

ABSTRACT
Alternative splicing (AS) analysis across the entire spectrum of human prostate cancer evolution reveals the unexpected findings that intron retention is a hallmark of stemness and tumor aggressiveness, and androgen receptor controls a splicing program distinct from its transcriptional regulation. Importantly, twisted activity of the spliceosome causing abnormal AS landscape represents a therapeutic vulnerability in aggressive prostate cancer.
KEYWORDS: Alternative splicing, prostate cancer, CRPC, spliceosome, E7107, cancer treatment
Prostate cancer (PCa) is a heterogeneous malignancy and still causes a significant mortality worldwide.1,2 Androgen deprivation therapy (ADT) is the main therapeutic regimen for advanced PCa patients. However, most treated patients invariably develop the castration-resistant PCa (CRPC), currently a lethal form of the disease. Within the CRPC category, although the majority of them histologically present as adenocarcinomas (Ad) (i.e., CRPC-Ad), but a significant fraction (up to 25%) of them evolve to an aggressive, androgen receptor negative (AR−) indifferent CRPC with neuroendocrine (NE) features called CRPC-NE.3 Generally, CRPC is less dependent on androgen/AR signaling and relatively stem-like. Mechanisms underlying CRPC development and maintenance remain poorly understood.
Splicing dysregulation is one of the molecular hallmarks of cancer.1 Recent pan-cancer studies have revealed that mis-splicing is prevalent in human cancers and many specific alternative splicing (AS) events (Figure 1a) contribute to tumor development in a context-dependent manner.4,5 In particular, studies on AR and AR variants have implicated splicing dysregulation in PCa resistance to ADT.1 Notably, although the global AS landscape of PCa has been recently reported by using The Cancer Genome Atlas (TCGA) data,4 these studies mainly focused on primary PCa (pri-PCa; versus normal tissues) and generally overlooked CRPC. The potential functions and clinical relevance of global splicing abnormality in PCa remain unclear. In order to understand the molecular basis underlying splicing misregulation and thus, to develop potential therapeutic strategies against ‘lethal’ CRPC, we characterized the first comprehensive AS landscape in the spectrum of human PCa evolution using RNA-sequencing datasets containing collectively >570 clinical PCa samples.6
Figure 1.
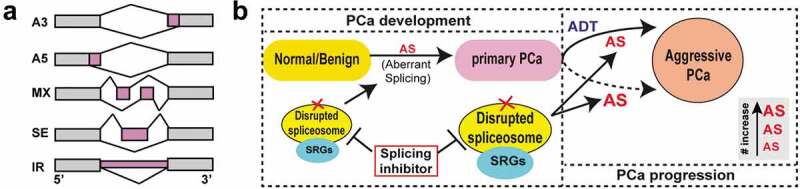
The spliceosome represents a therapeutic vulnerability in aggressive prostate cancer (PCa). (a) Five main types of alternative splicing (AS) patterns analyzed in our study. A3, alternative 3ʹ splice sites; A5, alternative 5ʹ splice sites; MX, mutually exclusive exons; SE, exon skipping; IR, intron retention. (b) The spliceosome machinery is disrupted due to mis-expression of splicing-regulatory genes (SRGs) in PCa evolution and contributes to PCa progression via modulating aberrant splicing events. The degree of global AS abnormities aggravates along with disease progression. In addition to treatment responsive emergence (solid arrow) of aggressive PCa (i.e., castration-resistant PCa (CRPC)), some primary PCa with small-cell PCa (SCPC) and/or neuroendocrine PCa (NEPC) features intrinsically resist androgen deprivation therapy (ADT) (dashed arrow), and will progress to advanced stages. Significantly, the tumor AS landscape differs before and after ADT, suggesting a treatment-induced reshaping of global AS pattern that might have contributed to therapy resistance. Collectively, targeting aberrant splicing via spliceosome modulators (e.g., E7107) offers a new way to combat CRPC.
By using two AS mapping algorithms, we revealed that the severity of global splicing dysregulation correlates with disease progression (Figure 1b).6 Biologically, the mis-splicing impacts genes enriched in cancer-related pathways in a tumor-stage dependent manner. For instance, gene ontology (GO) terms linked to ‘muscle and ion transport’, ‘lipid metabolism’, and ‘cell polarity’ were pri-PCa specific whereas GO terms ‘DNA damage’, ‘immunity’, and ‘nuclear pore’ were enriched in CRPC, consistent with recent reports.3,7 Interestingly, and as expected, terms ‘stem cell (SC) and development’ and ‘neuron and cell projection’ were greatly enriched in CRPC-NE, in line with its stem-like and neural-like properties. Functionally, aberrant splicing regulates PCa biology, at least partially, by switching isoform expression of key cancer-related genes. For example, multiple delta like non-canonical Notch ligand 1 (DLK1) splice variants (ENST00000341267.8, ENST00000331224.10, ENST00000556051.1) were found upregulated in CRPC-NE (vs. CRPC-Ad), and these isoforms expressed highly in DLK1+ cells, an SC population in human prostate,8 suggesting a functional link of DLK isoforms with stemness. Experimentally, knocking down clinically relevant isoforms of synaptotagmin 7 (SYT7) in two PCa lines inhibited cell proliferation and stemness,6 suggesting a causal role of mis-splicing in PCa pathogenesis. Interestingly, we found that the majority of AS events minimally changed the bulk gene expression, indicative of splicing dysregulation as an independent driver (vs. gene expression regulation) in oncogenesis.
RNA splicing can be dysregulated in cancer in many ways. In hematological cancer, recurrent point mutations in core spliceosome genes (e.g., splicing factor 3b subunit 1 (SF3B1), U2 small nuclear RNA auxiliary factor 1 (U2AF1)) have recognized as drivers for mis-splicing.5 Our genomic interrogation of 274 splicing-regulatory genes (SRGs) revealed copy number variants (CNV) as the main driver of AS alterations in PCa, which alter the expression of affected SRGs and illustrate cancer type-specific differences in mechanisms of splicing dysregulation. Regardless of mutational status, ~68% of SRGs were mis-expressed in various PCa stages. Many top altered SRGs are often co-mutated with either oncogenes (e.g., MYC proto-oncogene (MYC)) or tumor suppressors (e.g., RB transcriptional corepressor 1 (RB1)), warranting further dissection of their roles in MYC- or loss-of-RB1-driven tumorigenesis. Alternatively, mutations in splice sites may also cause AS abnormality.9 However, such mutations constitute the minority (~0.6%) of all somatic mutations in PCa,10 indicating that SRG dysregulation is the main mechanism underpinning mis-splicing. Importantly, we identified many unreported SRGs that can be linked, individually or in combination, to clinical features of advanced PCa, indicating a biomarker value.
One of our most significant findings is the link between the elevated intron retention (IR) level and PCa aggressiveness and stemness, which is evidenced by an increase in number of IR events detected in the spectrum of PCa evolution, in normal SC populations in various tissues, and in prostate cancer SCs.6 Consistently, IR in PCa impacts genes involved in stemness and cancer-promoting functions. As the least studied AS type, IR usually triggers nonsense-mediated RNA decay (NMD) to down-regulate gene expression. However, our data revealed, oppositely, that IR generally enhances gene expression and thus likely functions in PCa biology. In support, comparison of IR-affected genes in CRPC with that in human embryonic SC (hESC) and CD4+ T cells demonstrated that nearly 33% and 50% of the CRPC-IR genes also displayed IR in hESC and stem-like resting CD4+ T cells, respectively, directly suggesting a functional link of IR with stemness. Importantly, many IR-bearing transcripts exist in PCa cell lines. AR plays a central role in prostate tumorigenesis. We observed a generally negative association of AR activity with IR level in clinical datasets, but surprisingly, AR does not specifically regulate IR in androgen-sensitive PCa cells.
Of clinical relevance, aggressive PCa displays a preferential dependency on aberrant spliceosome activity and, E7107, the spliceosome modulator, effectively abolishes the growth of both Myc-driven autochthonous murine PCa and preclinical CRPC models in vivo.6 These findings offer a new therapeutic strategy for treating CRPC by targeting splicing misregulation (Figure 1b). Interestingly, our mechanistic study pointed out a potential action of E7107 to promote differentiation and reprogram PCa cells from an (aggressive) androgen-insensitive state to an (indolent) androgen-sensitive state. Animal studies revealed certain toxicities of E7107, highlighting the need to define intricate treatment window and doses for E7107. As we look forward to the future, we envision a potential treatment regimen in which CRPC is the first subject to a short-term splicing inhibition (to avoid toxicity and also to reprogram aggressive PCa cells) followed by anti-androgens treatment such as Enzalutamide to illuminate AR+ cells. Further characterization of the origins and consequences of aberrant AS in PCa will help enhance our understanding of PCa pathogenesis and accelerate novel drug development.
Funding Statement
This work was supported by the National Natural Science Foundation of China [81972418] and Wuhan Municipal Science and Technology Bureau (CN) [2019020701011490].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Paschalis A, Sharp A, Welti JC, Neeb A, Raj GV, Luo J, Plymate SR, de Bono JS.. Alternative splicing in prostate cancer. Nat Rev Clin Oncol. 2018;15(11):1–3. doi: 10.1038/s41571-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 2.Zhang D, Jeter C, Gong S, Tracz A, Lu Y, Shen J, Tang DG.. Histone 2B-GFP Label-Retaining Prostate Luminal Cells Possess Progenitor Cell Properties and Are Intrinsically Resistant to Castration. Stem Cell Rep. 2018;10(1):228–242. doi: 10.1016/j.stemcr.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S, Chen X, Liu X, Chao HP, Whitney P, et al. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun. 2016;7:10798. doi: 10.1038/ncomms10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research N , et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell. 2018;34(2):211–224e216. doi: 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SC, Abdel-Wahab O.. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976–986. doi: 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D, Hu Q, Liu X, Ji Y, Chao HP, Liu Y, Tracz A, Kirk J, Buonamici S, Zhu P, et al. Intron retention is a hallmark and spliceosome represents a therapeutic vulnerability in aggressive prostate cancer. Nat Commun. 2020;11(1):2089. doi: 10.1038/s41467-020-15815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Bravo V, Pippa R, Song WM, Carceles-Cordon M, Dominguez-Andres A, Fujiwara N, Woo J, Koh AP, Ertel A, Lokareddy RK, et al. Nuclear Pores Promote Lethal Prostate Cancer by Increasing POM121-Driven E2F1, MYC, and AR Nuclear Import. Cell. 2018;174(5):1200–1215e1220. doi: 10.1016/j.cell.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moad M, Hannezo E, Buczacki SJ, Wilson L, El-Sherif A, Sims D, Pickard R, Wright NA, Williamson SC, Turnbull DM, et al. Multipotent Basal Stem Cells, Maintained in Localized Proximal Niches, Support Directed Long-Ranging Epithelial Flows in Human Prostates. Cell Rep. 2017;20(7):1609–1622. doi: 10.1016/j.celrep.2017.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, Zhao Y, Climente-Gonzalez H, Chai S, Wang F, et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018;23(1):270–281 e273. doi: 10.1016/j.celrep.2018.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, Coleman I, Ng SB, Salipante SJ, Rieder MJ, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108(41):17087–17092. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]