulatory mechanism(s) [45]. Herein, we successfully uncover that Paf1C not only modulates both ATG32 and ATG11 expression, but that knockdown of Paf1C subunits also leads to instant mitophagy induction. Given that some subunits of Paf1C (such as Rtf1) have less effect on mitophagy, we cannot absolutely exclude another unknown regulatory mechanism as being responsible for accelerated mitophagy in Paf1-deficient cells. However, our present data largely support the concept that Paf1C regulates mitophagy by controlling the expression of ATG11 and ATG32.
In mammalian cells, mitophagy can be activated and mediated via multiple mechanisms and several receptors, which are not exactly the same as in budding yeast, in which a sole receptor, Atg32, is responsible for mitophagy. Although BCL2L13 was suggested as a mammalian homolog of yeast Atg32 and can compensate the function of Atg32 in yeast [17], we found that PAF1C does not affect BCL2L13 expression with or without mitophagy induction. Because many mitophagy-related receptors are functionally redundant to Atg32, we speculate that PAF1C regulates mitophagy in mammalian cells through controlling one of the other receptors. Indeed, we found that human PAF1C specifically regulates expression of the mitochondria-related receptor OPTN and induces mitophagy via a PINK1-PRKN-dependent pathway. Although we could not rule out the possibility that PAF1C might also modulate other mitophagy pathways independent of OPTN expression in the mammalian system, our data suggest that PAF1C primarily functions in mitophagy through the PINK1-PRKN-dependent pathway. Therefore, we provide evidence showing that the autophagic function of Paf1C is conserved from yeast to human, underscoring the significance of the Paf1 complex.
The higher expression of ATG genes after nutrient or energy starvation is required to support optimal autophagy activity. Elevated ATG gene expression correlates with more active autophagy events that are normally quiescent in particular physiological environments. A previous study on Rph1 highlights its important role in macroautophagy, as precise control of Rph1 activity is a prerequisite to the induction of ATG gene transcription and macroautophagy [21]. In this study, we identified another transcription factor, Paf1C, which particularly modulates expression of mitophagy-related ATG genes. During respiratory growth, Atg32 is induced and binds to Atg11, a scaffold/adaptor protein for selective types of autophagy, which recruits and imports mitochondria into vacuoles [9,14]. Concomitant with Atg32 sequestration, more ATG32 transcripts were induced so that a persistent mitophagy process can occur. This response was also observed in mammalian cells where OPTN expression was induced but the global OPTN protein levels were decreased upon mitophagy induction (Figure 7). Therefore, we can image that, upon mitophagy induction, a simultaneous upregulation of ATG32 and ATG11 is required for efficient mitochondria-phagophore association and subsequently an enhancement of mitophagy activity. Indeed, supporting this hypothesis, overexpression of ATG32 and ATG11 that are under the control of the ADH1 promoter obviously promoted mitophagy under glucose-starvation condition (Figure S4D-F).
Deficiencies in mitophagy have been linked to several pathologies, such as cancers and Parkinson disease [4,5,8]. Numerous missense mutations of PAF1 and CTR9 have been identified from the COSMIC database, and germline mutations in CTR9 predispose to Wilms tumor [25,46]. Thus, it is worth investigating whether regulating this novel regulatory pathway might be a potential target for the development of disease therapies.
Materials and methods
Yeast media and strains
Yeast strains used in this study are listed in Table S2. Gene disruption and integrated tagging were performed as described previously [47]. The generated strains were verified by PCR and western blot analysis. Yeast cells were grown at 30°C in YPD medium (1% yeast extract, 2% peptone and 2% dextrose) or SD medium (0.67% yeast nitrogen base without amino acids, supplemented with amino acids, and 2% glucose) with appropriate supplements. Bulk autophagy was induced by nitrogen starvation in SD-N (0.17% yeast nitrogen base without amino acids and ammonium sulfate, 2% glucose), SM-D (0.67% yeast nitrogen base without amino acids, supplemented with amino acids, without dextrose), or by treatment with 200 ng/mL rapamycin (Sangon Biotech, A606203) for the indicated time points. For inducing mitophagy, YPL (1% yeast extract, 2% peptone, and 2% lactic acid, pH 5.5) and SMG (0.67% yeast nitrogen base without amino acids, supplemented with amino acids, 0.1% dextrose and 3% glycerol) were used.
Western blotting
Western blot analysis was carried out following the protocol as described previously [47]. Anti-GFP (AE012) and anti-CALCOCO2/NDP52 (A7358) were purchased from Abclonal. Anti-mCherry (KM8017) was purchased from Sungene Biotech. Anti-HA (Y-11; sc-805), and anti-HSPD1/HSP60 (H-1; sc-13115) antibodies were purchased from Santa Cruz Biotechnology. Anti-phospho-PRKAA/AMPKα (Thr172; 2535S) antibody was purchased from Cell Signaling Technology. Anti-PAF1 (15441-1-AP), anti-CTR9 (21264-1-AP), anti-ATG5A (14676-1-AP), anti-OPTN (10837-1-AP), anti-histone H4 (16,047-1-AP), and anti-ACTB/β-Actin (60008-1-Ig) antibodies were purchased from Proteintech. Anti-TIMM23/Tim23 (611222) and anti-TOMM20/Tom20 (612278) antibodies were purchased from BD Biosciences. Anti-pan acetyl-histone H4 (Lys5, 8, 12; 04–557) was purchased from Merck. Anti-Zwf1/G6PDH (HPA000247) was purchased from Sigma-Aldrich. The anti-Atg8 antibody was generated following a protocol as described previously [48].
Plasmid construction and cell culture
The PAF1 gene from a human cDNA library was transferred to a pCS2-based Gateway vector containing a 3xHA tag via an LR reaction as described previously [49]. A one-step cloning method was used to generate the constructs list below following the standard protocol (Vazyme). The pairs of primers for generating gene fragments are described below: ATG11-BamHI-F + 3xHA-BamHI-R for pRS416-ADHp-Atg11; ATG32- BamHI-F + 3xHA-BamHI-R for pRS415-ADHp-Atg32. The primer sequences are listed in Table S3. The wild-type and D211A KD mutant of HA-Atg1 constructs were obtained from Dr. Cong Yi (Zhejiang University).
HeLa cells stably expressing GFP-PRKN were cultured in DMEM medium, supplemented with 10% fetal bovine serum with additional supplements. Cell transfection was performed using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s instruction. After 48 h, Cells were treated with DMSO or CCCP (Sigma, MKCD6029; final concentration of 10 μM). For immunostaining experiments, z-VAD (OME)-FMK (MedChemExpress, HY-16658; final concentration of 20 μM) was added to prevent cell apoptosis. The synthesized siRNA sequences (GenePharma Com. from Shanghai) are below: siPAF1: 5ʹ-UACCGAGGAAGAAUU-3ʹ; siCTR9: 5ʹ-GUGGCUCCAAACUUUAUU-3ʹ.
Vacuolar phosphatase assay
For monitoring nonspecific autophagy (Pho8Δ60) activity, cells were cultured in YPD to mid-log phase then shifted to SD-N for 4 h. For monitoring mitophagy activity, mitoPho8Δ60 cells were first cultured in YPD to mid-log phase then diluted to OD = 0.1 in YPL and then cultured for 12 h; finally, cells were shifted to SD-N for 4 h. Pho8∆60-dependent phosphatase activity was measured as described previously [34,50].
Oxygen consumption measurements
OCR was measured as described previously [51]. Yeast cells grown overnight were diluted and continued growing to OD600 = 0.3 and collected by centrifugation. Oxygen consumption rate (OCR) was determined using a XF96 Seahorse instrument (Agilent). Following initial calibration, basal OCR was measured for the indicated strains in n = 6 technical replica using a 2-min mix, 2-min measure cycle and was repeated 3 times before decoupling oxidative phosphorylation using the ionophore FCCP (carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone) (Sigma-Aldrich, C2920) at 3 μM as final concentration. Measurements were normalized to OD600 for each well.
Microscopy analysis
Immunofluorescence microscopy assays were performed as described previously [33]. Briefly, cells expressing fluorescent tag-fusion proteins were grown in YPD to mid-log phase (OD600 = 0.8 ~ 1.2), and then were shifted to SMG medium (0.67% yeast nitrogen base without amino acids, supplemented with amino acids, 0.1% dextrose and 3% glycerol) and cultured for 36 h. Mammalian cells grown on coverslips were fixed with cold methanol for 10 min at 4°C, and then permeabilized with 0.3% Triton X-100 (Sigma, T8787) on coverslips in PBS (136 mM NaCl, 2.6 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4) for 20 min. After blocking with 3% BSA (Baitg, 735094) in PBS at room temperature for 30 min, cells were incubated with primary antibody for 2 h followed by secondary antibodies (Proteintech Group, SA00009-1) for 1 h at room temperature. Fluorescence signals were visualized using a Leica TCS SP8 microscope equipped with a 60x objective lens. Images were acquired using QCapture Suite (QImaging Corporation, Canada).
RNA and RT-qPCR
Yeast RNA was extracted with Trizol reagent (Thermo, 15596018) and RT-qPCR was performed as described previously [52]. The abundance of mRNA was detected using Bio-Rad CFX Optics Module, and mRNA levels were normalized to the ACT1 gene. The primer sets used as RT-qPCR probes are listed in Table S3.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed as described previously [47]. Primers used for detection of DNA enrichment are listed in Table S3.
Quantification and statistical analysis
Unless otherwise stated, western blot data were quantified using ImageJ software to measure the relative intensity of each band. All quantification data were presented as the mean ± SD (standard deviation) from at least 3 independent experiments. Statistical differences were determined by two-tailed unpaired t-test, and a P value of less than 0.05, 0.01, 0.001 or 1 × 10−4 was considered statistically significant and marked as “*”, “**”, “***”, “****”, respectively. “NS” indicates “not significant”.
Supplementary Material
Acknowledgments
We thank Drs. Xuefeng Chen and Mingzhou Chen (Wuhan University), Li Yu (Tsinghua University), Cong Yi (Zhejiang University), and Zhiping Xie (Shanghai Jiaotong University) for sharing reagents. This work was supported by grants from the National Natural Science Foundation of China (31770843 and 31271369 to H.N.D., 91854107 to Z.S.), the Major State Basic Research Development Program of China (2013CB910700 to H.N.D.), and Wuhan University (2042018kf0217 to W.J. and H.N.D.). D.J.K. was supported by NIH grant GM131919. F.R. is supported by ZonMW VICI (016.130.606), ZonMW TOP (91217002), ALW Open Programme (ALWOP.310) and Marie Skłodowska-Curie Cofund (713660, 765912) grants. M.M. is supported by an ALW Open Programme (ALWOP.355).
Funding Statement
This work was supported by the Major State Basic Research Development Program of China [2013CB910700]; National Institutes of Health [GM131919]; National Natural Science Foundation of China [31271369]; National Natural Science Foundation of China [31770843]; National Natural Science Foundation of China [91854107];ZonMW VICI [016.130.606]; Kristi Yamaguchi and Always Dream Foundation (US) [ALWOP.355];ZonMW TOP [91217002];ALW Open Programme [ALWOP.310]; H2020 Marie Skłodowska-Curie Actions () [713660]; Marie Skłodowska-Curie Cofund [765912]; Wuhan University [2042018kf0217]; Wuhan University [2042018kf0217].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Carafoli E. Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem Sci. 2003;28(4):175–181. [DOI] [PubMed] [Google Scholar]
- [2].Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462(2):245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10(7):458–467. [DOI] [PubMed] [Google Scholar]
- [7].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. [DOI] [PubMed] [Google Scholar]
- [8].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17(1):87–97. [DOI] [PubMed] [Google Scholar]
- [10].Mochida K, Oikawa Y, Kimura Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522(7556):359–362. [DOI] [PubMed] [Google Scholar]
- [11].Liu L, Sakakibara K, Chen Q, et al. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24(7):787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. J Biol Chem. 2008;283(47):32386–32393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hutchins MU, Veenhuis M, Klionsky DJ. Peroxisome degradation in saccharomyces cerevisiae is dependent on machinery of macroautophagy and the Cvt pathway. J Cell Sci. 1999;112(Pt 22):4079–4087. [DOI] [PubMed] [Google Scholar]
- [14].Kanki T, Wang K, Cao Y, et al. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17(1):98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Farre JC, Burkenroad A, Burnett SF, et al. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 2013;14(5):441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Murakawa T, Yamaguchi O, Hashimoto A, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wei Y, Chiang WC, Sumpter R Jr., et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168(1–2):224–238 e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Delorme-Axford E, Klionsky DJ. Transcriptional and post-transcriptional regulation of autophagy in the yeast saccharomyces cerevisiae. J Biol Chem. 2018;293(15):5396–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jin M, He D, Backues SK, et al. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr Biol. 2014;24(12):1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bernard A, Jin M, Gonzalez-Rodriguez P, et al. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr Biol. 2015;25(5):546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lenstra TL, Benschop JJ, Kim T, et al. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell. 2011;42(4):536–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Van Oss SB, Cucinotta CE, Arndt KM. Emerging insights into the roles of the Paf1 complex in gene regulation. Trends Biochem Sci. 2017;42(10):788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta. 2010;1799(5–6):379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xie Y, Zheng M, Chu X, et al. Paf1 and Ctr9 subcomplex formation is essential for Paf1 complex assembly and functional regulation. Nat Commun. 2018;9(1):3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rodrigues J, Lydall D. Paf1 and Ctr9, core components of the PAF1 complex, maintain low levels of telomeric repeat containing RNA. Nucleic Acids Res. 2018;46(2):621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004;279(29):29889–29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huang WP, Shintani T, Xie Z. Assays for autophagy I: the Cvt pathway and nonselective autophagy. Methods Mol Biol. 2014;1163:153–164. [DOI] [PubMed] [Google Scholar]
- [29].Yi C, Tong J, Lu P, et al. Formation of a Snf1-Mec1-Atg1 module on mitochondria governs energy deprivation-induced autophagy by regulating mitochondrial respiration. Dev Cell. 2017;41(1):59–71 e54. [DOI] [PubMed] [Google Scholar]
- [30].Galluzzi L, Pietrocola F, Levine B, et al. Metabolic control of autophagy. Cell. 2014;159(6):1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ha J, Guan KL, Kim J. AMPK and autophagy in glucose/glycogen metabolism. Mol Aspects Med. 2015;46:46–62. [DOI] [PubMed] [Google Scholar]
- [32].Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nagumo S, Okamoto K. Investigation of yeast mitophagy with fluorescence microscopy and western blotting. Methods Mol Biol. 2018;1759:71–83. [DOI] [PubMed] [Google Scholar]
- [34].Yao Z, Liu X, Klionsky DJ. MitoPho8Delta60 assay as a tool to quantitatively measure mitophagy activity. Methods Mol Biol. 2018;1759:85–93. [DOI] [PubMed] [Google Scholar]
- [35].Noda T, Klionsky DJ. The quantitative Pho8Delta60 assay of nonspecific autophagy. Methods Enzymol. 2008;451:33–42. [DOI] [PubMed] [Google Scholar]
- [36].Chen S, Jing Y, Kang X, et al. Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic Acids Res. 2017;45(3):1144–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ng HH, Dole S, Struhl K. The Rtf1 component of the Paf1 transcriptional elongation complex is required for ubiquitination of histone H2B. J Biol Chem. 2003;278(36):33625–33628. [DOI] [PubMed] [Google Scholar]
- [38].Wood A, Schneider J, Dover J, et al. The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J Biol Chem. 2003;278(37):34739–34742. [DOI] [PubMed] [Google Scholar]
- [39].Zhu B, Zheng Y, Pham AD, et al. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell. 2005;20(4):601–611. [DOI] [PubMed] [Google Scholar]
- [40].Pruneski JA, Hainer SJ, Petrov KO, et al. The Paf1 complex represses SER3 transcription in Saccharomyces cerevisiae by facilitating intergenic transcription-dependent nucleosome occupancy of the SER3 promoter. Eukaryot Cell. 2011;10(10):1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jian F, Chen D, Chen L, et al. Sam50 regulates PINK1-parkin-mediated mitophagy by controlling PINK1 stability and mitochondrial morphology. Cell Rep. 2018;23(10):2989–3005. [DOI] [PubMed] [Google Scholar]
- [42].Allen GF, Toth R, James J, et al. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14(12):1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Penheiter KL, Washburn TM, Porter SE, et al. A posttranscriptional role for the yeast Paf1-RNA polymerase II complex is revealed by identification of primary targets. Mol Cell. 2005;20(2):213–223. [DOI] [PubMed] [Google Scholar]
- [44].Crisucci EM, Arndt KM. Paf1 restricts Gcn4 occupancy and antisense transcription at the ARG1 promoter. Mol Cell Biol. 2012;32(6):1150–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aihara M, Jin X, Kurihara Y, et al. Tor and the Sin3-Rpd3 complex regulate expression of the mitophagy receptor protein Atg32 in yeast. J Cell Sci. 2014;127(Pt 14):3184–3196. [DOI] [PubMed] [Google Scholar]
- [46].Hanks S, Perdeaux ER, Seal S, et al. Germline mutations in the PAF1 complex gene CTR9 predispose to wilms tumour. Nat Commun. 2014;5:4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Du HN, Fingerman IM, Briggs SD. Histone H3 K36 methylation is mediated by a trans-histone methylation pathway involving an interaction between Set2 and histone H4. Genes Dev. 2008;22(20):2786–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Huang WP, Scott SV, Kim J, et al. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem. 2000;275(8):5845–5851. [DOI] [PubMed] [Google Scholar]
- [49].Wang HY, Li Y, Xue T, et al. Construction of a series of pCS2+ backbone-based Gateway vectors for overexpressing various tagged proteins in vertebrates. Acta Biochim Biophys Sin. 2016;48(12):1128–1134. [DOI] [PubMed] [Google Scholar]
- [50].Noda T, Klionsky DJ. Chapter 3 the quantitative Pho8Δ60 assay of nonspecific autophagy. Methods Enzymol. 2008;451:33–42. [DOI] [PubMed] [Google Scholar]
- [51].Madsen CT, Sylvestersen KB, Young C, et al. Biotin starvation causes mitochondrial protein hyperacetylation and partial rescue by the SIRT3-like deacetylase Hst4p. Nat Commun. 2015;6:7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li F, Zheng LD, Chen X, et al. Gcn5-mediated Rph1 acetylation regulates its autophagic degradation under DNA damage stress. Nucleic Acids Res. 2017;45(9):5183–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.