

ABSTRACT
Myocardial infarction (MI) is a vital cause of death and disability globally. The primary treatment for diminishing acute myocardial ischemic injury is myocardial reperfusion, which may induce cardiomyocyte death. Our aim is to unravel the mechanism of sevoflurane (Sev) in microRNA-219a (miR-219a)-mediated regulation of absent in melanoma 2 (AIM2) and TLR4/MyD88 pathway during myocardial ischemia/reperfusion (I/R). The area of MI and apoptosis of cardiomyocytes of the developed mouse model were evaluated by TTC staining and TUNEL, respectively. After the determination of miR-219a as our target using microarray analysis, miR-219a atagomiR was used to treat the mouse model. The luciferase assay verified whether miR-219a targeted AIM2, and the miR-219a and AIM2 expression in myocardial tissues was detected by RT-qPCR and Western blot. miR-219a was significantly increased in myocardial tissues from mice treated with Sev, and the area of MI and cardiomyocyte apoptosis were decreased as a consequence. The miR-219a inhibitor reversed the action of Sev. Moreover, overexpression of AIM2 or induction of the TLR4 pathway aggravated myocardial I/R injury alleviated by miR-219a. All in all, the treatment of Sev upregulated miR-219a expression, which blocked the TLR4 pathway by targeting AIM2 and attenuated cardiomyocyte apoptosis in myocardial I/R mouse model.
KEYWORDS: Sevoflurane, myocardial ischemia/reperfusion, microRNA-219a, AIM2, TLR4/MyD88 pathway
1. Introduction
As the leading cause of death and disability in a world range, coronary heart disease may culminate in acute myocardial ischemia/reperfusion injury (IRI) [1]. Principally, no blood flow to the heart leads to an inequality between oxygen demand and supply, called ischemia, however, reinstating blood flow to the ischemic heart, termed as reperfusion by thrombolytic therapy or primary percutaneous coronary intervention may also cause injury [2]. Sevoflurane (Sev) is extensively applied in cardiac surgery due to its advantages of shorter induction and recovery time and higher safety level relative to other anesthetics [3]. The beneficial role of Sev in alleviating lung [4], brain [5] as well as liver IRI [6] has been reported. Moreover, Sev has been observed to alleviate myocardial IRI via the suppression of caveolin-3-mediated cyclooxygenase-2 [7]. More recently, Sev preconditioning has been implicated in protecting myocardial IRI through the regulation of microRNA-370 (miR-370) [8], which highlights the connection between Sev and microRNAs in progression of IRI.
miRNAs are a group of non-coding RNAs with 20–23 nucleotides in length, which perform to modulate gene expression, principally by repressing transcription or degrading messenger RNA (mRNA), and the relevance of miRNAs in mediating the cascade of myocardial IRI has been underscored previously [9]. miR-219a-1-3p was found to be one of the ten downregulated miRNAs in coronary artery disease [10]. Besides, protocatechuic acid-induced miR-219a-5p expression ameliorated alcoholic liver disease by lowering p66shc-mediated formation of reactive oxygen species [11]. Most importantly, age-related miR-219a-5p mitigated hepatic IRI by directly targeting TP53BP2 and repressing TP53-dependent apoptosis of hepatic cells in a mouse model [12]. Among the predicted potential miR-219a-targeting mRNAs, we found the binding sites of absent in melanoma 2 (AIM2) in miR-219a through Starbase (http://starbase.sysu.edu.cn/). AIM2 was accountable for the activation of caspase-1 in hepatic I/R [13]. Moreover, steatohepatitis in the methionine-choline-deficient mouse model was also linked to enhanced AIM2 expression and activated inflammasome in a MyD88-dependent way in hepatocytes and bone marrow-derived cells [14]. In addition, High mobility group box 1 protein was involved in lipopolysaccharide (LPS)-evoked acute lung injury in mice by inducing AIM2 in macrophages and activating polarization of M1 macrophages through toll‑like receptor 4 (TLR4) [15]. Our aim here is to assess the role of miR-219a in the Sev-against myocardial IRI and to test the hypothesis that the miR-219a/AIM2/TLR4/MyD88 pathway participates in Sev regulation of cardiomyocyte apoptosis.
2. Materials and methods
2.1. Ethics statement
All protocols involving animals in this work were conducted with the approval of the Ethics Committee for Animal Experimentation of the First Affiliated Hospital of Zhengzhou University.
2.2. Animals and experiment setup
A total of 168 healthy male BALB/c mice were purchased from the Experimental Animal Center of Chongqing Medical University (Chongqing, China). All mice were allowed to acclimatize for 10 weeks before any treatment was initiated. These mice were kept in specific-pathogen-free animal rooms under controlled environmental conditions (at 23°C; 65% relative humidity; 12 hours light/dark cycle) with free access to food and water. Initially, all mice were selected for either sham operation (n = 24) or I/R establishment. The induced myocardial I/R mice were injected with Sev alone, with AtagomiR Mock, AgomiR-219a (100 nmol), dimethylsulfoxide (DMSO), or LPS plus Sev (65139, Sigma Aldrich, SF, CA, USA) or without any treatment (n = 24). The mice were subjected to intraperitoneal injection of 50 μg/kg TLR4 agonist LPS or the same volume of control DMSO. Another 48 mice were intramyocardially injected with AtagomiR Mock or AgomiR-219a. Afterward, all mice were placed in a box (25 cm × 18 cm × 18 cm) with a hole on each side. One side of the box was attached to a machine that produces oxygen and anesthetic gas (60 mg/kg pentobarbital sodium), while the other side of the box was designed to be the pump side connected to the Datex anesthetic gas detector. Sev (2%, 1.0 minimum alveolar concentration) was pumped into the box for 60 min [7]. Next, a mouse model with myocardial IR was established within 60 min after the termination of Sev treatment. First, an intraperitoneal injection of 0.1 mg/kg atropine inhibited the formation of respiratory secretions in mice. Subsequently, the mice were anesthetized for 10 min on the supine position and connected to a rodent ventilator (DH-140B, Medical Experimental Factory, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China). The respiratory rate was regulated by a nasal/mask placed on a mouse to assist in respiration (tidal volume: 6 mL/kg; respiratory rate: 81–90 times/min; inspiratory-to-expiratory ratio: 1:2). A 3 cm longitudinal incision was then made between the second and third intercostal space to reveal the heart and pericardium. At the lower edge of the left atrial appendage at 1.5 cm, the anterior descending branches of the left coronary artery were ligated using a hollow plastic tube with a 6–0 suture. After 30 min of ischemia, the hollow plastic tube was cut open to restore blood flow, and the skin was then stitched. The sham-operated mice received the same method of thoracotomy, but the left coronary artery was not ligated. After surgery, a total of 200 U/kg heparin was injected into the I/R-treated mice to prevent thrombosis. Heart rate (HR), left ventricular end-diastolic pressure (LVEDP), left ventricular developmental pressure (LVDP), maximum rate of rise of left ventricular pressure [dp/dtmax (+)], and maximum rate of decline of the left ventricular pressure [dp/dtmax (-)] were recorded at a time interval of 30 min: at the end of reperfusion (T0), 30 min after the beginning of reperfusion (T1), 60 min after the beginning of reperfusion (T2), 90 min after the beginning of reperfusion (T3) and 120 min after the beginning of reperfusion (T4). After each experiment, all mice were kept warm, and standard rodent food and water were freely accessible.
2.3. Hematoxylin-eosin (HE) staining
The myocardial tissues of euthanized mice (pentobarbital sodium at 100 mg/kg) were extracted for HE staining [16] and observed and photographed under a light fluorescence microscope (PrimoStar iLEDTM, Zeiss, Oberhochen, Germany).
2.4. Triphenyltetrazolium chloride (TTC) staining
Fresh tissues were sliced and frozen in a − 20°C refrigerator for 30 min. The heart tissues were sectioned (2 mm) along the short axis of the heart from the apex (3 to 4 slices each heart). The sliced sections were incubated in a fresh 2% TTC solution for 0.5 h, treated with 4% paraformaldehyde, and photographed.
2.5. Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) staining
Mouse myocardial sections were treated with protein kinase K for 0.5 h and reacted with TUNEL at 37°C for 1 h. Following an incubation at 37°C for 1 h with specific antibodies labeled with horseradish peroxidase, the sections were treated with diaminobenzidine (Solarbio, Beijing, China) at room temperature for 10 min. After hematoxylin nucleus staining, the cells were photographed and counted using an optical microscope.
2.6. Microarray analysis
Total RNA was extracted by Trizol (Thermo Fisher) in 6 I/R mice without other treatment and I/R mice treated with Sev, respectively. Next, 0.5 μg RNA was used for hybridization using Human miRNA Expression Array V4.0 (Arraystar Inc., Rockville, MD, USA). Microarray was subsequently washed and scanned using GeneChipTM Scanner3000 7 G system (Thermo Fisher Scientific).
2.7. Quantitative real-time polymerase chain reaction (qRT-PCR)
The total RNA of the cells was extracted by Trizol (Thermo Fisher Scientific). Then, the synthesis of the first cDNA strand was performed using the high-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) kit. After dilution (10 ×), cDNA was used for qRT-PCR experiments. Primer sequences of miR-219a (#RP300127), U6 (#MP3001), AIM2 (#MP200186), and GAPDH (#MP205604) were purchased from OriGene Technologies (Beijing, China).
2.8. Dual‐luciferase reporter gene assay
Starbase (http://starbase.sysu.edu.cn/) was used to predict target genes for mmu-miR-219a. 293 T cells (AT-1592, ATCC, Manassas, VI, USA) were plated into a 24-well plate and incubated for 24 h. Synthesized AIM2-3ʹuntranslated region (3ʹUTR)-wild-type (WT) plasmid and AIM2-3ʹUTR-mutant (MT) plasmid were co-transfected with miR-219a mimic or mimic negative control (NC) into HEK-293T cells. The cells were collected and lysed 48 h later. The firefly and Renilla luciferase activities were measured using the dual-luciferase® reporter assay system (E1910, Promega Biotechnology Corporation, Beijing, China).
2.9. Immunoblotting
The primary antibodies against AIM2 (1:1000, ab119791, Abcam, NY, USA), TLR4 (1:1000, ab13867), and MyD88 (1:1000, ab133739) were utilized for immunoblotting. The experimental procedure was carried out in accordance with a previous report [17].
2.10. Culture and treatment of cardiomyocytes
The isolation and culture of mouse cardiomyocytes were carried out according to a previous literature [8]. Next, miR-219a mimic, mimic control (Mock), pcDNA3.1-AIM2, pcDNA3.1 were transfected into cardiomyocytes using the Lipofectamine 2000 kit (Thermo Fisher) strictly according to the instructions. Subsequently, the TLR4-specific agonist LPS was added to cardiomyocytes (Selleck, USA). The cells were collected for subsequent experiments.
2.11. Determination of cardiomyocyte proliferation
We used EdU staining to detect cellular activity using an EdU kit (Roche, Mannheim, Germany) in strict accordance with the instructions.
2.12. Flow cytometry
We used TUNEL staining and propidium iodide (PI)/Annexin V-labeled flow cytometry to detect cell apoptosis levels, all of which were performed as previously reported [18].
2.13. Statistical analysis
SPSS 21.0 (IBM Corp, Armonk, NY, USA) was used for data analysis. Measurement data are expressed by mean ± standard deviation (SD) after normally distribution evaluation using Kolmogorov-Smirnov. Measurement data between two groups were analyzed by paired t test. Comparison among multiple groups was analyzed by one‐way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test. The level of significance was accepted as p < 0.05 (two‐tailed).
3. Results
3.1. Sev treatment reduces myocardial IR injury in mice
We initially measured hemodynamic parameters as indicators of the evaluation of I/R model establishment as well as changes in cardiac function in I/R mice treated with Sev. We found that after I/R treatment in mice, HR, LVDP, dp/dtmax (+), and dp/dtmax (-) were significantly decreased, whereas LVEDP was significantly increased. However, Sev significantly elevated HR, LVDP, dp/dtmax (+), and dp/dtmax (-) compared with mice only treated with I/R (Table 1). Furthermore, we observed that Sev treatment significantly reduced MI size in mice treated with I/R (Figure 1(a)). We then used HE staining to analyze the myocardial pathological structure of mice (Figure 1(b)). The cell edge of the sham-operated mice was clear with intact nuclear membrane and uniformly aligned myocardial fibers, and there was no interstitial edema. In the I/R mice, the cells showed swelling, obvious interstitial edema, blurred boundaries, and some cross stripes, whereas in Sev-pretreated I/R mice, the cardiomyocytes were similarly disordered, but the degree of lesion was alleviated relative to I/R mice. We further found that Sev treatment significantly reduced apoptosis levels of cells in myocardial tissue after I/R treatment (Figure 1(c)).
Table 1.
Sev ameliorates cardiac dysfunction induced by myocardial IRI at T0 and T4.
| Index | Time | Sham group | I/R group | Sev + I/R group |
|---|---|---|---|---|
| HR (beat/min) | T0 | 161.00 ± 13.24 | 159.32 ± 14.81 | 157.32 ± 10.37 |
| T4 | 136.63 ± 11.52 | 91.84 ± 6.27 | 120.31 ± 11.28 | |
| LVDP (mmHg) | T0 | 103.24 ± 9.68 | 106.57 ± 10.55 | 114.72 ± 9.63 |
| T4 | 96.85 ± 5.74 | 34.30 ± 6.79 | 64.37 ± 4.41 | |
| LVEDP (mmHg) | T0 | 8.01 ± 0.69 | 6.15 ± 0.57 | 7.11 ± 0.38 |
| T4 | 6.18 ± 1.10 | 42.94 ± 5.67 | 26.38 ± 2.58 | |
| dp/dtmax(+)(mmHg/s) | T0 | 2879.65 ± 169.82 | 2910.44 ± 231.33 | 2967.01 ± 197.61 |
| T4 | 2769.04 ± 215.07 | 1236.97 ± 147.26 | 1617.53 ± 138.64 | |
| dp/dtmax(-)(mmHg/s) | T0 | 2317.65 ± 157.30 | 2323.12 ± 251.11 | 2297.45 ± 177.21 |
| T4 | 2226.34 ± 134.79 | 413.47 ± 27.88 | 963.29 ± 39.46 |
Note: Sev, Sevoflurane; IRI, ischemia/reperfusion injury; HR, heart rate; LVEDP, left ventricular end-diastolic pressure; LVDP, left ventricular developmental pressure; dp/dtmax (+), maximum rate of rise of left ventricular pressure; dp/dtmax (-), maximum rate of decline of the left ventricular pressure.
Figure 1.
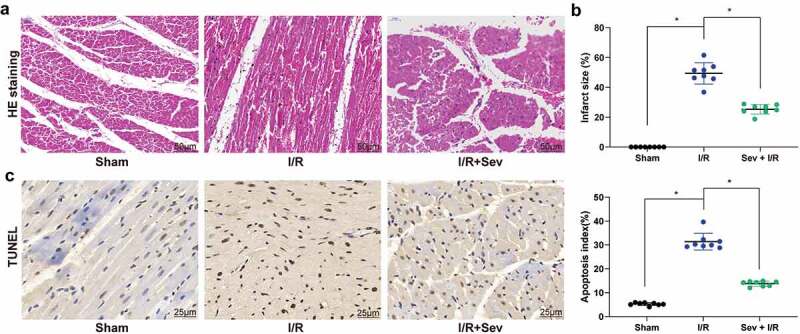
Sev ameliorates cardiac dysfunction. a, myocardial tissues of mice in each group examined by HE staining. b, effects of Sev pretreatment on the MI size of mice in each group determined by TTC staining. c, cardiomyocyte apoptosis of mice in each group examined by TUNEL staining. Each group includes 8 mice and each spot represents for a single mouse. One-way ANOVA and Tukey’s multiple comparison test were used to determine statistical significance, *p < 0.05.
3.2. Sev increases expression of miR-219a in myocardial tissues of I/R mice
To further determine the role of Sev in myocardial IRI, miRNA microarray analysis was utilized, which revealed that a total of 49 miRNAs were significantly up-regulated after Sev treatment and 34 miRNAs were down-regulated. The heatmap showed some differentially expressed miRNAs (Figure 2(a); Supplementary Table S1), where miR-219a was enhanced after Sev treatment. We subsequently examined the expression of miR-219a in each group of mice. The results showed that miR-219a was significantly reduced after I/R treatment, whereas the expression of miR-219a in the myocardium could be partially restored after Sev preconditioning (Figure 2(b)). Therefore, we administrated miR-219a atagomiR and the corresponding control, atagomiR Mock into the mice, followed by Sev pre-treatment and finally performed I/R surgery. RT-qPCR results showed that miR-219a atagomiR was successfully delivered into mouse myocardial tissues (Figure 2(c)).
Figure 2.
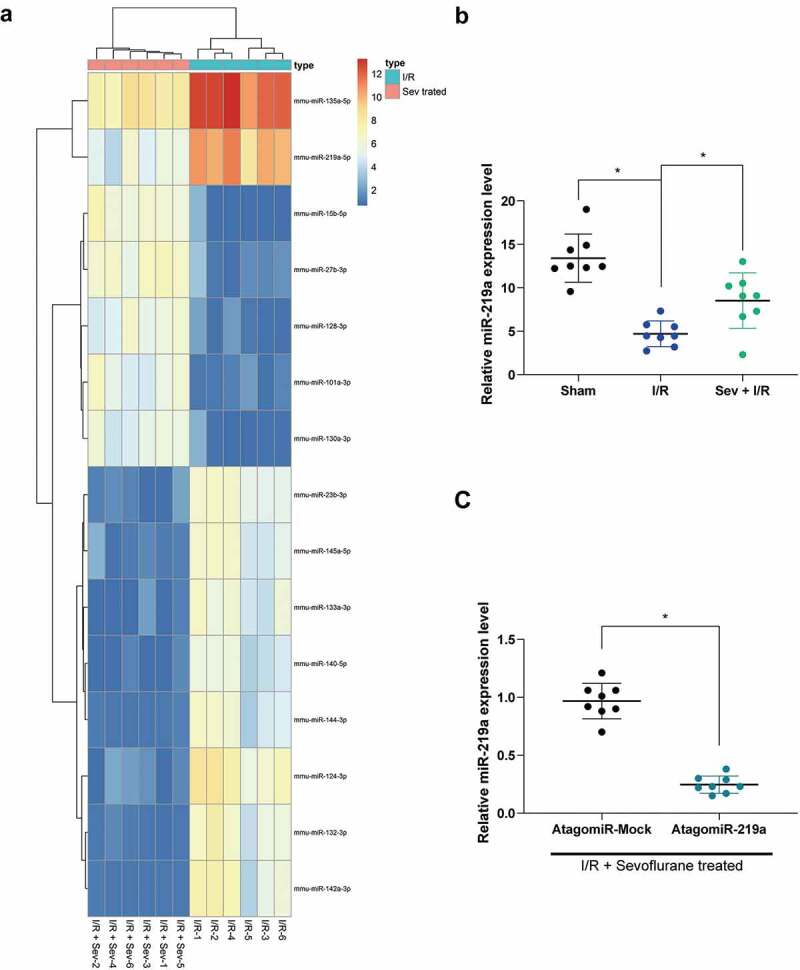
miR-219a is elevated in Sev-treated I/R mice. a, the heatmap of differentially expressed miRNAs. b, miR-219a expression among sham-, I/R- and I/R + Sev-treated mice determined by RT-qPCR. Then, miR-219a atagomiR and its negative control atagomiR Mock was injected into myocardial tissues. c, miR-219a expression determined by RT-qPCR. Each group includes 8 mice and each spot represents for a single mouse. One-way ANOVA and Tukey’s multiple comparison test or unpaired t test were used to determine statistical significance, *p < 0.05.
3.3. MiR-219a atagomiR weakens the preventive effect of Sev on I/R
Hemodynamic parameters indicated that miR-219a atagomiR inhibited the myocardial protective effect of Sev on I/R mice (Table 2) and increased the infarct size of myocardial tissue (Figure 3(a)), as well as weakened the maintenance of Sev on myocardial tissue structure (Figure 3(b)). Then, TUNEL staining showed that reducing the expression of miR-219a partially reversed the inhibition of Sev on cardiomyocyte apoptosis after I/R treatment (Figure 3(c)).
Table 2.
MiR-219a silencing atagomiR blocks the protective role of Sev on cardiac dysfunction.
| Index | Time | Sev + I/R + AntagomiR-Mock group | Sev + I/R + AntagomiR-219a group |
|---|---|---|---|
| HR (beat/min) | T0 | 159.67 ± 11.41 | 154.90 ± 10.41 |
| T4 | 129.16 ± 13.55 | 106.36 ± 15.12 | |
| LVDP (mmHg) | T0 | 114.72 ± 9.63 | 104.16 ± 6.97 |
| T4 | 68.83 ± 5.32 | 47.32 ± 4.13 | |
| LVEDP (mmHg) | T0 | 7.86 ± 0.75 | 7.94 ± 1.28 |
| T4 | 29.84 ± 3.02 | 36.57 ± 3.10 | |
| dp/dtmax(+)(mmHg/s) | T0 | 2994.01 ± 197.61 | 2919.65 ± 199.27 |
| T4 | 1617.53 ± 138.64 | 1313.44 ± 114.28 | |
| dp/dtmax(-)(mmHg/s) | T0 | 2303.91 ± 213.69 | 2316.88 ± 216.22 |
| T4 | 1010.42 ± 103.77 | 654.47 ± 34.87 |
Note: Sev, Sevoflurane; IRI, ischemia/reperfusion injury; HR, heart rate; LVEDP, left ventricular end-diastolic pressure; LVDP, left ventricular developmental pressure; dp/dtmax (+), maximum rate of rise of left ventricular pressure; dp/dtmax (-), maximum rate of decline of the left ventricular pressure.
Figure 3.
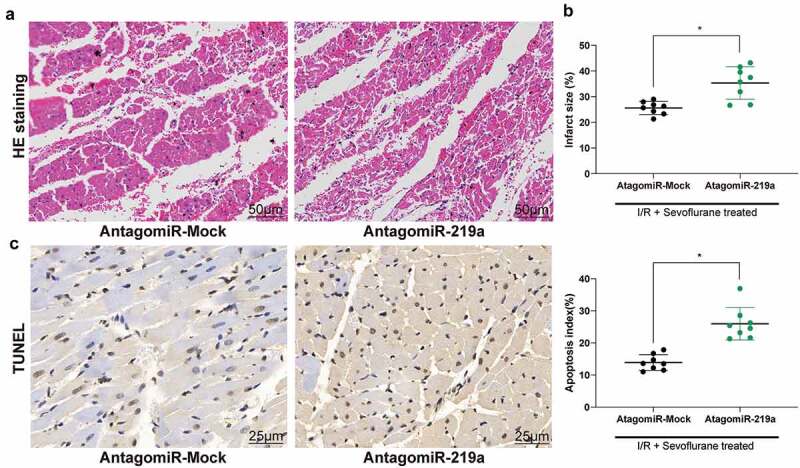
MiR-219a atagomiR blocks the preventive effect of Sev on cardiac dysfunction. a, myocardial tissues of mice in each group examined by HE staining. b, effects of Sev treatment on the infarction size of mice determined by TTC staining. c, cardiomyocyte apoptosis of mice in each group examined by TUNEL staining. Each group includes 8 mice and each spot represents for a single mouse. Unpaired t test was used to determine statistical significance, *p < 0.05.
3.4. MiR-219a inhibits activation of the TLR4/MyD88 pathway by targeting AIM2
To further identify the targeting mRNAs of miR-219a, we verified that miR-219a could target AIM2 (Figure 4(a)) through Starbase prediction as well as dual-luciferase reporter gene assays. Next, AIM2 expression was monitored to be significantly increased in mouse myocardial tissues after I/R treatment, while AIM2 expression was significantly decreased after Sev treatment, but simultaneous intervention of miR-219a in myocardial tissues restored AIM2 expression (Figure 4(b-c)). We further found that the trend in the expression of AIM2 was consistent with that of the TLR4/MyD88 pathway (Figure 4(d)).
Figure 4.
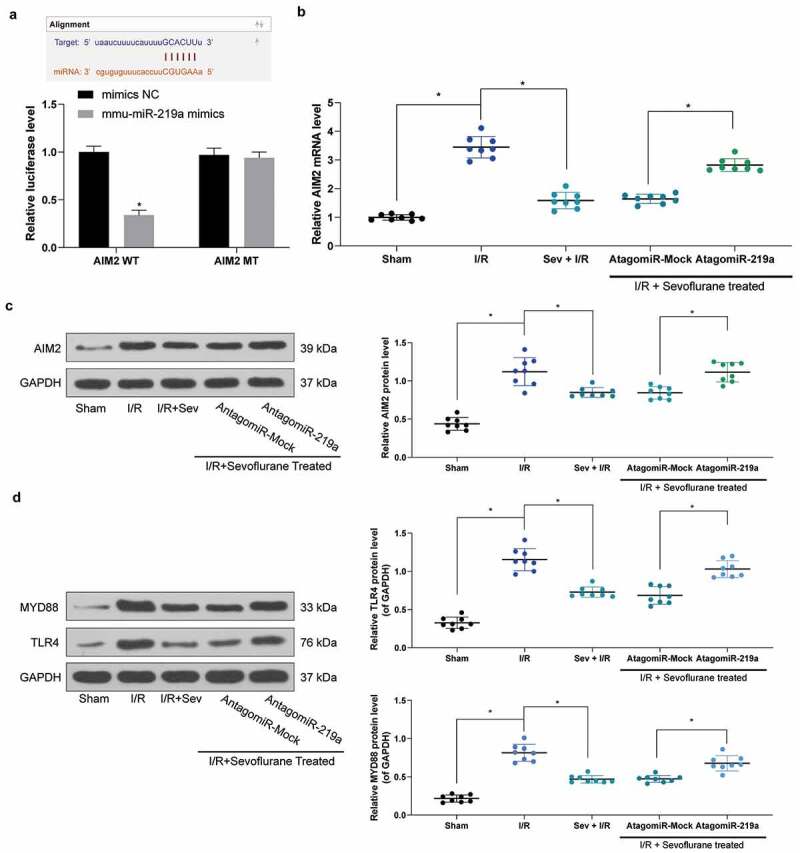
miR-219a inhibits the activation of the TLR4/MYD88 pathway by targeting AIM2. a, AIM2 mRNA, and miR-219a combination determined by dual-luciferase assays. b, AIM2 mRNA expression determined by RT-qPCR. c, AIM2 protein expression determined by western blot. d, TLR4 and MYD88 protein expression in mice determined by western blot. Each group includes 8 mice and each spot represents for a single mouse. Unpaired t test was used to determine statistical significance, *p< 0.05.
3.5. TLR4 agonist LPS treatment inhibits the effect of Sev on I/R
To determine the role of the TLR4/MyD88 pathway in IRI in Sev-treated I/R mice, we added the TLR4/MyD88 agonist LPS in Sev-treated I/R mice. We monitored that the symptoms of I/R mice were further exacerbated after the addition of LPS, and the number of apoptotic cells in myocardial tissues was also significantly increased (Table 3, Figure 5(a-d)).
Table 3.
TLR4 agonist LPS treatment blocks the protection of Sev on cardiac dysfunction.
| Index | Time | Sev + I/R + DMSO group | Sev + I/R + LPS group |
|---|---|---|---|
| HR (beat/min) | T0 | 162.24 ± 12.23 | 157.68 ± 12.59 |
| T4 | 125.24 ± 9.65 | 101.27 ± 9.24 | |
| LVDP (mmHg) | T0 | 119.06 ± 10.32 | 112.69 ± 10.55 |
| T4 | 68.69 ± 7.73 | 41.62 ± 3.11 | |
| LVEDP (mmHg) | T0 | 9.44 ± 1.25 | 8.24 ± 0.97 |
| T4 | 31.27 ± 3.54 | 42.32 ± 4.17 | |
| dp/dtmax(+)(mmHg/s) | T0 | 2971.81 ± 301.36 | 2937.87 ± 225.49 |
| T4 | 1622.03 ± 153.17 | 1201.15 ± 98.44 | |
| dp/dtmax(-)(mmHg/s) | T0 | 2302.06 ± 200.64 | 2288.61 ± 154.37 |
| T4 | 967.76 ± 93.37 | 489.57 ± 28.95 |
Note: Sev, Sevoflurane; IRI, ischemia/reperfusion injury; HR, heart rate; LVEDP, left ventricular end-diastolic pressure; LPS, lipopolysaccharide; DMSO, dimethylsulfoxide; LVDP, left ventricular developmental pressure; dp/dtmax (+), maximum rate of rise of left ventricular pressure; dp/dtmax (-), maximum rate of decline of the left ventricular pressure.
Figure 5.
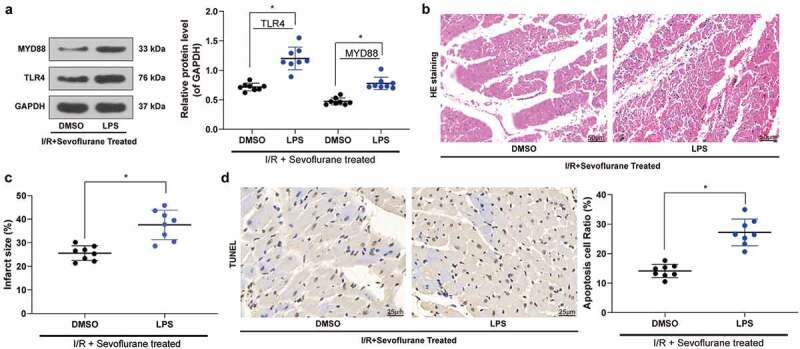
TLR4 agonist LPS treatment blocks the effective role of Sev on cardiac dysfunction. a, TLR4 and MYD88 protein expression in mice was determined by western blot. b, myocardial tissues of mice in each group examined by HE staining. c, effects of Sev pretreatment on the MI size of mice in each group determined by TTC staining. d, cardiomyocyte apoptosis of mice in each group examined by TUNEL staining. Each group includes 8 mice and each spot represents for a single mouse. Unpaired t test was used to determine statistical significance, *p < 0.05.
3.6. MiR-219 overexpression boosts cardiomyocyte viability
To further determine the effect of miR-219a on cardiomyocytes, we transfected miR-219a mimic into cardiomyocytes and carried out RT-qPCR to verify the successful transfection (Figure 6(a)). miR-219a mimic enhanced the number of EdU-positive cells and inhibited the number of PI/Annexin V-positive cells and TUNEL-positive cells, while further overexpression of AIM2 or addition of TLR4 agonist LPS treatment attenuated the effect of miR-219a on cardiomyocytes (Figure 6(b-d)).
Figure 6.
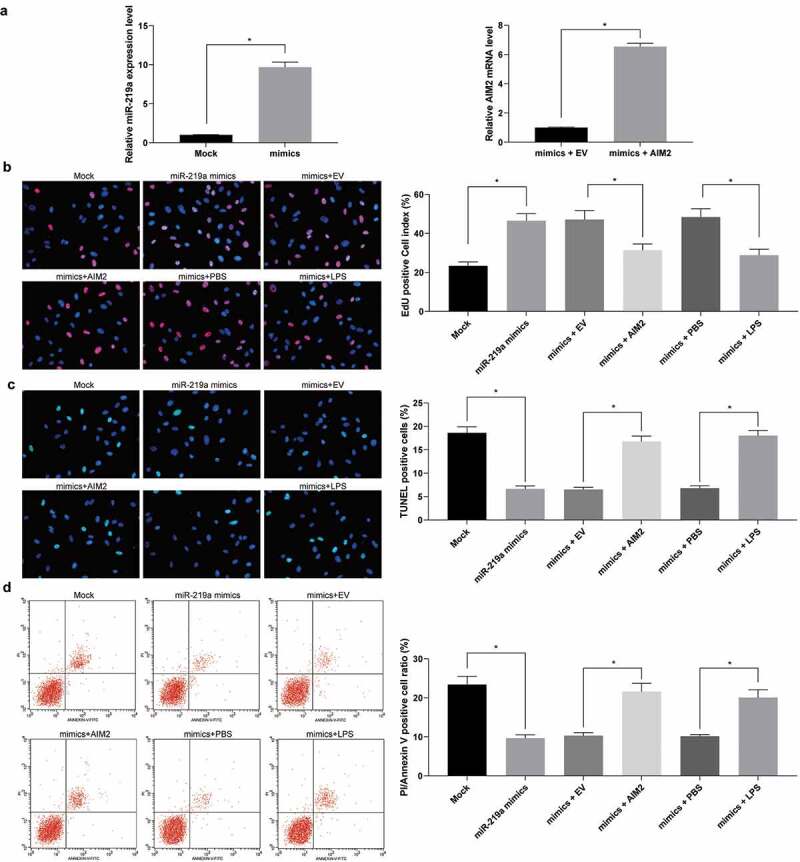
Upregulation of miR-219a induces cardiomyocyte viability. a. miR-219a expression and the mRNA expression of AIM2 in cardiomyocytes determined by RT-qPCR. b, EdU staining of cardiomyocyte viability. c, TUNEL staining of cardiomyocyte apoptosis. d, cardiomyocyte apoptosis index evaluated by PI/Annexin V with flow cytometry. The data are displayed as the mean ± SD. One-way ANOVA and Tukey’s multiple comparison test were applied to determine statistical significance, *p < 0.05.
4. Discussion
Mechanical and drug involvements are the most effective strategies to swiftly recover the blood flow in occlusive coronary artery, minimize the infarction area in myocardium, and improve the outcome for patients after acute MI [19]. Nevertheless, reperfusion may also result in other cardiac cell death and elevate infarction area in the heart, the main factors leading to which include inflammation, oxidative stress, as well as apoptosis [20]. Herein, we demonstrated that upregulated miR-219a reduced the expression of AIM2 through the TLR4/MyD88 pathway using a myocardial I/R mouse model, whereby the cardio-protection of Sev was reinforced against myocardial IRI.
A mouse model with myocardial I/R was developed by ligating the anterior descending branches of the left coronary artery, followed by validation with cardiac functions with hemodynamic parameter assessment. The decline in HR after I/R treatment indicated impaired cardiac function, reduced dp/dtmax suggested significantly repressed myocardial contractility, while increased LVEDP indicated a significant increase in left ventricular pressure load, implying a significant impairment of cardiac function after I/R treatment [21,22]. The administration of Sev increased cardiac functions with increased HR, LVDP, dp/dtmax (+), and dp/dtmax (−), while reduced LVEDP, which suggested that Sev treatment significantly protected myocardial tissues from IRI. Furthermore, Sev ameliorated pathological injuries in the heart and diminished infarct size and cardiomyocyte apoptosis. Similarly, Sev was observed to protect the myocardium in post-ischemic guinea pigs from promoted coronary leakage and endothelial dysfunction [23]. Mechanistically, Sev preconditioning enhanced Bcl-2 expression in the intestinal tissues and repressed caspase-3 expression in intestinal IRI [24]. Besides, Sev postconditioning hampered the activation of caspase-3 and caspase-9, mediators of apoptosis in myocardial IRI [25]. The detail mechanism involving Sev treatment in cardiomyocyte apoptosis was not investigated due to time and fund limitation, which deserves further studies. With the treatment of miR-219a atagomiR, the effects of Sev on maintaining myocardium structure and suppressing cardiomyocyte apoptosis were inhibited. In mice with I/R, miR-219a expressed at a poor level, and its target AIM2 expressed at a high level. miR-219a-5p exerted an anti-inflammatory role in inflammatory bowel disease in vitro and in vivo [26]. Also, exosomes secreted from neural stem cells under exposure to IGF-1 suppressed apoptosis and encouraged nerve regeneration at least partly through an miR-219a-2-3p-mediated mechanism following the spinal cord injury [27].
Further bioinformation prediction and dual-luciferase reporter assays revealed that miR-219a directly targeted and reversely regulated the expression pattern of AIM2. Previously, maternally expressed gene 3 facilitated cerebral IRI through AIM2 by sponging miR-485 [28]. AIM2 was first identified as a novel gene that is differentially expressed in a melanoma cell line UACC903 that had received chromosome 6, contributing to resumption of AIM2 expression and reversion of the malignant behaviors [29]. Moreover, silencing of AIM2 alleviated cardiac dysfunction caused by metabolic disorder and ventricular remodeling as well as reduced pyroptosis in H9c2 cardiomyoblasts [30]. The anti-inflammatory function of AIM2 in an inflammasome-independent way was also reported in cardiomyocytes [31]. Thus, the functional relevance of AIM2 in cardiomyocytes may be established. While in the present study, upregulation of AIM2 could reverse the inhibitive role of miR-219a on cardiomyocyte apoptosis. Furthermore, the TLR4/MyD88 pathway was monitored to closely participate in regulating cardiomyocyte apoptosis in the myocardial IR mouse model. In line with our findings, miR-203 agonist has the potency to elevate the action of Sev by inhibiting the expression of MyD88 and neuroinflammation in cerebral I/R [32]. Asmussen et al. detected an enhancement of TLR4 in monocytes immediately after a restoration of spontaneous circulation, which may resemble the inflammatory activation induced by the systematic IRI [33]. Accordingly, Khan et al. demonstrated that elevated brain injury and neurological deficits were mitigated in mice treated with a specific TLR4 inhibitor [34]. In a similar manner, we provided forceful evidences supporting that the TLR4 agonist LPS significantly abrogated the protective role of Sev on the apoptosis of cardiomyocytes. The direct interaction between LPS and Sev has been determined in a mouse model with cognitive impairments [35]. Meanwhile, Sev was capable of curtailing the cytokine cascade initiation and protecting against myocardial injury or dysfunction with the involvement of cardiomyocyte-expressed TLR4 [36].
Taken together, this study illustrated that miR-219a bound to and negatively regulated AIM2 expression via the TLR4/MyD88 pathway, through which the cardio-protection of Sev functioned against myocardial IRI in mice. MiR-219a may present as a novel target for enhancing Sev-induced myocardial protection. Additional efforts should be made to gain more insights about Sev and to develop new treatment strategies for myocardial IRI.
Supplementary Material
Disclosure statement
All authors declare that there is no conflict of interests in this study.
Author contributions
Yan Li is the guarantor of integrity of the entire study and contributed to the concepts and design of this study; Na Xing contributed to the experimental studies and clinical studies; Jingjing Yuan contributed to the data and statistical analysis; Jianjun Yang took charge of the manuscript preparation; Yan Li contributed to the manuscript review. All authors read and approved the final manuscript.
References
- [1].Hausenloy DJ, Yellon DM.. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Frank A, Bonney M, Bonney S, et al. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cao J, Xie H, Sun Y, et al. Sevoflurane post-conditioning reduces rat myocardial ischemia reperfusion injury through an increase in NOS and a decrease in phosphorylated NHE1 levels. Int J Mol Med. 2015;36:1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rancan L, Huerta L, Cusati G, et al. Sevoflurane prevents liver inflammatory response induced by lung ischemia-reperfusion. Transplantation. 2014;98:1151–1157. [DOI] [PubMed] [Google Scholar]
- [5].Wen XR, Fu YY, Liu HZ, et al. Neuroprotection of Sevoflurane Against Ischemia/Reperfusion-Induced Brain Injury Through Inhibiting JNK3/Caspase-3 by Enhancing Akt Signaling Pathway. Mol Neurobiol. 2016;53:1661–1671. [DOI] [PubMed] [Google Scholar]
- [6].Cavalcante FP, Coelho AM, Machado MC, et al. Mechanisms of the beneficial effect of sevoflurane in liver ischemia/reperfusion injury. Acta Cir Bras. 2015;30:749–755. [DOI] [PubMed] [Google Scholar]
- [7].Zhao J, Wang F, Zhang Y, et al. Sevoflurane preconditioning attenuates myocardial ischemia/reperfusion injury via caveolin-3-dependent cyclooxygenase-2 inhibition. Circulation. 2013;128:S121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao YB, Zhao J, Zhang LJ, et al. MicroRNA-370 protects against myocardial ischemia/reperfusion injury in mice following sevoflurane anesthetic preconditioning through PLIN5-dependent PPAR signaling pathway. Biomed Pharmacother. 2019;113:108697. [DOI] [PubMed] [Google Scholar]
- [9].Siebert V, Allencherril J, Ye Y, et al. The Role of Non-coding RNAs in Ischemic Myocardial Reperfusion Injury. Cardiovasc Drugs Ther. 2019;33:489–498. [DOI] [PubMed] [Google Scholar]
- [10].Zhong Z, Zhong W, Zhang Q, et al. Circulating microRNA expression profiling and bioinformatics analysis of patients with coronary artery disease by RNA sequencing. J Clin Lab Anal. 2019;34:e23020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fu R, Zhou J, Wang R, et al. Protocatechuic Acid-Mediated miR-219a-5p Activation Inhibits the p66shc Oxidant Pathway to Alleviate Alcoholic Liver Injury. Oxid Med Cell Longev. 2019;2019:3527809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xiao Y, Zhang S, Li Q, et al. miR-219a-5p Ameliorates Hepatic Ischemia/Reperfusion Injury via Impairing TP53BP2. Dig Dis Sci. 2019;64:2177–2186. [DOI] [PubMed] [Google Scholar]
- [13].Kim HY, Kim SJ, Lee SM. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J. 2015;282:259–270. [DOI] [PubMed] [Google Scholar]
- [14].Csak T, Pillai A, Ganz M, et al. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 2014;34:1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang J, Li R, Peng Z, et al. HMGB1 participates in LPSinduced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NFkappaB signaling pathways. Int J Mol Med. 2020;45:61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dickinson RJ, Delavaine L, Cejudo-Marin R, et al. Phosphorylation of the kinase interaction motif in mitogen-activated protein (MAP) kinase phosphatase-4 mediates cross-talk between protein kinase A and MAP kinase signaling pathways. J Biol Chem. 2011;286:38018–38026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hu Z, Cai M, Zhang Y, et al. miR-29c-3p inhibits autophagy and cisplatin resistance in ovarian cancer by regulating FOXP1/ATG14 pathway. Cell Cycle. 2020;19:193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhou L, Kong Q, Zhang Y, et al. Glucose deprivation promotes apoptotic response to S1 by enhancing autophagy in human cervical cancer cells. Cancer Manag Res. 2018;10:6195–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: from Inflammation to Fibrosis. Circ Res. 2016;119:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang Y, Wang L, Li JH, et al. Morphine alleviates myocardial ischemia/reperfusion injury in rats by inhibiting TLR4/NF-kappaB signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23:8616–8624. [DOI] [PubMed] [Google Scholar]
- [21].Guo Y, Yang Q, Weng XG, et al. Shenlian Extract Against Myocardial Injury Induced by Ischemia Through the Regulation of NF-kappaB/IkappaB Signaling Axis. Front Pharmacol. 2020;11:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Noroozzadeh M, Raoufy MR, Bidhendi Yarandi R, et al. The effects of prenatal androgen exposure on cardiac function and tolerance to ischemia/reperfusion injury in male and female rats during adulthood. Life Sci. 2019;229:251–260. [DOI] [PubMed] [Google Scholar]
- [23].Annecke T, Chappell D, Chen C, et al. Sevoflurane preserves the endothelial glycocalyx against ischaemia-reperfusion injury. Br J Anaesth. 2010;104:414–421. [DOI] [PubMed] [Google Scholar]
- [24].Liu C, Ding R, Huang W, et al. Sevoflurane Protects against Intestinal Ischemia-Reperfusion Injury by Activating Peroxisome Proliferator-Activated Receptor Gamma/Nuclear Factor-kappaB Pathway in Rats. Pharmacology. 2020;105:231–242. [DOI] [PubMed] [Google Scholar]
- [25].Inamura Y, Miyamae M, Sugioka S, et al. Sevoflurane postconditioning prevents activation of caspase 3 and 9 through antiapoptotic signaling after myocardial ischemia-reperfusion. J Anesth. 2010;24:215–224. [DOI] [PubMed] [Google Scholar]
- [26].Shi Y, Dai S, Qiu C, et al. MicroRNA-219a-5p suppresses intestinal inflammation through inhibiting Th1/Th17-mediated immune responses in inflammatory bowel disease. Mucosal Immunol. 2020;13:303–312. [DOI] [PubMed] [Google Scholar]
- [27].Ma K, Xu H, Zhang J, et al. Insulin-like growth factor-1 enhances neuroprotective effects of neural stem cell exosomes after spinal cord injury via an miR-219a-2-3p/YY1 mechanism. Aging (Albany NY). 2019;11:12278–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liang J, Wang Q, Li JQ, et al. Long non-coding RNA MEG3 promotes cerebral ischemia-reperfusion injury through increasing pyroptosis by targeting miR-485/AIM2 axis. Exp Neurol. 2019;325:113139. [DOI] [PubMed] [Google Scholar]
- [29].Choubey D, Duan X, Dickerson E, et al. Interferon-inducible p200-family proteins as novel sensors of cytoplasmic DNA: role in inflammation and autoimmunity. J Interferon Cytokine Res. 2010;30:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang X, Pan J, Liu H, et al. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019;221:249–258. [DOI] [PubMed] [Google Scholar]
- [31].Furrer A, Hottiger MO, Valaperti A. Absent in Melanoma 2 (AIM2) limits pro-inflammatory cytokine transcription in cardiomyocytes by inhibiting STAT1 phosphorylation. Mol Immunol. 2016;74:47–58. [DOI] [PubMed] [Google Scholar]
- [32].Zhong H, Chen H, Gu C. Sevoflurane Post-treatment Upregulated miR-203 Expression to Attenuate Cerebral Ischemia-Reperfusion-Induced Neuroinflammation by Targeting MyD88. Inflammation. 2020;43:651–663. [DOI] [PubMed] [Google Scholar]
- [33].Asmussen A, Fink K, Busch HJ, et al. Inflammasome and toll-like receptor signaling in human monocytes after successful cardiopulmonary resuscitation. Crit Care. 2016;20:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Khan MM, Gandhi C, Chauhan N, et al. Alternatively-spliced extra domain A of fibronectin promotes acute inflammation and brain injury after cerebral ischemia in mice. Stroke. 2012;43:1376–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Satomoto M, Sun Z, Adachi YU, et al. Sevoflurane preconditioning ameliorates lipopolysaccharide-induced cognitive impairment in mice. Exp Anim. 2018;67:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Li J, Liu P, Li H, et al. Sevoflurane Preconditioning Prevents Septic Myocardial Dysfunction in Lipopolysaccharide-Challenged Mice. J Cardiovasc Pharmacol. 2019;74:462–473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.