

ABSTRACT
Macroautophagy/autophagy is a conserved catabolic recycling pathway involving the sequestration of cytoplasmic components within double-membrane vesicles termed autophagosomes. The autophagy-related (Atg) protein Atg13 is a key member of the autophagy initiation complex. The Atg13 C terminus is an intrinsically disordered region (IDR) harboring a binding site for the vacuolar membrane protein Vac8. Recent reports suggest Atg13 acts as a hub to assemble the initiation complex, and also participates in membrane recognition. Here we show that the Atg13 C terminus directly binds to lipid membranes via electrostatic interactions between positively charged residues in Atg13 and negatively charged phospholipids as well as a hydrophobic insertion of a Phe residue. We identified 2 sets of residues in the Atg13 IDR that affect its phospholipid-binding properties; these residues overlap with the Vac8-binding domain of Atg13. Our data indicate that Atg13 binding to phospholipids and Vac8 is mutually exclusive, and both are required for efficient autophagy.
Abbreviations
Atg: autophagy-related; CD: circular dichroism; Cvt: cytoplasm-to-vacuole targeting; IDR: intrinsically disordered region; ITC: isothermal calorimetry; MIM: MIT-interacting motif; MKO: multiple-knockout; PAS: phagophore assembly site; PC: phosphatidylcholine; PS: phosphatidylserine; PtdIns: phosphatidylinositol; PtdIns3P: phosphatidylinositol-3-phosphate
KEYWORDS: Autophagy, intrinsically disordered region, membrane binding, phospholipids, structure
Introduction
Macroautophagy, hereinafter referred to as autophagy, is a catabolic process marked by the formation of the phagophore, a transient cup-shaped structure that matures into a double-membrane compartment called the autophagosome. In yeast, the autophagosome forms at the phagophore assembly site (PAS), where autophagy-related (Atg) proteins, donor membranes, and cytoplasmic cargo coalesce in preparation for sequestration of superfluous and aberrant cellular materials. The process of autophagy can be subdivided into 4 ordered steps: initiation/nucleation, elongation/closure, docking/fusion, and degradation/efflux [1].
The Atg1 kinase complex, comprised of Atg1, Atg13, and the Atg17-Atg31-Atg29 subcomplex, is a fundamental unit for autophagy initiation. This complex promotes autophagosome formation through post-translational regulation of downstream core autophagy proteins [2,3] and by providing the structural backbone for the growing autophagosome [4]. In recent years, multiple structural and biochemical studies involving the Atg1 kinase complex revealed its highly dynamic self-assembling propensity that presents a comprehensive model for recognizing and integrating Atg9-positive vesicles (donor membranes) at the PAS [5–8].
In the center of all the interacting proteins is Atg13, a regulatory protein of the Atg1 kinase complex. In nutrient-rich conditions, Atg13 is hyperphosphorylated in a TOR-dependent manner, whereas nutrient depletion leads to TOR inhibition, Atg13 dephosphorylation, and the activation of Atg1 [9]. Atg13 contains an N-terminal HORMA domain, which has been suggested to recruit Atg14 [10,11]. The C-terminal domain of Atg13 was demonstrated to be an intrinsically disordered region (IDR) by high-speed atomic force microscopy [8]. This C-terminal domain harbors multiple protein binding sites. Specifically, the Atg13 IDR was reported to contain at least 2 Atg17 binding motifs: the Atg17-linking region (17LR) (359–389) and Atg17-binding region (17BR) (424–436) [8], a microtubule interacting and transport (MIT)-interacting motif (MIM) that binds to Atg1 (460–521) [12], and the C-terminal (567–738) region occupied by the vacuolar peripheral membrane protein Vac8 [13]. Furthermore, recent in vitro experiments by Rao et al. [2] showed that recombinant purified Atg13 from S. cerevisiae directly binds yeast polar lipids, suggesting that Atg13 acts as a molecular hub to mediate the intricate spatiotemporal organization necessary for Atg1 kinase complex assembly as well as membrane recognition.
Some insights have been gained into assembly of the Atg1 complex [2,4,8], but the molecular mechanism of yeast Atg13 lipid binding capability remains to be discovered. In contrast, lipid binding by human ATG13 is mediated by a cluster of lysine residues, at the N terminus of the HORMA domain, which are responsible for electrostatic interactions with negatively charged phospholipids. These membrane-binding lysines are evolutionarily conserved in mouse and zebrafish, but not in yeast [14], suggesting that a membrane-binding motif of yeast Atg13 is located in a different region than the N terminus of the HORMA domain.
Here we show that the C terminus of yeast Atg13 directly binds to liposomes via electrostatic interactions, between positively charged residues in Atg13 and negatively charged phospholipids, as well as insertion of a Phe side chain. We identified 2 sets of positively charged residues in the IDR of Atg13 from S. cerevisiae that affect the phospholipid binding properties of Atg13 when mutagenized. These residues reside in short phenylalanine-containing motifs within the amino acid sequence that overlaps with the Vac8-binding domain of Atg13. Our data show that phospholipid- and Vac8-binding of Atg13 are mutually exclusive and both are required for efficient autophagy.
Results
Atg13[571-700] is intrinsically disordered
The C-terminal tail of Atg13, spanning residues 269–738, is intrinsically disordered [8]. Several regions of this disordered domain bind proteins including Atg17, Atg1 and Vac8 (Figure 1A). In addition to supporting protein-protein interactions, disordered regions bind liposomes and in some cases even alter membrane curvature [15]. As such, we wondered if the previously observed membrane-binding behavior of Atg13 was carried out by some portion of the predicted disordered C terminus of Atg13. After screening several C-terminal constructs for expression and solubility in E. coli, we succeeded in purifying Atg13[571–700]. As expected, this region was intrinsically disordered as observed by 2D 1H-15N HSQC spectroscopy and circular dichroism (CD) (Figures S1 and S2).
Figure 1.
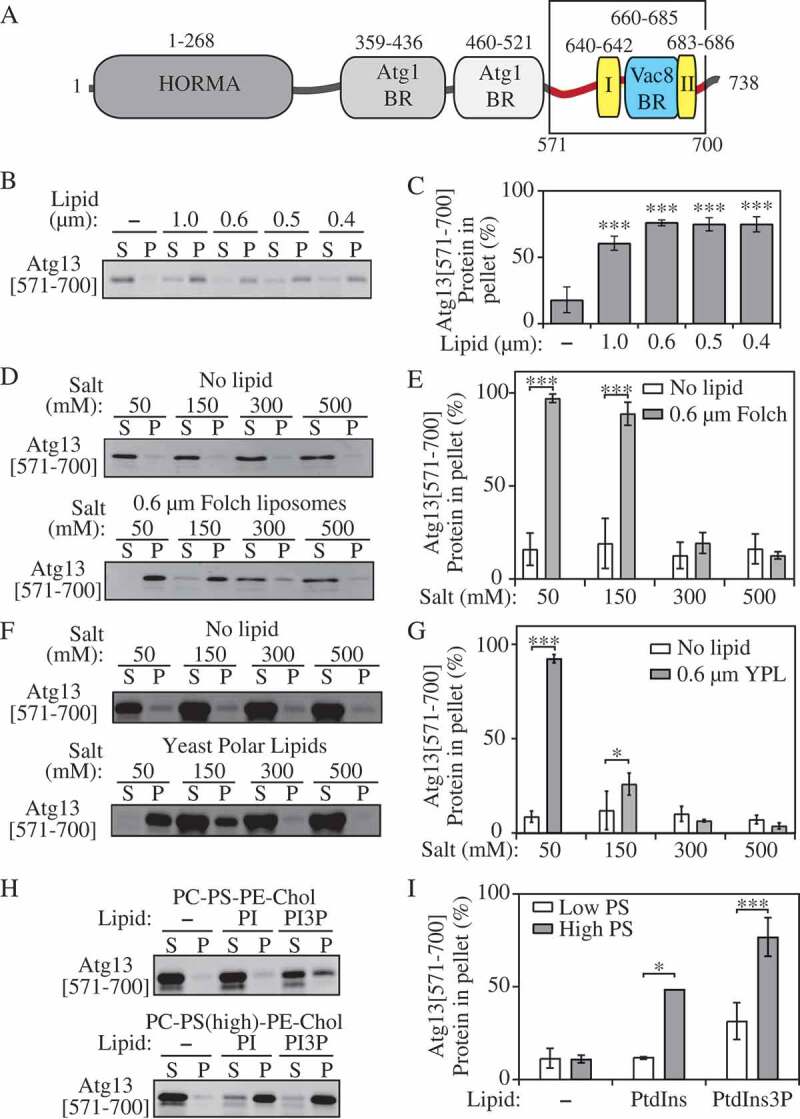
Atg13[571–700] binds negatively charged membranes. (A) Domain representation of the Atg13 protein from S. cerevisiae. In the C terminal IDR, following the HORMA domain (dark gray), the Atg17 (gray), Atg1 (light gray), and Vac8 (blue) binding regions are denoted. Phospholipid binding site I and II discovered in this study are denoted (maize). (B) Liposome sedimentation assay of Atg13[571–700] with Folch liposomes containing diameters from 1.0 µm to 0.4 µm. (C) Densitrometric quantification of panel B. (D) Liposome sedimentation assay of Atg13[571–700] with 0.6-µm Folch liposomes at varying concentrations of NaCl. (E) Densitrometric quantification of panel D. (F) Liposome sedimentation assay of Atg13[571–700] with yeast polar lipids. (G) Densitrometric quantification of panel F. (H) Liposome sedimentation assay of Atg13[571–700] with liposomes composed of PC, PS, PE, Chol, PtdIns and PtdIns3P. (I) Densitrometric quantification of panel H. Error bars represent the SD from 3 experiments. Statistical analysis by ANOVA: * P < 0.05; *** P < 0.001.
Atg13[571-700] binds negatively charged liposomes
To test whether Atg13[571–700] interacts with membranes, we performed liposome sedimentation assays with Folch-derived unilamellar vesicles. In liposome sedimentation assays, protein and liposomes are incubated together and subsequently subjected to centrifugation. Due to their size, liposomes will pellet during centrifugation. In the absence of liposomes or if the protein does not interact with liposomes, the protein will remain in the supernatant fraction after centrifugation. However, if the protein interacts with membranes it will pellet along with the liposomes. Folch vesicles were generated with diameters of 1.0, 0.6, 0.5, and 0.4 µm, incubated with Atg13[571–700] and subjected to centrifugation. Atg13[571–700] appeared in the supernatant fraction in the absence of liposomes but pelleted when incubated with all sizes of liposome tested (Figure 1B,C). This finding demonstrates that Atg13[571–700] interacts with Folch liposomes and appears to have little specificity for the size of the vesicle.
Full-length Atg13 was shown to bind yeast polar lipids [2], suggesting that electrostatic interactions may play an important role in liposome binding by the Atg13[571–700] peptide. To test if this is the case, we performed liposome sedimentation assays in the presence of increasing concentrations of NaCl (Figure 1D,E). If electrostatic interactions are required for membrane binding, then increasing NaCl concentrations will disrupt this interaction. We found that Atg13[571–700] bound to Folch liposomes in buffer containing either 50 or 150 mM NaCl, but that binding was almost completely abolished in buffer containing 300 or 500 mM NaCl. This result suggests that Atg13 binding to liposomes is mediated primarily through electrostatic interactions. In agreement with this result, Atg13[571–700] also bound yeast polar lipids, and this binding was severely disrupted by a NaCl concentration higher than 50 mM (Figure 1 F,G).
Intrinsically disordered proteins can bind to membranes using hydrophobic amino acids that fold to form an amphipathic helix upon membrane binding [16]. While our liposome sedimentation data suggested that this is not the case for Atg13[571–700] we wanted to further confirm this by monitoring the secondary structure of Atg13[571–700] using CD in the absence and presence of liposomes. We did not observe any increase in helix formation upon liposome binding by Atg13[571–700], further supporting the idea that electrostatic interactions are the main mechanism of liposome binding (Figure S2).
We next sought to determine which phospholipids are required for Atg13[571–700] binding and if Atg13[571–700] recognized liposomes based solely on their negative charge. For this purpose, we generated liposomes containing PC, PS, phosphatidylinositol (PtdIns), or phosphatidylethanolamine (PE), which are the main phospholipids in yeast polar extracts, and cholesterol. Liposomes were generated with 53.25% PC and 19% PS or 29.75% PC and 42.50% PS while keeping the concentrations of PtdIns and PE constant. Atg13[571–700] did not bind to liposomes containing 19% PS but did bind to liposomes containing 42.5% PS (Figure 1H,I). In addition, we tested if PtdIns3P would alter the binding of Atg13[571–700] in these vesicles. Substituting PtdIns3P for PtdIns in liposomes led to enhanced binding of Atg13. However, further increased negative charge, using PtdIns4P, PtdIns5P, PtdIns(3,5)P2 or PtdIns(4,5)P2, yielded binding of Atg13[571–700] to liposomes comparable to or slightly lower than that with PtdIns3P (Figure S3), suggesting that binding is not highly PtdIns3P specific and is primarily electrostatic. The increased binding of Atg13[571–700] to PtdIns3P-containing liposomes with a higher content of PS at the expense of PC is likely due to fulfilling the requirement for a lipid with a rather smaller head group. Taken together, our data suggest that Atg13[571–700] binds negatively charged lipids via electrostatic forces that are accompanied by an interaction facilitated by a small-headed phospholipid, such as a hydrophobic insertion.
Atg13[571-700] contains 2 distinct liposome binding regions
Due to the requirement of negatively charged phospholipids for Atg13[571–700] binding, we hypothesized that liposome binding by Atg13[571–700] would be mediated by positively charged amino acids. The Atg13[571–700] peptide has 2 conserved positively charged motifs, 640KFK and 683KFHK (Figure 1A and S4). To elucidate if and how these motifs are responsible for liposome binding, we first generated a series of truncations. These constructs included 1) 571–682, which lacks the highly conserved 683KFHK motif; 2) 571–640, which lacks both motifs; and 3) 647–700 which contains the 683KFHK motif, but lacks the 640KFK motif. Liposome sedimentation assays were performed using these constructs in 50, 150, 300 and 500 mM NaCl. The only truncation construct that had substantially reduced liposome binding at low NaCl concentrations was Atg13[571–640] (Figure 2A, Figure S4), suggesting that the 641–700 region is important for liposome binding. In addition, Atg13[571–682] and [647–700] displayed nearly identical binding to Atg13[571–700], further supporting a role for the 641–700 region and its 2 motifs in membrane binding.
Figure 2.
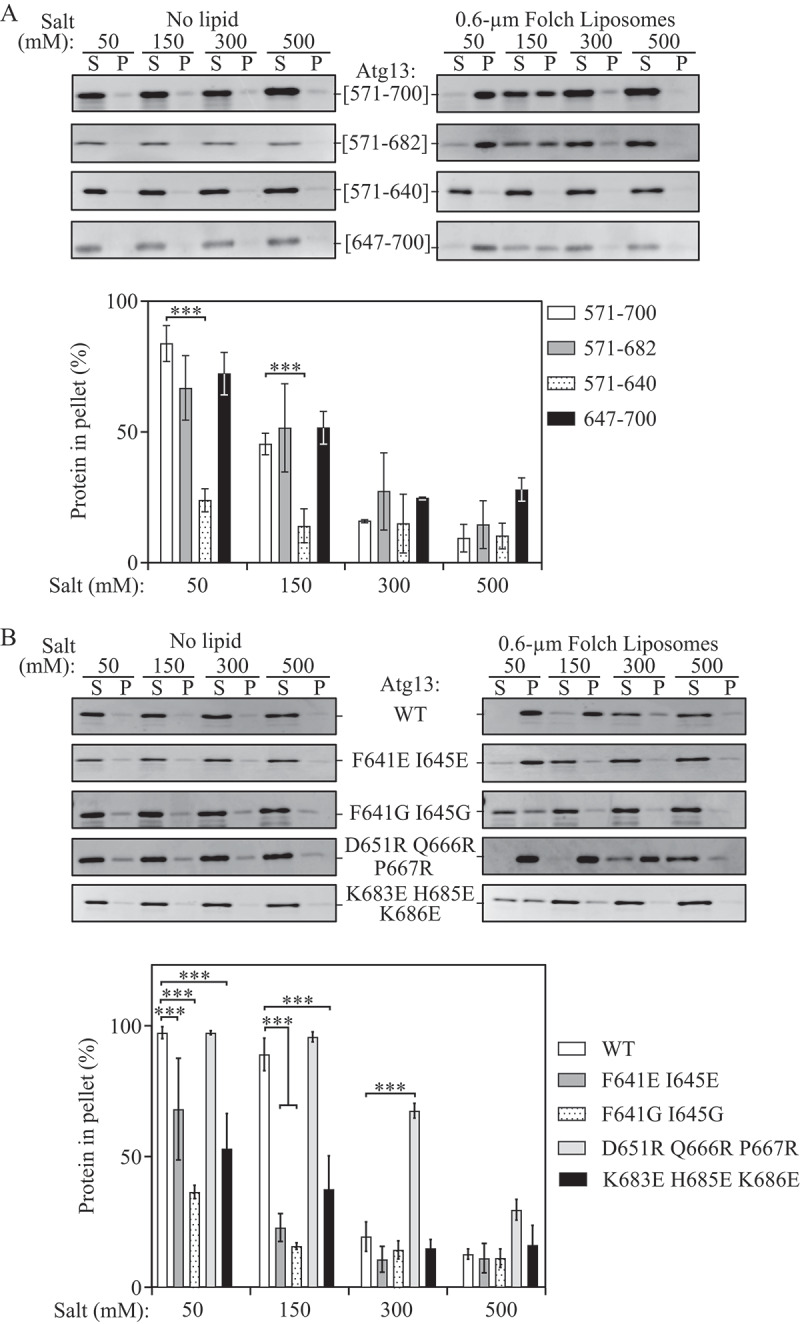
Atg13 utilizes 2 distinct regions for liposome binding. (A) Liposome sedimentation assay of recombinant Atg13[571–700], [571–682], [571–640], and [647–700] in the absence of lipid or with 0.6-µm Folch liposomes at varying concentrations of NaCl (top) and densitrometric quantification (bottom). (B) Liposome sedimentation assay of recombinant Atg13 mutants F641E I645E, F641G I645G, K683E H685E K686E, and D651R Q666R P667R, in the absence of lipid or with 0.6-µm Folch liposomes at varying concentrations of NaCl (top) and densitrometric quantification (bottom). Error bars represent the SD from 3 individual trials. Statistical analysis by ANOVA: *** P < 0.001.
To further examine the role of the 640KFK and 683KFHK motifs in liposome binding, we mutated the Atg13[571–700] sequence to F641G I645G, F641E I645E, or K683E H685E K686E. Liposome sedimentation assays were performed in 50, 150, 300 and 500 mM NaCl. All 3 mutants were significantly defective in membrane binding at a physiological concentration of 150 mM NaCl, with more than 60% reduction relative to wild type (Figure 2B). A summary of all truncation and mutagenesis data is presented in Figure S5. These data suggest that Atg13[571–700] contains 2 distinct liposome-binding motifs, 640KFK (site I) and 683KFHK (site II) (Figure 1A) and that electrostatic force is not a sole factor responsible for lipid binding of the Atg13[571–700] region. A hydrophobic insertion of aromatic residues in the motifs plays a role in binding efficiency, in agreement with data in Figure 1.
Two phospholipid-binding motifs of Atg13 are within the Vac8-binding domain
In yeast, Vac8 interacts with the Atg13 C-terminal sequence, which does not have homology to human ATG13 [13,17]. A segment of the Atg13 C-terminal region has homology with Nvj1, a nuclear membrane protein that also interacts with Vac8 [17]. The Vac8-Nvj1 interaction forms the nucleus-vacuole junction that is essential for piecemeal microautophagy of the nucleus [18–20]. Vac8 interaction with Nvj1 or Atg13 is exclusive, and competitive, and relies on positively charged residues in the Vac8 groove [17]. The recently solved crystal structure of Atg13 in the complex with Vac8 revealed that Atg13 employs a relatively long region to interact with Vac8. In the region of Atg13 that was used for crystallization (residues 567–695), the segment S660-H685 is stabilized in the Vac8 groove (Figure S6A), and reveals the Vac8-Atg13 interface (Park et al., co-submitted manuscript), the N terminus of which interacts more weakly than the C terminus. The phospholipid-binding motifs identified in our study are within the region 571–700, a very similar region to that used by Park et al. (co-submitted manuscript) in the Vac8-Atg13 crystals. When compared to the Atg13-Vac8 crystal structure, the first phospholipid-binding motif of Atg13 (640KFK; site I) is near, but outside, the stabilized Atg13-Vac8 interface. The second phospholipid-binding motif (683KFHK, site II) is at the very end of the interface (Figure S6A).
To investigate whether mutations in the phospholipid-binding sites I and II of Atg13 (640KFK and 683KFHK) affect binding with Vac8, we tested the binding affinity of the Atg13 glycine and glutamate mutants with Vac8 relative to wild type. As indicated above, we also constructed a mutant with a presumably disrupted interaction between the Atg13 peptide and Vac8. The D651R Q666R P667R mutant carries cationic residues in the Vac8-binding region to repulse the peptide from the positively charged Vac8 groove. We applied isothermal calorimetry (ITC) to measure dissociation constants for Atg13[571–700] binding to recombinant purified Vac8[10–515]. The WT and all of the mutated Atg13[571–700] peptides, except for Atg13[571–700]D651,Q666,P667R, bound to Vac8 with dissociation constants from 227–736 nM (Table 1), confirming that the Atg13[571–700] region carries the Vac8-binding domain, and that the Atg13 residues required for liposome binding are not required for Vac8 binding (Figure 3A). Atg13[571–700]D651,Q666,P667R bound to Vac8 with the lowest affinity (Kd of 1885 nM; Table 1), demonstrating that the Atg13 region between the phospholipid site I and II (Figure S6A) is involved in Vac8 binding, in agreement with the crystallographic data.
Table 1.
Dissociation constants for Atg13[571–700] WT and mutant proteins titrated into Vac8 [10–515] as determined by ITC.
| Titrant | Kd (nM) | ||
|---|---|---|---|
| Atg13[571–700] | 736 ± 31 | ||
| Atg13[571–700] F641E I645E | 673 ± 9 | ||
| Atg13[571–700] F641G I645G | 227 ± 12 | ||
| Atg13[571–700] D651R Q666R P667R | 1885 ± 440 | ||
| Atg13[571–700] K683E H685E K686E | 680 ± 422 | ||
Figure 3.
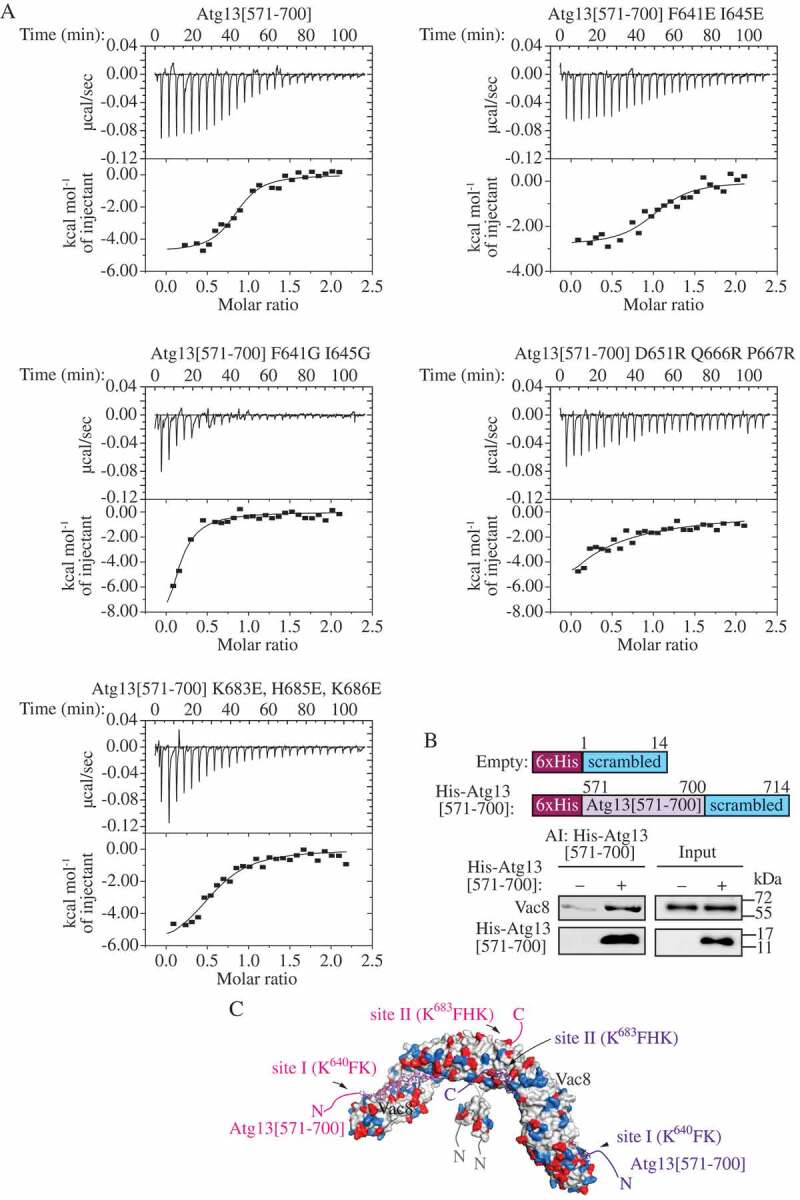
Atg13[571–700] interacts with Vac8. (A) Raw ITC data (top) and binding isotherms (bottom) for Atg13[571–700] WT, F641G I645G, F641E I645E, K683E H685E K686E, and D651R Q666R P667R titrated into Vac8 [10–515]. (B) His-tag affinity-isolation experiment. Top panels show schematic representation of the constructs that were immobilized on the Ni-NTA agarose to detect the interaction with Vac8 after affinity isolation (AI). MKO cells transformed with the pVac8(426) plasmid and pCuHis6-Ss(424) or pCuHis6-Atg13[571–700]-Ss(424) plasmid were cultured in nutrient-rich conditions to OD600 ~ 1 and then shifted to nitrogen-starvation medium for 1 h. The proteins were separated by SDS-PAGE and detected with anti-Vac8 antiserum or anti-His antibody (lower panels). (C) Atg13[571–700] (magenta and purple) modeled on the Vac8 crystal structure (PDB ID: 5XJG). Positively (blue) and negatively (red) charged residues in the Vac8 (gray) homodimer are highlighted. The positions of phospholipid-binding site I (640KFK) and site II (683KFHK) are denoted.
To confirm the interaction between Atg13[571–700] and Vac8 in yeast cells, we overexpressed a His6-tagged Atg13[571–700] along with Vac8 in yeast multiple-knockout (MKO) cells [21] and tested the presence of Vac8 after affinity isolation of Atg13[571–700] using Ni-NTA agarose. Empty vector with a His6-tagged nonspecific (scrambled) sequence of 14 amino acid residues was used as a negative control. The affinity-isolation experiment showed that His6-Atg13[571–700] interacted with Vac8 (Figure 3B). In the MKO cells, the levels of His6-Atg13[571–700] positively correlated with the level of Vac8 (Figure S6B), further confirming the interaction between Atg13[571–700] and Vac8 in vivo, and showing that the His6-Atg13[571–700] peptide level detected by western blot is a readout of the interaction in yeast cells. Taking advantage of this finding, we analyzed the peptide level of wild-type and mutated His6-Atg13[571–700]. In agreement with the ITC data (Figure 3A), we found that the cellular level of the His6-Atg13[571–700] peptide, and thus the interaction with Vac8, was not affected by sequence alterations, except for the D651R Q666R P667R mutation that rendered His6-Atg13[571–700]D651,Q666,P667R nearly undetectable (Figure S6C). The co-immunoprecipitation experiments yielded a similar result (Figure S6D). To test how 3 additional arginine residues in the Atg13[571–700] sequence affect the interaction with phospholipids, we subjected the mutant to a sedimentation assay with Folch liposomes in the absence of Vac8. As expected, the recombinant cationic mutant bound Folch liposomes similar to wild type at 50 and 150 mM NaCl and more efficiently than wild type at 300 and 500 mM NaCl (Figure 2B, S5).
Together, our results suggest that the liposome-binding region in Atg13 overlaps with the Vac8-binding region (Figure 3C), but both require distinct residues. This observation opens the possibility that the interaction of Atg13[571–700] with Vac8 and phospholipid membrane is mutually exclusive.
Atg13[571-700] exclusively interacts with either phospholipid membranes or Vac8
If Atg13[571–700] interacts exclusively with either phospholipid membranes or Vac8, the lipid-binding motifs should be masked upon Vac8 binding, and the liposome binding of Atg13[571–700] that we observed (Figures 1 and 2) should be reduced when Atg13 is bound to Vac8. To investigate this possibility, we carried out liposome sedimentation assays using recombinant purified Atg13[571–700] and Vac8[10–515] at 50 and 150 mM NaCl. Atg13 was incubated with either an equimolar amount of Vac8[10–515] or buffer prior to mixing with liposomes to allow for Atg13[571–700]-Vac8[10–515] complex formation. Vac8[10–515] did not pellet in the presence of liposomes at any salt concentration tested, indicating that this construct did not bind liposomes (Figure 4). At 50 mM NaCl, Vac8[10–515] reduced Atg13[571–700] liposome binding by 45% (Figure 4A,B). At 150 mM NaCl, Vac8[10–515] reduced the amount of Atg13[571–700] in the pellet essentially to background levels, demonstrating that there was no liposome binding by Atg13[571–700] in the presence of Vac8[10–515] (Figure 4C,D). These data demonstrate that Vac8[10–515] binding to Atg13[571–700] inhibits liposome binding by the latter, and suggest that the Vac8 and liposome binding sites on Atg13 are mutually exclusive despite utilizing distinct amino acids.
Figure 4.
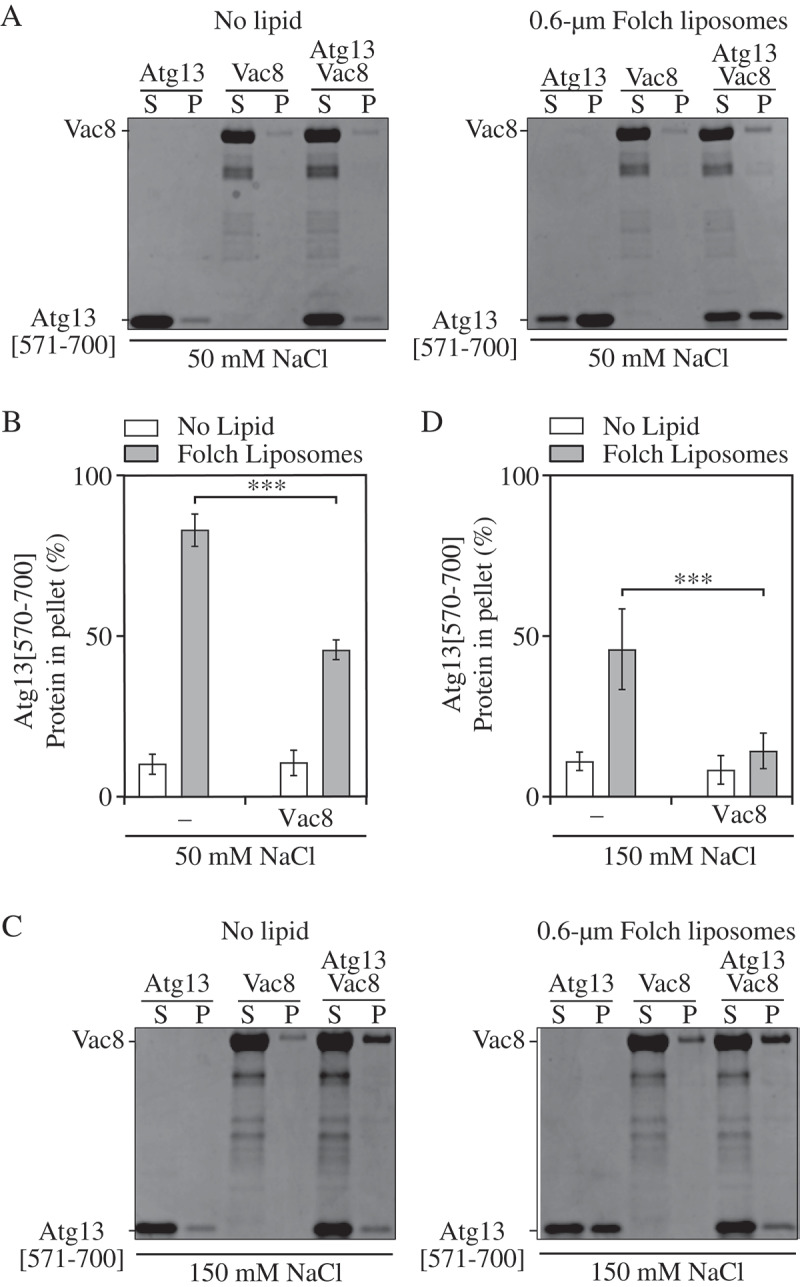
Vac8 inhibits Atg13[571–700] liposome binding. (A,B) Atg13[571–700], Vac8 [10–515] and a mixture of both proteins were incubated with Folch liposomes in buffer containing 50 mM NaCl and subjected to centrifugation. Pellet and supernatant fractions were collected and analyzed by western blot. The percent of Atg13[571–700] in the pellet was quantified using densitometry. (C,D) Same as A,B except performed in the presence of 150 mM NaCl. Error bars indicate the standard deviation of 4 independent experiments. Statistical analysis by ANOVA: *** P < 0.001.
The lipid- and Vac8-binding capability of Atg13 are required for efficient autophagy
To determine if nonselective autophagy activity is affected by a chromosomal mutation in the phospholipid- and Vac8-binding motifs of the full-length Atg13 protein, we performed the quantitative Pho8Δ60 assay, in which Pho8Δ60 is a cytosolic zymogen of the vacuolar Pho8 phosphatase that lacks the first 60 amino acids of its N terminus. Vacuolar localization and activation of this zymogen relies on nonselective autophagy [22]. Cells with wild-type (WT) Atg13 exhibited a substantial increase in Pho8Δ60 activity after 3 h of nitrogen starvation (Figure 5A). In comparison, the F641E I645E, K683E H685E K686E, and D651R Q666R P667R mutants showed an ~30% decrease in Pho8Δ60 activity; the F641G I645G mutant was even more defective, as it exhibited an ~50% decrease in autophagy activity under the same experimental conditions.
Figure 5.
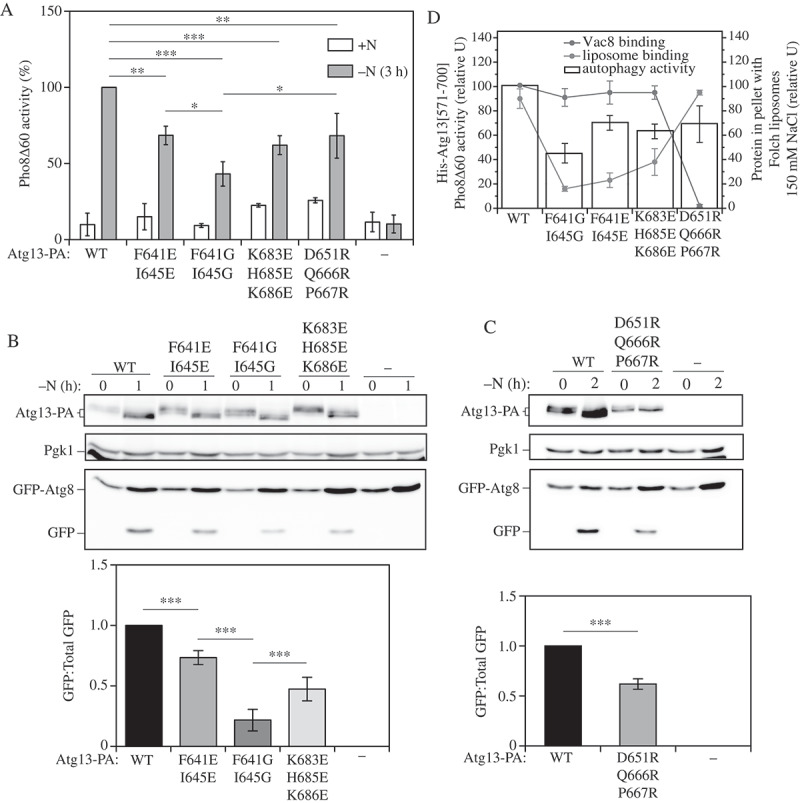
Atg13 IDR mutants reduce autophagy activity. (A) Autophagy activity was measured by the Pho8Δ60 assay in WT and atg13∆ strains, and in cells expressing the F641E I645E, F641G I645G, K683E H685E K686E, or D651R Q666R P667R mutants under nutrient-rich conditions (+N) and after 3 h of nitrogen starvation (-N 3 h). Error bars indicate the standard deviation of 3 independent experiments. Statistical analysis by Student’s t-test: * P < 0.05; ** P < 0.01; *** P < 0.001. (B,C) Autophagy was measured using the GFP-Atg8 processing assay in the same strains as in (A) under nutrient-rich conditions and after 1 or 2 h of nitrogen starvation. A representative image is shown. Error bars indicate the standard deviation of 3 independent experiments. Statistical analysis by ANOVA: *** P < 0.001. (D) Summary diagram combining the results on liposome binding (Figure 2B), Vac8 interaction in MKO yeast cells (Fig. S6C) and the Pho8Δ60 activity (Figure 5A) of the Atg13 mutants. For Pho8∆60 activity and the amount of His6-Atg13[571–700], where the latter was normalized to the loading control (Dpm1), the value for the wild type was set to 100; other values are presented relative to the WT.
To further test the nonselective autophagy activity of Atg13 mutants, we used the GFP-Atg8 processing assay that relies on a higher stability of free GFP relative to Atg8 in the vacuole after its fusion with the autophagosome [23,24]. In WT cells, a strong band corresponding to free GFP could be observed by western blot after 1 or 2 h of nitrogen starvation (Figure 5B). In contrast, the chromosomal mutations F641E I645E and K683E H685E K686E in full-length Atg13 rendered the yeast cells significantly defective in autophagy flux, as the level of free GFP for these mutants decreased by 30–50%. Combining 5 mutagenic replacements into a quintuple glutamate mutant did not lead to any further decrease in auto-phagy activity (Figure S7A). The GFP-Atg8 processing assay showed that the F641G I645G mutant was most defective, yielding an ~ 75% decrease in the level of free GFP (Figure 5B). This phenotype is in agreement with this mutant displaying the weakest binding to liposomes (Figures 2B and 5A–C). In comparison, Atg13D651,Q666,P667R followed slightly different kinetics. The latter mutant required 2 h of nitrogen starvation to exhibit a 40% defect in the GFP-Atg8 processing assay (Figure 5C). These data suggest that disruption of the interaction between Atg13 and lipid membrane has a more imminent effect on production of free GFP than interfering with the Vac8-Atg13 interaction, which in turn potentiates a more efficient binding of Atg13 to phospholipids (Figures 2, 3, and 5). Thus, both insufficient and excessive interaction of the Atg13 IDR with lipid membrane leads to a decreased auto-phagy flux, but manifestation of the latter requires a longer time to detect experimentally.
To test how the Atg13 mutants that have normal Vac8 interaction (F641G I645G, F641E I645E, and K683E H685E K686E) behave in the absence of their Vac8 binding partner, we expressed these mutants in a vac8Δ strain and measured precursor aminopeptidase I (prApe1) processing (Figure S7B,C). In this background, the cytoplasm-to-vacuole targeting (Cvt) pathway is blocked due to VAC8 deletion and prApe1 can only be delivered to the vacuole through autophagy. Without Vac8, Atg13F641,I645E was completely unable to transport prApe1 to the vacuole, whereas Atg13K683,H685,686E functioned similar to the wild-type protein, and Atg13F641,I645G was only partially defective (Figure S7B,C). This result reveals that there is an additional layer of regulation of the phospholipid-binding motifs in the absence of Vac8.
The summary diagram of the lipid- and Vac8-binding capability and autophagy activity of the Atg13 mutants (Figure 5D) shows that the Atg13 protein promotes efficient autophagy only when its interaction with both phospholipids and Vac8 is intact and thereby balanced.
The region 571-700 is necessary for efficient organization of Atg13 during starvation
Our finding that the Atg13 region 571–700 alternates binding between phospholipids and the vacuole membrane-associated protein Vac8 opens a possibility that efficient organization of Atg13 in proximity to the vacuole could be maintained by the 571–700 region. To probe such a role of this region in Atg13, we constructed Atg13-GFP yeast strains expressing either WT, F641E I645E, F641G I645G, K683E H685E K686E or D651R Q666R P667R mutants and compared their ability to form peri-vacuolar puncta after 1 h of nitrogen starvation. Whereas Atg13-GFP WT and the D651R Q666R P667R mutant showed a similar number of cells with the ability to form Atg13-GFP peri-vacuolar puncta, the F641E I645E, F641G I645G and K683E H685E K686E mutants showed an ~50% decrease in the number of cells with this ability (Figure 6A,B). This result correlates with the liposome binding ability of each mutant (Figures 2B, 5D), suggesting that lipid-binding capability of Atg13 is needed for efficient organization of the protein in the peri-vacuolar space during nitrogen starvation. Quantification of the number of Atg13-GFP puncta formed per cell revealed that the D651R Q666R P667R mutant was able to form 2 or at times even 3 Atg13-GFP puncta per cell, as compared to wild type and glycine or glutamate mutants, which formed 1 punctum per cell in most cases (Figure 6C). The D651R Q666R P667R mutant exhibited decreased binding to Vac8 (Figure 3 and S6) but retained, or even exceeded liposome-binding capability relative to wild type (Figure 2). Again, these data suggest that the Atg13-phospholipd interaction needs to be effective and in concert with the Atg13-Vac8 interaction to maintain an optimal balance in localization of Atg13 in proximity to the vacuole; too little or too much binding to either component is detrimental to autophagy (Figures 5 and 6).
Figure 6.
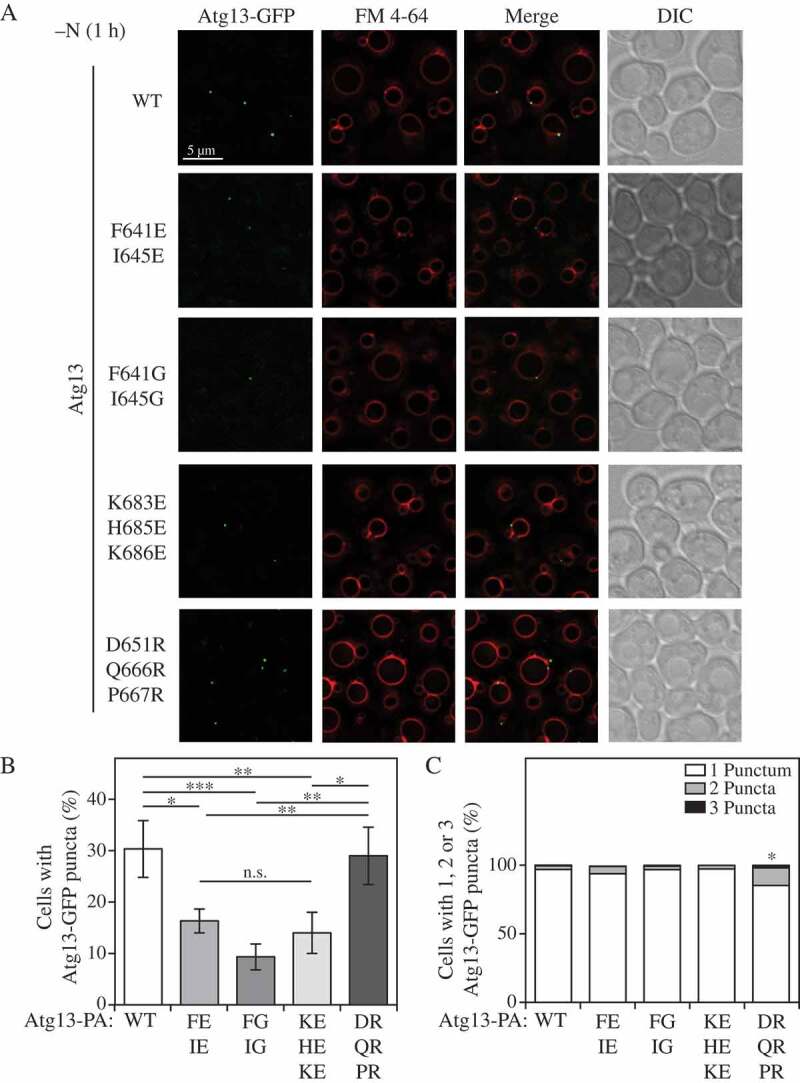
The Atg13 IDR mutants exhibit different abilities to form Atg13-GFP puncta. (A) Atg13-GFP puncta formation was monitored through fluorescence microscopy after 1 h of nitrogen starvation (-N 1 h). Representative image planes are shown. (B) Quantification of the number of cells with the ability to form Atg13-GFP puncta. The ratio of the total number of Atg13-GFP puncta was divided by the total number of cells for WT, F641E I645E, F641G I645G, K683E H685E K686E and D651R Q666R P667R forms of Atg13-GFP. Statistical analysis by ANOVA: * P < 0.05; ** P < 0.01; *** P < 0.001; n.s, no significant difference. (C) Quantification of the number of cells forming 1, 2, or 3 puncta per cell for WT, F641E I645E, F641G I645G, K683E H685E K686E and D651R Q666R P667R forms of Atg13-GFP. Statistical analysis by ANOVA: * P < 0.05.
Discussion
Numerous cellular processes rely on optimal spatiotemporal organization of biological molecules (proteins, DNA, RNA, or membranes). Because intricate assembly and efficient interaction of structurally diverse components must be free of steric hindrances, these processes are often mediated by a backbone or hub that exhibits a high architectural plasticity, which is an embedded feature of intrinsically disordered regions [25,26]. Multiple studies in recent years showed that the IDR from yeast Atg13 functions as such a hub. So far, the Atg13 IDR was shown to be essential for assembly of 2 different Atg17 dimers, the early autophagy targeting and tethering (EAT) domain of the Atg1 kinase, and the peripheral vacuolar protein Vac8 [8,12,13,17].
Here we show that the Atg13 IDR also incorporates phospholipid membranes into the protein network assembly. The interaction between lipid membrane and the Atg13 IDR is mediated by 2 Phe-containing motifs (640KFK, site I; and 683KFHK, site II) within the 571–700 region of Atg13. These motifs employ electrostatic forces, between positively charged residues and negatively charged phospholipids head groups, and a small hydrophobic insertion mediated by aromatic residues in the motifs (Figures 1 and 2). Mutagenic alteration in Atg13 that puts glutamic acid or glycine in these motifs results in substantially weakened liposome binding (Figure 2), despite normal binding to Vac8 [13] (Figure 3).
The linear motifs for phospholipid binding in the C terminal IDR of Atg13 overlap with the Vac8-binding domain, but Vac8 binding is mediated by a different set of residues than phospholipid binding (Figure 3). Overlapping binding regions are not unusual in IDRs. A well-known hub, TP53/p53, carries multiple overlapping motifs in its disordered C terminal sequence [27]. Another example is the disordered N terminal domain of KNL1 (kinetochore scaffold 1), a protein that binds microtubules at 2 sites, which overlap with the PPP1 (protein phosphatase 1) binding sites [28]. A purpose of these overlapping regions is to function as fast molecular switches, because they exhibit a mutually exclusive interaction.
Indeed, in analogy to TP53 and KNL1, we found that the phospholipid- and Vac8-binding of Atg13 are mutually exclusive (Figure 4), suggesting that the Atg13 C terminus reversibly switches between attachment to Vac8 and membrane. This is consistent with our finding that mutagenic replacement of a few Vac8-binding residues with arginine disrupts the Atg13-Vac8 interaction and makes the protein prone to bind lipids more efficiently. The glutamate or glycine and arginine mutants of Atg13 are all defective in autophagy flux (Figure 5), but for different underlying reasons (Figure 2, 3, 5, 6). The data presented in this work lead to a proposed model (Figure 7) for the hub-like function of the Atg13 IDR. According to this model, the Atg13 IDR binds the Vac8 groove, where cationic residues are essential for the Atg13-Vac8 interaction. This would mask the phospholipid-binding motifs and prevent them from attaching to a membrane, and thereby keeping Atg13 constitutively bound to Vac8 under growing conditions. At the same time, Atg13 binds constitutively with a weak affinity to the 17BR motif (424–436) of Atg17 and to the MIT domain of Atg1 via the MIM (460–521) [12]. Upon starvation, a regulatory mechanism, for example, a posttranslational modification such as dephosphorylation, could release the positively charged phospholipid-binding motifs (640KFK and 683KFHK) from Vac8, and thereby facilitate their attachment to a negatively charged membrane, in analogy to the membrane-binding mechanism known for the myristoylated alanine rich protein kinase C substrate (MARKCS) [29]. Along with an increased binding affinity of Atg13 to 17BR of Atg17 and MIT of Atg1, Atg13 triggers its operation as an organizing hub near the vacuole during starvation. This model would explain why efficient hub-like activity of the Atg13 IDR in the peri-vacuolar space during starvation is, at least partially, dependent on its membrane binding capability. Insufficient binding of Atg13 to lipid membrane, manifested here by glutamate and glycine mutants, fails to fulfill this activity, which is also reflected in a low amount of cells with the capability to form Atg13-GFP puncta (Figure 6). Conversely, excessive lipid binding due to a decreased Vac8 binding, manifested here by the arginine mutant, can trigger too much of the hub-like activity of Atg13 and the nucleation process, which would explain an occasional accumulation of more than 1 Atg13-GFP punctum per cell. Therefore, phospholipid and Vac8 binding by the Atg13 IDR must be in a carefully regulated balance, disruption of which leads to inefficient autophagy flux. Elucidation of a regulatory switching mechanism in more detail remains a task for future research. At this point, we can only exclude a possibility that the Atg13 IDR is released during starvation from Vac8 via a backfolding mechanism, where a basic stretch of residues in the IDR would bind to an acidic stretch of residues. This mechanism would sequester the basic phospholipid binding motifs (640KFK and 683KFHK), rendering them unavailable to membrane and, thereby, decrease rather than increase autophagy activity. In Atg13, the conserved acidic stretch of residues is both upstream and downstream of the fungi membrane-binding site I and II (Figure S4). We speculate that these acidic stretches in the Atg13 sequence might perhaps help in navigating the IDR away from the membrane.
Figure 7.

Model of proposed mechanism for hub-like activity of the Atg13 IDR. Under growing conditions, the 571–700 region of Atg13 from S. cerevisiae binds constitutively to the Vac8 groove where cationic residues are critical for interaction. This interaction masks lipid-binding motifs and prevents them from attaching to the membrane. A regulatory mechanism, possibly dephosphorylation, could facilitate a release of the Atg13 positively charged motifs (640KFK in site I, and 683KFHK in site II) from Vac8, and allow a reversible switch to facilitate binding to a negatively charged phospholipid membrane. Along with an increased binding affinity of Atg13 to the MIT domain of Atg1 and 17BR domain of Atg17, Atg13 organizes a protein-lipid network during starvation. The following crystal structures were used in the model: 4J2G, Horma domain of Atg13; 4HPQ, Atg17-Atg31-Atg29 dimer; 4P1N, MIT domain of Atg1; 5XJG, Vac8. Inset: A detailed model of the Atg13 IDR interacting with phospholipid membrane. Lysine residues (blue spheres) are electrostatically attracted to negatively charged head groups, and bulky phenylalanine residues (gray spheres) are inserted into the hydrophobic membrane core. This proposed mechanism is analogous to the known interaction between the phosphatidylserine-enriched plasma membrane and the effector domain (ED) of the MARCKS (myristoylated alanine rich protein kinase C substrate) protein [29], an intrinsically disordered polypeptide. The ED of MARCKS binds lipids reversibly in a random coil conformation via ionic forces of lysines, and hydrophobic forces of phenylalanines. Phosphorylation of the ED by protein kinase C inhibits this protein-membrane interaction.
Materials and methods
Overexpression, purification, and characterization of recombinant Atg13[571-700] and mutants
S. cerevisiae Atg13[571–700] was subcloned into pHis2 and the pET His6 tobacco etch virus ligation-independent cloning vector (1B) (Addgene, 29,653; deposited by Scott Gradia). Atg13[647–700], Atg13[571–682], and Atg13[571–640] were also subcloned into this same vector. All mutants were generated from Atg13[571–700] pHis2 using Q5 mutagenesis (NEB, E0554). Atg13 constructs were transformed into E. coli BL21 (DE3) RIL cells (Invitrogen/ThermoFisher Scientific, 230,245). Cells were grown in TB medium at 37°C to an OD600 of 1.2, protein expression was induced with 1 mM isopropyl-beta-D-thiogalactopyranoside, and cells were subsequently grown for 18 h at 18°C. Cells were harvested and pellets were stored at −80°C. Cell pellets were thawed and resuspended in 50 mM Tris, pH 7.4, 500 mM NaCl, 0.1% Triton X-100 (Sigma, T8787) containing an EDTA-free mini protease inhibitor tablet (Roche, 05056489001). Cells were lysed with 3 passes through a French press (Thermo Electron). Lysates were cleared by centrifugation and the cleared supernatant was added to TALON resin (Clontech, 635,504) and incubated at 4°C for 1 h. The resin was washed with 50 mM Tris, pH 7.4, 500 mM NaCl, and the protein was eluted using 50 mM Tris, pH 7.4, 500 mM NaCl, 200 mM imidazole. Elutions containing protein were pooled and further purified using a HiLoad 16/600 Superdex 75-pg column (GE Healthcare, 28–9893-33) equilibrated in 20 mM Tris, pH 7.4, 100 mM NaCl, 0.2 mM Tris(2-carboxyethyl)phosphine (TCEP; VWR, K831).
Liposome sedimentation assay
Folch fraction type I lipids isolated from bovine brain (Sigma-Aldrich, B1502) and synthetic liposomes made using L-α-phosphatidylcholine (Avanti, 840,051), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (Avanti, 840,034), L-α-phosphatidylethanolamine (Avanti, 840,022), L-α-phosphatidylinositol (Avanti, 840,042), 1,2-dioleoyl-sn-glycero-3-phospho-(1ʹ-myo-inositol-3ʹ-phosphate) (Avanti, 850,150), and cholesterol (Avanti, 700,000) were dried under a nitrogen stream for 30 min and then for 18 h in a vacuum oven. Dried Folch was resuspended in 20 mM Tris, pH 7.4, 200 mM NaCl, 0.2 mM TCEP to a final concentration of 0.5 mg/ml. Synthetic liposomes were resuspended in 20 mM Tris, pH 7.4, 50 mM NaCl, 0.2 mM TCEP to a final concentration of 1.9 mg/ml. Lipids were subjected to freeze-thaw by incubating at −80°C for 10 min and then thawing in a water bath. Liposomes were extruded using an Avanti Mini Extruder with the appropriate size membrane. Purified Atg13[571–700] (25 μl of 5 μM) was mixed with 25 μl of 0.5 mg/ml folch liposomes or 1.9 mg/ml synthetic liposomes. For Atg13[571–700] liposome binding in the presence of Vac8[10–515], 12.5 µl of 24 µM Atg13 was mixed with either 12.5 µl of 24 µM Vac8[10–515] or buffer and incubated for 30 min on ice to allow for complex formation. Folch liposomes (25 µl) were then added at a final concentration of 0.25 mg/ml liposomes and 6 µM protein. Liposome and protein mixture were incubated at 4°C for 1 h, followed by centrifugation at 45,000 rpm (91,287 x g) for 40 min at 4°C using a TLA45 rotor. Supernatants were removed, and an equal volume of buffer was added to resuspend the pellets. Samples were subjected to SDS-PAGE for analysis and bands were quantified using Image Lab v 5.1 (Bio-Rad). Densitometry was performed on both the pellet and supernatant fractions. The supernatant and pellet band intensity were added together to determine the total intensity of Atg13 in each sample. The percentage of Atg13 bound to liposomes was determined by taking the ratio of the pellet band intensity over the total intensity. Each experiment was performed in triplicate. These results were averaged and plotted with error bars representing the standard deviations of these 3 experiments.
Yeast in vivo assays
Saccharomyces cerevisiae strains WLY176 and JMY347 were used to generate atg13∆, full-length ATG13-PA, and ATG13-GFP strains as previously described [30,31]. Genomic point mutations ATG13-PA F641G I645G, F641E I645E, K683E H685E K686E and D651R Q666R P667R were generated as previously described [][32]. Yeasts cultures were grown in YPD medium (1% [w:v] yeast extract (ForMedium, YEM04), 2% [w:v] peptone ((ForMedium, PEP04), 2% [w:v] glucose) to mid-log phase and then samples were collected. Strains grown in YPD were shifted to minus nitrogen medium (0.17% yeast nitrogen base without ammonium sulfate or amino acids [ForMedium, CYN0501], 2% [w:v] glucose) for the indicated time points and then collected. Pho8Δ60 and western blot analyses were performed as previously described [22,24]. Western blot densitometry quantification was performed using ImageJ software.
Fluorescence microscopy
Yeast cells were grown to OD600 ~ 0.5 in YPD medium and shifted to minus nitrogen medium for autophagy induction. For vacuolar staining, cells were grown in YPD with 30 μM FM 4–64 (Molecular Probes/Fisher Scientific, T3166) for 30 min and then washed to remove excess dye. Images were collected on a DeltaVision Elite deconvolution microscope (GE Healthcare/Applied Precision) with a 100x objective and a CCD camera (CoolSnap HQ; Photometrics). For Atg13-GFP puncta quantification, stacks of 20 image planes were collected with a spacing of 0.2 mm to cover the entire yeast cell. Analysis was performed on an average projection of the imaging planes on ImageJ software.
His-tag affinity isolation experiments
For His-tag affinity isolation, the pCuHis6-Ss(424) or pCuHis6-Atg13[571–700]-Ss(424) plasmids with the CUP1 promoter were constructed by fast cloning. The non-specific scrambled sequence (Ss) of 14 amino acids (DIKLIDTVDLESCN) was used in both plasmids to produce an empty vector and the fused Atg13 peptide with the same non-specific properties. Cells (60 OD600 units) starved for 1 h in SD-N medium were lysed in 0.7 ml of lysis buffer (1x PBS [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM K2HPO4, pH 7.4], 0.2 M sorbitol, 1 mM MgCl, 0.5% Triton X-100, 1 mM PMSF, Complete EDTA-free protease inhibitor) with glass beads in 6 cycles of vortexing for 45 s with a 2-min interval of incubation on ice between each vortexing cycle. Cell debris were removed by centrifugation at 4000 x g for 5 min. An aliquot of the supernatant fraction (20%) was TCA precipitated as the Input. The remaining supernatant was incubated with 100 µl Ni-NTA agarose (Qiagen, 30,210) for 3 h at 4°C. After 3 washes with ice-cold lysis buffer, the proteins were eluted by incubating the agarose for 15 min at 55°C with SDS-PAGE buffer containing 150 mM imidazole. The eluted proteins were analyzed by western blot with anti-polyhistidine monoclonal antibodies (Sigma, H1029) and anti-Vac8 polyclonal antiserum [33].
Isothermal titration calorimetry
ITC was performed using a VP-ITC (Microcal). All proteins were prepared in 25 mM Tris, pH 7.5, 150 mM NaCl, 5 mM beta-mercaptoethanol for ITC experiments. Recombinant purified Atg13[571–700] WT and mutants at approximately 50 µM were titrated into Vac8[10–515] at 5.1 µM at 24°C. Data were analyzed using Origin (Microcal). Each ITC experiment was performed a minimum of two times and standard deviations were calculated for these repeats.
Statistics and reproducibility
Analysis and image processing of microscopy data was carried out using softWoRx software (GE Healthcare). Sample sizing for cellular imaging was chosen to be the minimum number of independent experiments required for statistically significant results. Western blot images were quantified using ImageJ software. Statistical analyses were performed using GraphPad Prism 6. Statistical significance was determined in all cases from at least 3 independent experiments using either Student’s t-test or ANOVA. Differences with a P value <0.05 or lower were considered significant. *p < 0.05, **p < 0.01, ***p < 0.001. Number of independent experiments (n), statistical test utilized, dispersion of measurements and significance is described in the figure legends.
Supplementary Material
Funding Statement
This work was supported by the National Institute of General Medical Sciences [GM053396 and GM131919 to DJK]; National Institute of General Medical Sciences [GM128663 and GM113132 to MJR]; Protein Folding Diseases FastForward Initiative, University of Michigan [DJK].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
supplementary data for this article can be accessed here.
References
- [1].Wen X, Klionsky D.. An overview of macroautophagy in yeast [Review]. J Mol Biol. 2016. May 8;428(9):1681–1699. PubMed PMID: WOS:000376695000003; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rao Y, Perna MG, Hofmann B, et al. The Atg1-kinase complex tethers Atg9-vesicles to initiate autophagy. Nat Commun. 2016. January 12;7:10338. PubMed PMID: 26753620; PubMed Central PMCID: PMC4729957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sanchez-Wandelmer J, Kriegenburg F, Rohringer S, et al. Atg4 proteolytic activity can be inhibited by Atg1 phosphorylation. Nat Commun. 2017. August 18;8(1):295. PubMed PMID: 28821724; PubMed Central PMCID: PMC5562703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell. 2012. December 21;151(7):1501–1512. PubMed PMID: 23219485; PubMed Central PMCID: PMC3806636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chew LH, Lu S, Liu X, et al. Molecular interactions of the Saccharomyces cerevisiae Atg1 complex provide insights into assembly and regulatory mechanisms. Autophagy. 2015;11(6):891–905. PubMed PMID: 25998554; PubMed Central PMCID: PMC4502708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Popelka H, Klionsky DJ. The molecular mechanism of Atg13 function in autophagy induction: what is hidden behind the data? Autophagy. 2017. March 4;13(3):449–451. PubMed PMID: 28118060; PubMed Central PMCID: PMC5361603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stjepanovic G, Davies CW, Stanley RE, et al. Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc Natl Acad Sci U S A. 2014. September 2;111(35):12793–12798. PubMed PMID: 25139988; PubMed Central PMCID: PMC4156731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yamamoto H, Fujioka Y, Suzuki SW, et al. The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev Cell. 2016. July 11;38(1):86–99. PubMed PMID: 27404361. [DOI] [PubMed] [Google Scholar]
- [9].Kamada Y, Funakoshi T, Shintani T, et al. Tor-mediated induction of autophagy via an Apg1 protein kinase complex [Article]. J Cell Biol. 2000. September 18;150(6):1507–1513. PubMed PMID: WOS:000089454500027; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Suzuki S, Yamamoto H, Oikawa Y, et al. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation [Article]. Proc Natl Acad Sci U S A. 2015. March 17;112(11):3350–3355. PubMed PMID: WOS:000351060000064; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jao C, Ragusa M, Stanley R, et al. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy [Article]. Proc Natl Acad Sci U S A. 2013. April 2;110(14):5486–5491. PubMed PMID: WOS:000318037800058; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fujioka Y, Suzuki SW, Yamamoto H, et al. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat Struct Mol Biol. 2014. June;21(6):513–521. PubMed PMID: 24793651. [DOI] [PubMed] [Google Scholar]
- [13].Scott SV, Nice DC 3rd, Nau JJ, et al. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem. 2000. August 18;275(33):25840–25849. PubMed PMID: 10837477. [DOI] [PubMed] [Google Scholar]
- [14].Karanasios E, Stapleton E, Manifava M, et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci. 2013. November 15;126(Pt 22):5224–5238. PubMed PMID: 24013547. [DOI] [PubMed] [Google Scholar]
- [15].Busch DJ, Houser JR, Hayden CC, et al. Intrinsically disordered proteins drive membrane curvature. Nat Commun. 2015. July 24;6:7875. PubMed PMID: 26204806; PubMed Central PMCID: PMC4515776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bartels T, Ahlstrom LS, Leftin A, et al. The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys J. 2010. October 6;99(7):2116–2124. PubMed PMID: 20923645; PubMed Central PMCID: PMC3042581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jeong H, Park J, Kim HI, et al. Mechanistic insight into the nucleus-vacuole junction based on the Vac8p-Nvj1p crystal structure. Proc Natl Acad Sci U S A. 2017. June 6;114(23):E4539–E4548. PubMed PMID: 28533415; PubMed Central PMCID: PMC5468681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kvam E, Goldfarb DS. Nucleus-vacuole junctions and piecemeal microautophagy of the nucleus in S. cerevisiae. Autophagy. 2007. March–Apr;3(2):85–92. PubMed PMID: 17204844. [DOI] [PubMed] [Google Scholar]
- [19].Roberts P, Moshitch-Moshkovitz S, Kvam E, et al. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 2003. January;14(1):129–141. PubMed PMID: 12529432; PubMed Central PMCID: PMC140233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krick R, Muehe Y, Prick T, et al. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell. 2008;19:4492–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cao Y, Cheong H, Song H, et al. In vivo reconstitution of autophagy in Saccharomyces cerevisiae. J Cell Biol. 2008. August 25;182(4):703–713. PubMed PMID: 18725539; PubMed Central PMCID: PMC2518709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Noda T, Klionsky D. THE QUANTITATIVE PHO8 Delta 60 ASSAY OF NONSPECIFIC AUTOPHAGY [Review|Book Chapter]. Autophagy. 2008;451:33–42. PubMed PMID: WOS:000262255500003; English. [DOI] [PubMed] [Google Scholar]
- [23].Klionsky D, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) [Review]. Autophagy. 2016;12(1):1–222. PubMed PMID: WOS:000373595400001; English. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shintani T, Klionsky DJ. Cargo proteins facilitate the formation of transport vesicles in the cytoplasm to vacuole targeting pathway. J Biol Chem. 2004. July 16;279(29):29889–29894. PubMed PMID: 15138258; PubMed Central PMCID: PMC1712665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006. April 3;580(8):2041–2045. PubMed PMID: 16542654. [DOI] [PubMed] [Google Scholar]
- [26].Habchi J, Tompa P, Longhi S, et al. Introducing protein intrinsic disorder. Chem Rev. 2014. July 9;114(13):6561–6588. PubMed PMID: 24739139. [DOI] [PubMed] [Google Scholar]
- [27].Gibson TJ. Cell regulation: determined to signal discrete cooperation. Trends Biochem Sci. 2009. October;34(10):471–482. . PubMed PMID: 19744855. [DOI] [PubMed] [Google Scholar]
- [28].Bajaj R, Bollen M, Peti W, et al. KNL1 Binding to PP1 and Microtubules Is Mutually Exclusive. Structure. 2018. October 2;26(10):1327–1336 e4. PubMed PMID: 30100357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kastelowitz N, Tamura R, Onasoga A, et al. Peptides derived from MARCKS block coagulation complex assembly on phosphatidylserine. Sci Rep. 2017. June 27;7(1):4275. PubMed PMID: 28655899; PubMed Central PMCID: PMC5487340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gueldener U, Heinisch J, Koehler GJ, et al. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002. March 15;30(6):e23. PubMed PMID: 11884642; PubMed Central PMCID: PMC101367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Toulmay A, Schneiter R. A two-step method for the introduction of single or multiple defined point mutations into the genome of Saccharomyces cerevisiae. Yeast. 2006. August;23(11):825–831. . PubMed PMID: 16921548. [DOI] [PubMed] [Google Scholar]
- [32].Wang Y-X, Catlett NL, Weisman LS. Vac8p, a vacuolar protein with armadillo repeats, functions in both vacuole inheritance and protein targeting from the cytoplasm to vacuole. J Cell Biol. 1998;140:1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Longtine MS, McKenzie A, Demarini DJ, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in saccharomyces cerevisiae. Yeast. 1998. July;14(10):953–961. PubMed PMID: 9717241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.