

Abstract
PURPOSE
Hereditary cancer genetic testing can inform personalized medical management for individuals at increased cancer risk. However, many variants in cancer predisposition genes are individually rare, and traditional tools may be insufficient to evaluate pathogenicity. This analysis presents data on variant classification and reclassification over a 20-year period.
PATIENTS AND METHODS
This is a retrospective analysis of > 1.9 million individuals who received hereditary cancer genetic testing from a single clinical laboratory (March 1997 to December 2017). Variant classification included review of evidence from traditional tools (eg, population frequency databases, literature) and laboratory-developed tools (eg, novel statistical methods, in-house RNA analysis) by a multidisciplinary expert committee. Variants may have been reclassified more than once and with more than one line of evidence.
RESULTS
In this time period, 62,842 unique variants were observed across 25 cancer predisposition genes, and 2,976 variants were reclassified. Overall, 82.1% of reclassification events were downgrades (eg, variant of uncertain significance [VUS] to benign), and 17.9% were upgrades (eg, VUS to pathogenic). Among reclassified variants, 82.8% were initially classified as VUS, and 47.5% were identified in ≤ 20 individuals (allele frequency ≤ 0.001%). Laboratory-developed tools were used in 72.3% of variant reclassification events, which affected > 600,000 individuals. More than 1.3 million patients were identified as carrying a variant that was reclassified within this 20-year time period.
CONCLUSION
The variant classification program used by the laboratory evaluated here enabled the reclassification of variants that were individually rare. Laboratory-developed tools were a key component of this program and were used in the majority of reclassifications. This demonstrates the importance of using robust and novel tools to reclassify rare variants to appropriately inform personalized medical management.
INTRODUCTION
As our knowledge of hereditary cancer risk has evolved, multigene panel testing of cancer predisposition genes has become an important component of patient care. The National Comprehensive Cancer Network provides gene-specific recommendations for changes in medical management on the basis of the identification of pathogenic variants.1 To this end, accurate and definitive variant classification is a critical component of clinical genetic testing. The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) provide guidelines for clinical variant classification2; however, there is often insufficient evidence to support a definitive classification of pathogenic or benign when a variant is first detected. In addition, ACMG/AMP guidelines are intentionally broad to support variant classification for any genetic condition. As a result, clinical testing laboratories may vary in their interpretation of ACMG/AMP guidelines for hereditary cancer genetic testing and, ultimately, in their variant classifications.3-6
CONTEXT
Key Objective
With the increasing use of hereditary cancer genetic testing and introduction of multigene panels, robust and accurate methods for variant re/classification of cancer predisposition genes are of utmost importance. We reviewed a variant re/classification program developed by a single laboratory over 20 years, with data from nearly 2 million patients.
Knowledge Generated
A total of 2,976 unique variants were reclassified during this time period, the majority of which were initially classified as a variant of uncertain significance and were individually rare (allele frequency < 0.01%). Laboratory-developed methods were the most common lines of evidence used in variant reclassification, with Pheno analysis (a clinical history weighting algorithm) being used in approximately 40% of cases.
Relevance
Many tools traditionally used for variant re/classification are not informative for rare variants; however, rare variants collectively affect millions of patients. As such, it is critical that classification programs evolve to address knowledge gaps and provide informative test results for patients at increased cancer risk.
With the increased use of multigene panels, the number of variants of uncertain significance (VUSs) detected has also increased.7,8 In addition to a larger number of genes, many cancer predisposition genes have been more recently incorporated into clinical testing, and there is less evidence available to support variant classification. This growing number of VUSs presents a challenge for classification/reclassification because most VUSs are individually rare. Although ACMG/AMP guidelines for variant classification include a variety of methods, many of these tools are of limited value for rare variants. Some variants can be classified after a single observation on the basis of ACMG/AMP guidelines when there is a clear and severe biologic implication; however, most rare variants suffer from a lack of classification tools and methodologies. For example, there is often a lack of data in the literature, limited data in population databases, and the inability to apply traditional statistical methods.
Although VUSs are individually rare, a large proportion of tested patients are identified as carrying a VUS. Therefore, to ensure appropriate patient care, it is crucial for clinical testing laboratories to improve VUS reclassification. This includes the development of new tools to address the reclassification of rare variants as well as efficient systems that allow for real-time evaluation of newly released data. Here, we describe a reclassification program for hereditary cancer genetic testing developed over a 20-year period by a single laboratory.
PATIENTS AND METHODS
Clinical Cohort and Genetic Testing
A retrospective analysis was performed for > 1.9 million individuals who received genetic testing (Myriad Genetics Laboratories, Salt Lake City, UT) from March 1997 through December 2017. In 1997, the test offering for hereditary breast and ovarian cancer (HBOC) syndrome became available (BRCA1 and BRCA2). This was followed by testing for Lynch syndrome (MLH1, MSH2, MSH6, and PMS2) and melanoma-pancreatic cancer syndrome (CDKN2A) in 2002 and for familial adenomatous polyposis (FAP)/attenuated FAP/MUTYH-associated polyposis (APC and MUTYH) in 2003. In 2013, a hereditary cancer multigene panel was introduced, which was analytically validated and described in detail previously.9 The panel included 25 genes: APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A (p16INK4a and p14ARF), CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53. In 2017, POLD1, POLE, and GREM1 were added to the panel. Individuals who had single-site or founder mutation testing were excluded from this analysis.10-12 Testing was performed on DNA extracted from blood or saliva samples, as described previously.9 Clinical information was obtained from provider-completed test requisition forms, and patient data were de-identified.
Reclassification Program
The reclassification program used by this testing laboratory included a multidisciplinary committee of laboratory directors, genetic counselors, PhD-level scientists, and supporting variant specialists. The committee met daily to consider the classification of new variants and once a week for variants that required an in-depth discussion. Variant classification and reclassification followed a stringent, well-documented protocol as previously described.13 On the basis of ACMG/AMP guidelines, variants were classified into five categories of increasing clinical severity: Benign (B), Likely Benign (LB), VUS, Likely Pathogenic (LP), and Pathogenic (P).2,14,15 Automated processes proactively monitored and gathered all available evidence to expedite reclassification. Variant re-evaluation and reclassification were based on new evidence that coincided with computerized review queues founded on the variant type and new information received. Evidence was derived from continuously evaluated external and internal sources. External sources included published literature and population database frequencies, while internal sources consisted of laboratory-developed statistical methods. Multiple classification tools could have been used to evaluate the pathogenicity of a variant at the time of original classification and/or reclassification (Fig 1). If a variant was reclassified, an amended report was sent to health care providers for all patients who were carriers of the variant. Genetic counselors at the testing laboratory contacted health care providers to inform them when a variant was upgraded from VUS to LP/P.
FIG 1.
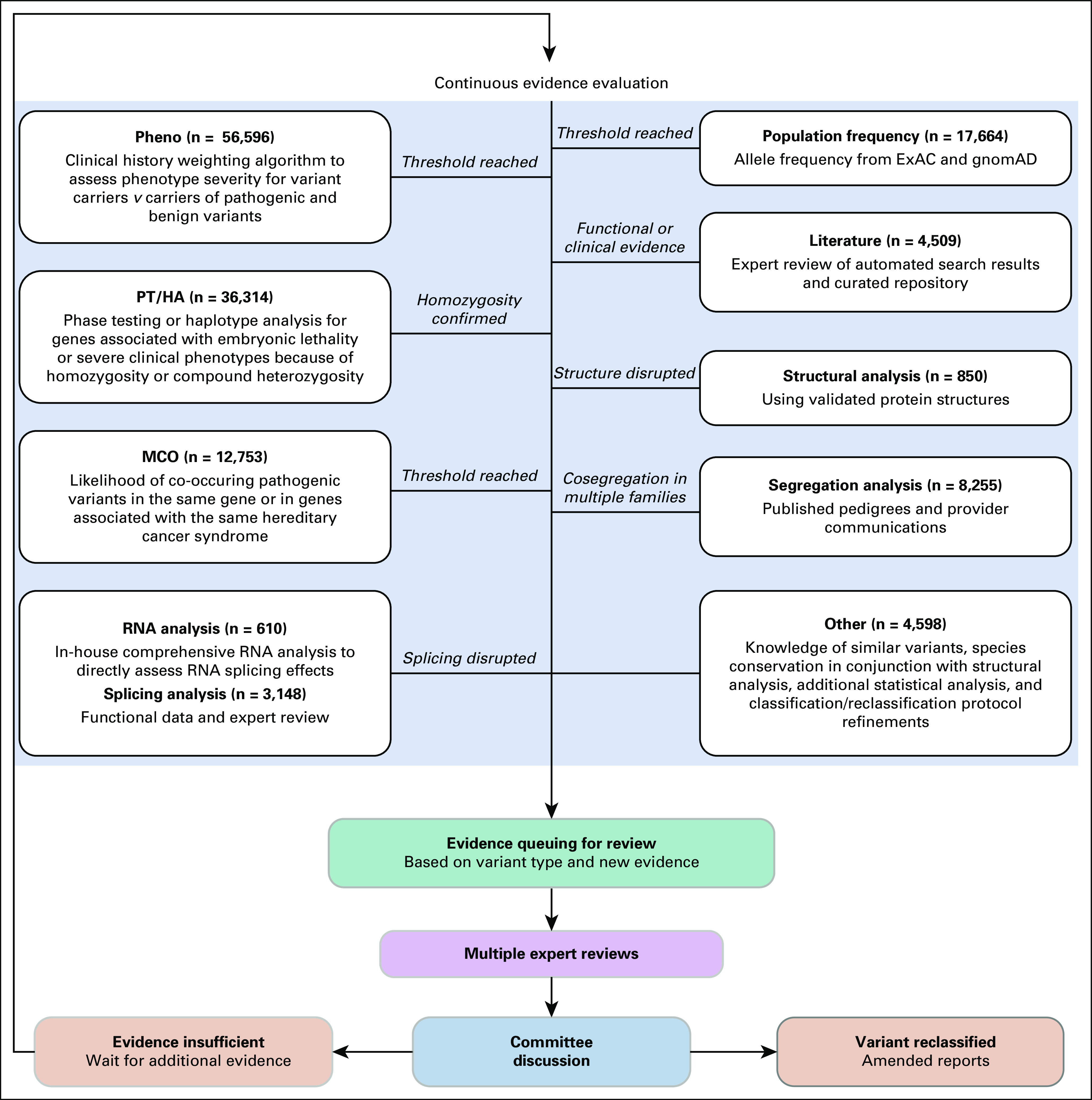
Reclassification process used by the testing laboratory. The number of patients identified as carrying a variant that was later reclassified using evidence from a given reclassification tool is provided for each reclassification tool. Variants may be classified using multiple lines of evidence. MCO, mutation co-occurrence; PT/HA, phase testing/homozygosity analysis.
Pheno analysis.
Pheno analysis is a validated cancer history weighting algorithm that measured the severity of personal and family cancer history associated with a specific variant and then assigned a variant-associated statistical score.16,17 Pheno scores were compared with pathogenic and benign control variants to determine potential pathogenicity for the variant of interest (positive predictive value > 99.5%, negative predictive value > 99.5% for relevant genes).
Phase testing/homozygosity analysis.
Phase testing/homozygosity analysis (PT/HA) was only applied to genes associated with autosomal dominant cancer risks that may also be associated with embryonic lethality or severe clinical phenotypes in individuals carrying pathogenic variants in the affected gene on different parental chromosomes as a result of either homozygosity or compound heterozygosity. Common single nucleotide polymorphisms (SNPs) observed in the patient population were used to develop gene-specific haplotypes, which were automatically assigned to determine the variants’ phase (ie, in cis or in trans), whenever possible. If a VUS and pathogenic variant were determined to be in trans, the variant was eligible for downgrade.
Mutation co-occurrence.
Mutation co-occurrence (MCO) analysis was based on an assumption of a low likelihood that an individual will carry two autosomal dominant pathogenic variants that cause an increased risk for the same or a similar cancers.18 MCO analysis measured the statistical significance of a variant co-occurring multiple times with one or more pathogenic variants in the same patient (in the same gene or in a different gene for the same syndrome). Control individuals were matched to patients on the basis of ethnicity and time of testing. MCO was used in downgrading variants, which was driven by a threshold and negative predictive value of > 99.5%.
RNA splicing analysis and protein structural analysis.
Assessment of in silico RNA splicing predictions was used to initially evaluate the potential effects of a variant on RNA splicing.19 Published reports of biochemical splicing analysis, which used high-quality data and appropriate controls, were considered for variant classification. In some cases where evidence was unavailable or of insufficient quality or detail to be conclusive, quantitative RNA splicing analyses were performed in house, using informative SNPs, to assess potential RNA splicing effects resulting from the variant of interest.20 Expert protein structural analysis was also used to assess the impact of variants predicted to affect protein structure at the amino acid level. Pathogenicity assignment was determined by evidence of a fully penetrant splice defect or no splicing.
Literature review.
Daily comprehensive reviews of the variant literature were performed for each gene tested by the laboratory, facilitated by an automated search algorithm. References and associated variants were stored in a curated, searchable repository. All publications relevant to variant classification were thoroughly assessed and discussed by the laboratory’s classification committee during the reclassification process. This included in vitro studies, protein functional studies, and segregation analyses. Literature with appropriate controls and significant findings was applied to reclassification in conjunction with additional supporting evidence.
Population frequency.
Population frequencies were established by identifying how often a variant is observed in the general population. However, population frequencies can be difficult to use when assessing rare variants specifically. An in-house analysis of population databases, such as ExAC and gnomAD, was performed to establish gene-specific allele frequency (AF) thresholds, which were validated against previously classified variants.21
Other tools.
Family testing was offered, when appropriate. However, segregation analysis was of limited use in the overall testing population because of restricted access to extended families. In addition, published pedigrees and personal communications with providers were considered, when available, as evidence for upgrading variants. Other methods used in variant reclassification included reclassification on the basis of similar variants, species alignments, and earlier versions of statistical tools (Pheno and MCO). Classification criteria were stringent, well-documented rules that were based on ACMG/AMP guidelines, which were enhanced and refined by the reclassification committee’s expertise and experience for clinical laboratory use.
Statistical Analysis
The proportion of variants that were initially identified and later reclassified in the analysis time period was determined for all tested genes. Because GREM1, POLE, and POLD1 were added to the panel test near the end of the evaluated time period, no variants had been reclassified at the time of this analysis. As such, they were excluded from analysis, and data are presented for the remaining 25 tested genes. Variant type (eg, frameshift, in-frame indel, missense, nonsense) and variant-specific AFs (the number of instances of a unique variant in the patient cohort) were also calculated. Initial classification occurred at the first identification of each unique variant. A reclassification event was defined as any subsequent change in classification. A variant may have undergone more than one reclassification event (eg, VUS to LP and LP to P). The type(s) of evidence used was evaluated for each reclassification event. This included an evaluation of each individual tool as well as the subtotal for laboratory-developed tools (Pheno, PT/HA, MCO, RNA analysis). All analyses were performed using R version 3.4.1 software.22
RESULTS
Identified and Reclassified Variants
In this 20-year time period, > 1.9 million individuals had genetic testing from a single testing laboratory. A total of 62,842 unique variants were observed across 25 genes. Overall, 19.0% (11,930 of 62,842) of variants were initially classified as LP/P, 24.3% (15,250 of 62,842) as LB/B, and 56.7% (35,662 of 62,842) as VUS (Table 1). Missense mutations were the most common type of variant detected, representing 48.7% (30,628 of 62,842) of unique variants (Table 1).
TABLE 1.
Distribution of Initial Variant Classification at the Time of Identification, Variant Type, Allele Frequency, and Gene Group for All Unique, Detected Variants and All Reclassified Variants
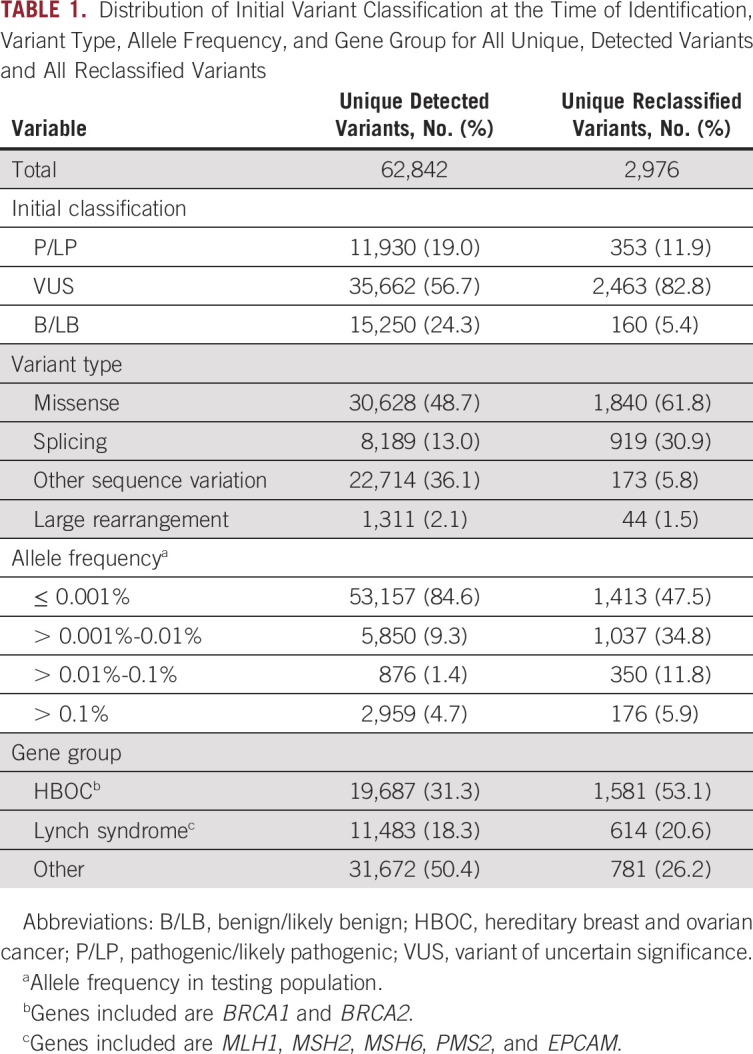
Of the total unique variants detected, 4.7% (2,976 of 62,842) were reclassified in a total of 3,538 reclassification events. This reflects the fact that some variants were reclassified more than once. Among reclassified variants, 82.8% (2,463 of 2,976) were initially classified as VUS (Table 1). The majority of reclassified variants that were initially classified as benign or pathogenic were reclassified within the same clinical category (eg, LP to P or LB to B), with only 0.2% (26 of 15,250) and 0.7% (85 of 11,930) of benign and pathogenic variants, respectively, being reclassified to VUS. Overall, 82.1% (2,906 of 3,538) of reclassification events were downgrades (eg, VUS to LB/B). This corresponds with 4.2% (2,675 of 62,842) unique, detected variants being downgraded in this time period. The remaining 17.9% (632 of 3,538) of reclassification events were upgrades (eg, VUS to LP/P), which corresponds with 0.5% (298 of 62,842) unique variants being upgraded in this time period.
The most common type of reclassified variant was missense (61.8%; 1,840 of 2,976), which represents an enrichment from the proportion of missense variants identified during testing. Similarly, variants that had the potential to result in abnormal splicing (30.9%; 919 of 2,976) were enriched among the reclassified variants compared with the identified variants (Table 1).
The majority (84.6%; 53,157 of 62,842) of sequence variants identified during testing were rare (AF ≤ 0.001%; Table 1). In the subset of variants that were reclassified in this time period, 47.5% (1,413 of 2,976) were identified in ≤ 20 individuals in this cohort (AF ≤ 0.001%). Furthermore, 21.7% (646 of 2,976) of reclassified variants were found in ≤ 5 individuals (AF ≤ 0.0002%). The proportion of reclassified variants that were in genes other than BRCA1 and BRCA2 increased over time (Fig 2A). This is not unexpected because of the inclusion of additional genes by the testing laboratory (Fig 2B) and reflects the successful application of the variant classification program to a growing number of genes. In particular, the incorporation of panel testing in 2013 is accompanied by an increase in reclassifications in genes outside the common hereditary cancer syndromes (HBOC and Lynch).
FIG 2.
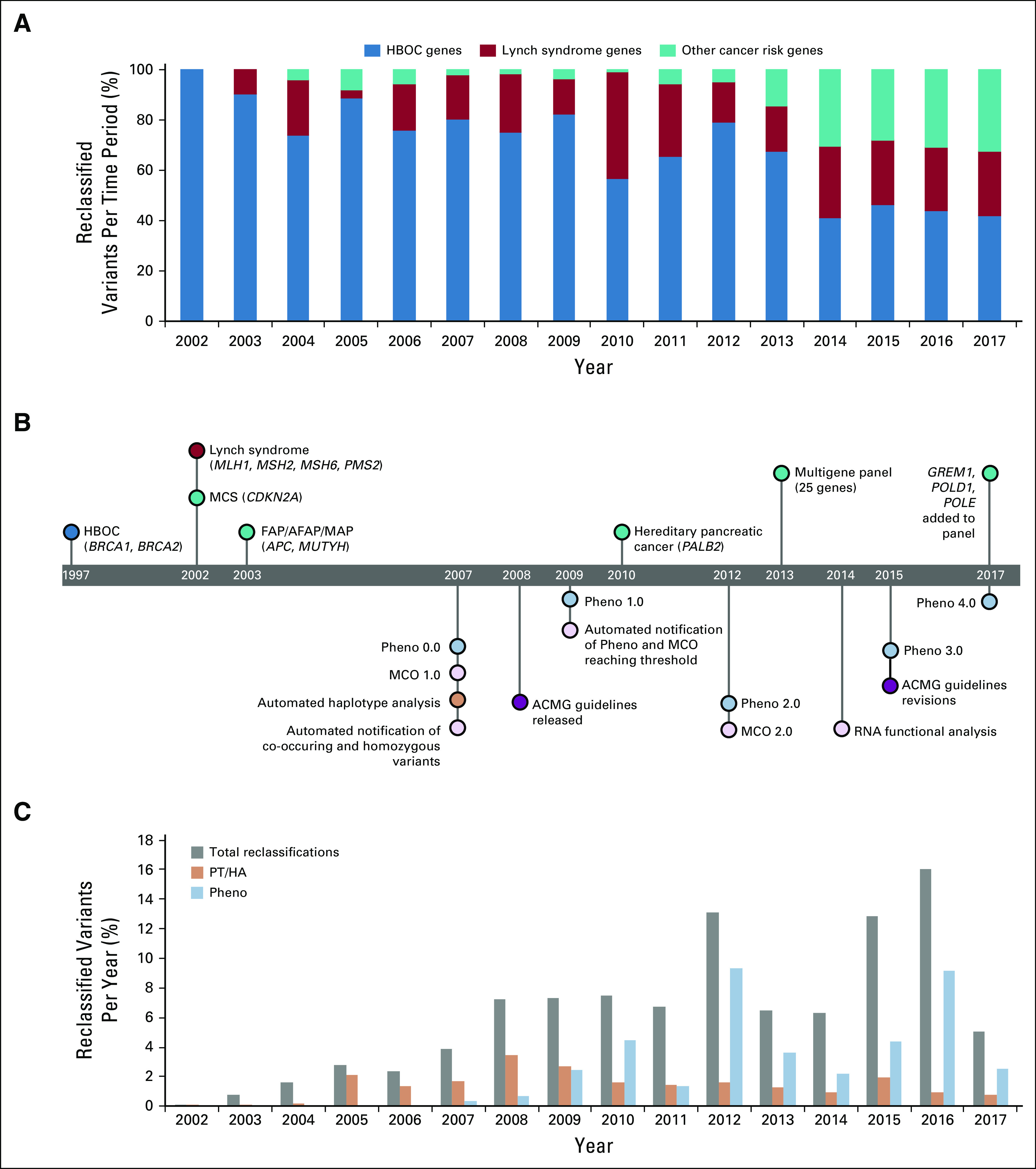
Timeline for the development of genetic tests and classification methods. (A) Reclassified variants by year, shown as a proportion of reclassified variants by gene group. (B) Timeline of changes in genetic test offering and advances in the reclassification program. (C) Reclassified variants by year, shown as a proportion of all variants identified in each year. ACMG, American College of Medical Genetics and Genomics; FAP/AFAP/MAP, familial adenomatous polyposis/attenuated familial adenomatous polyposis/MUTYH-associated polyposis syndrome; HBOC, hereditary breast and ovarian cancer; MCO, mutation co-occurrence; MCS, melanoma-pancreatic cancer syndrome; PT/HA, phase testing/homozygosity analysis.
Tools Used for Variant Reclassification
Figure 3 shows the frequency with which individual tools provided evidence that contributed to variant reclassification. The most common tools used in reclassification of unique variants were Pheno analysis (40.5% of reclassification events) and PT/HA (22.8% of reclassification events). In all, laboratory-developed tools were used in 72.3% of variant reclassification events. Multiple reclassification tools were generally used across all tested genes (Appendix Table A1). For example, BRCA2 c.6821G>T (p.Gly2274Val) is a missense variant that was initially classified as a VUS in 1999 and has been observed in a total of 63 patients. In 2008, the variant was reclassified to B on the basis of MCO and PT/HA (variant was identified in trans with a pathogenic variant). MLH1 c.245C>T (p.Thr82Ile) was initially classified as VUS in 2003 and has been observed in 25 patients. The variant was reclassified as LP in 2011 on the basis of structural evidence combined with literature and segregation analysis. In 2015, this variant was reclassified from LP to P on the basis of evidence from Pheno analysis. CDH1 c.1774G>A (p.Ala592Thr) has been observed in 853 patients and was initially classified as a VUS in 2013. It was reclassified as LB in less than a year on the basis of validated population frequency data from ExAC.21 In 2015, the variant was reclassified to B on the basis of evidence from PT/HA.
FIG 3.

The impact of individual classification tools on variant reclassification. Multiple tools may be used to reclassify a single variant. The “Other” category includes knowledge of similar variants, species conservation in conjunction with structural analysis, additional statistical analysis, and classification/reclassification protocol refinements. MCO, mutation co-occurrence; PT/HA, phase testing/homozygosity analysis.
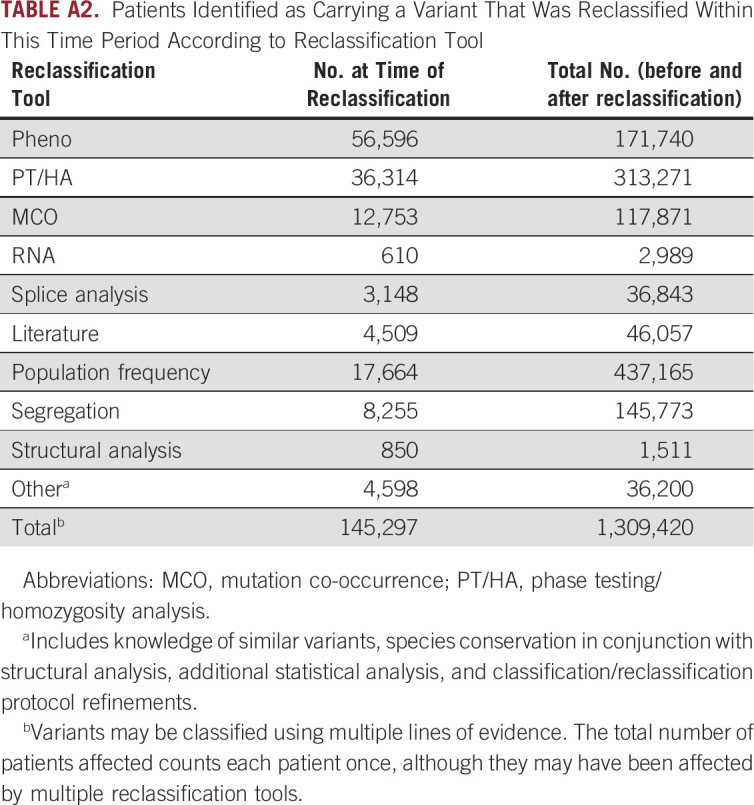
Overall, 1.31 million patients were found to have a reclassified variant (Appendix Table A2). In the majority (88.9%; 1.16 million of 1.31 million), the variant was reclassified before the patient was tested such that the patient’s initial test report included the new classification. When the distribution of tools used in reclassification was assessed, > 600,000 tested individuals carried a variant that was reclassified using a laboratory-developed tool (Appendix Table A2). Although the proportion of unique variants that were reclassified using population frequency was relatively low, this tool affected the most (437,165) patients (Appendix Table A2). This is consistent with population frequency being applicable to more common variants.
Evolution of Testing and Reclassification Program
The variant reclassification program has been revised over the past 15 years as available hereditary cancer tests have changed (Fig 2B). The increased proportion of variants reclassified was a direct result of increased test volume and the introduction of new statistical analysis tools, such as Pheno and MCO. In addition, existing tools were improved and revised as more data became available. As a result, the overall percentage of variants that were reclassified each year has increased steadily from 2003 to 2016 (Fig 2C). The dip in reclassifications in 2013 corresponds with the introduction of panel testing. This reflects the increased VUS rate associated with adding many new genes to the test as well as the time needed to acquire sufficient data to use many of the laboratory-developed reclassification tools. Overall, a substantial proportion of variants were reclassified using information from PT/HA or Pheno through an automated program (Fig 2C).
DISCUSSION
Hereditary cancer genetic testing is a critical component of patient care because the results are used to inform medical management.23 The number of rare variants identified with hereditary cancer genetic testing has grown with the increased use of multigene panels, and improved methods are needed to provide accurate and definitive classifications. In the 20-year time period evaluated here, the majority of reclassified variants were initially classified as VUS, and nearly half were individually rare, with an AF ≤ 0.001%. This equates to variants being observed in ≤ 20 patients in the testing population of 1.9 million patients. Historically, little emphasis has been placed on the reclassification of these rare variants because of the challenge in obtaining evidence through traditional approaches, such as segregation analysis or case-control studies.24 As the number of patients who receive genetic testing continues to rise, so will the number of rare variants identified, driving the need for clinical laboratories to develop innovative reclassification programs.
In the 20-year time period evaluated here, > 1.3 million patients were affected by variant reclassification. Providing a patient with a definitive classification can aid in medical management decisions with regard to appropriate risk-reducing and treatment strategies.25,26 The majority of reclassified variants were downgraded from VUS to B/LB. Downgrades can play an important role in patient care by helping to avoid unnecessary surveillance and surgical interventions.6,17,25 In addition, for some patients, a definitive classification to B/LB may result in reduced anxiety. Variants upgraded from VUS to P/LP are of greater clinical consequence because these patients may be candidates for increased screening or prophylactic interventions.1,27 While this represents critical information to support appropriate patient management, it is important for clinicians to also consider how often variant reclassification occurs to ensure appropriate patient counseling. Mersch et al25 demonstrated that it is very rare for variants that were initially classified as benign or pathogenic to be reclassified to a different clinical category, which accounts for only 0.1% of all B/LB variants and 0.3% of all P/LP variants. In addition, although approximately 10% of all unique VUSs were upgraded to P/LP, only 3% of all observed VUSs (ie, accounting for the same variant being observed in multiple individuals) were upgraded.25
The goal of the reclassification program used by the laboratory evaluated here was to ensure accurate and informative results for patients by providing a definitive classification. The data presented here show that the tools developed by the testing laboratory were the most commonly used tools in variant reclassification. As variant classification continues to evolve, additional technologies and approaches may become important. For example, testing for loss of heterozygosity may become relevant as tumor testing becomes more common for patients with cancer. In addition, large in vitro studies may become increasingly important as panel testing continues to grow. This may include a system to evaluate the quality of data from the growing number of CRISPR studies.
In summary, many variants observed in this analysis were individually rare but collectively affected millions of patients. This highlights the importance of developing and validating nontraditional tools to enable the reclassification of rare variants. It is critical that clinical testing laboratories use robust classification programs that have the capability to evolve as new information is introduced and technologies change. As this reclassification program continues to be used and improved, it will continue to benefit patients and their families through appropriate medical management for hereditary cancer.
ACKNOWLEDGMENT
We thank Stephanie Meek, PhD, and Anna Gardiner, PhD, for their assistance in manuscript preparation.
Appendix
TABLE A1.
Proportion of Variants Reclassified According to Gene and Reclassification Tools

TABLE A2.
Patients Identified as Carrying a Variant That Was Reclassified Within This Time Period According to Reclassification Tool
SUPPORT
Supported in part by National Institutes of Health Grant No. R01 HL132251-02, the Lucy and Henry Billingsley Fund, the Jeanne Ann Pitt Professorship in Breast Cancer Research (T.S.R.), and the H. Ben and Isabelle T. Decherd Chair in Internal Medicine at University of Texas Southwestern Medical Center (T.S.R.).
AUTHOR CONTRIBUTIONS
Conception and design: Lisa Esterling, Dmitry Pruss, Susan Manley, Karla R. Bowles, Theodora S. Ross
Collection and assembly of data: Lisa Esterling, Theodora S. Ross
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Lisa Esterling
Employment: Myriad Genetics
Stock and Other Ownership Interests: Myriad Genetics
Krystal Brown
Employment: Myriad Genetics
Stock and Other Ownership Interests: Myriad Genetics
Brian Morris
Employment: Myriad Genetics
Stock and Other Ownership Interests: Myriad Genetics
Elisha Hughes
Employment: Myriad Genetics
Stock and Other Ownership Interests: Myriad Genetics
Dmitry Pruss
Employment: Myriad Genetics
Stock and Other Ownership Interests: Myriad Genetics
Susan Manley
Employment: Myriad Genetics
Stock and Other Ownership Interests: Myriad Genetics
Karla R. Bowles
Employment: Myriad Genetic Laboratories
Leadership: Myriad Genetic Laboratories
Stock and Other Ownership Interests: Myriad Genetics
Theodora S. Ross
Employment: Merck & Co.
No other potential conflicts of interest were reported.
REFERENCES
- 1. Daly MB, Pilarski R, Berry M, et al. NCCN Guidelines Version 1.2020 Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Plymouth Meeting, PA, National Comprehensive Cancer Network, 2019. [DOI] [PubMed] [Google Scholar]
- 2.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gradishar W, Johnson K, Brown K, et al. Clinical variant classification: A comparison of public databases and a commercial testing laboratory. Oncologist. 2017;22:797–803. doi: 10.1634/theoncologist.2016-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vail PJ, Morris B, van Kan A, et al. Comparison of locus-specific databases for BRCA1 and BRCA2 variants reveals disparity in variant classification within and among databases. J Community Genet. 2015;6:351–359. doi: 10.1007/s12687-015-0220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenthal ET, Bowles KR, Pruss D, et al. Exceptions to the rule: Case studies in the prediction of pathogenicity for genetic variants in hereditary cancer genes. Clin Genet. 2015;88:533–541. doi: 10.1111/cge.12560. [DOI] [PubMed] [Google Scholar]
- 6.Mundt E, Nix P, Brown K, et al. Complexities of variant classification in clinical hereditary cancer genetic testing. J Clin Oncol. 2017;35:3796–3799. doi: 10.1200/JCO.2017.74.5182. [DOI] [PubMed] [Google Scholar]
- 7. Eggington JM, Saam J, Bowles K, et al: Segregation analysis offers a mechanism for variant reclassification in a small subset of cases but is especially powerful in classifying deleterious mutations. Presented at the ACMG Annual Clinical Genetics Meeting, Phoenix, AZ, March 19-23, 2013. https://s3.amazonaws.com/myriad-web/myriadpro.com/publications/ACMG+Segregation+Analysis+Eggington+Presented+22MAR2013.pdf. [Google Scholar]
- 8.Balmaña J, Digiovanni L, Gaddam P, et al. Conflicting interpretation of genetic variants and cancer risk by commercial laboratories as assessed by the prospective registry of multiplex testing. J Clin Oncol. 2016;34:4071–4078. doi: 10.1200/JCO.2016.68.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Judkins T, Leclair B, Bowles K, et al. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer. 2015;15:215. doi: 10.1186/s12885-015-1224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 11.Oros KK, Ghadirian P, Greenwood CM, et al. Significant proportion of breast and/or ovarian cancer families of French Canadian descent harbor 1 of 5 BRCA1 and BRCA2 mutations. Int J Cancer. 2004;112:411–419. doi: 10.1002/ijc.20406. [DOI] [PubMed] [Google Scholar]
- 12.Sampson JR, Jones N. MUTYH-associated polyposis. Best Pract Res Clin Gastroenterol. 2009;23:209–218. doi: 10.1016/j.bpg.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Eggington JM, Bowles KR, Moyes K, et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. 2014;86:229–237. doi: 10.1111/cge.12315. [DOI] [PubMed] [Google Scholar]
- 14.Kazazian H, Boehm C, Seltzer W. ACMG recommendations for standards for interpretation of sequence variations. Genet Med. 2000;2:302–303. [Google Scholar]
- 15.Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 16.Pruss D, Morris B, Hughes E, et al. Development and validation of a new algorithm for the reclassification of genetic variants identified in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat. 2014;147:119–132. doi: 10.1007/s10549-014-3065-9. [DOI] [PubMed] [Google Scholar]
- 17.Morris B, Hughes E, Rosenthal E, et al. Classification of genetic variants in genes associated with Lynch syndrome using a clinical history weighting algorithm. BMC Genet. 2016;17:99. doi: 10.1186/s12863-016-0407-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavtigian SV, Deffenbaugh AM, Yin L, et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahashi K, Masuda A, Matsuura T, et al. In vitro and in silico analysis reveals an efficient algorithm to predict the splicing consequences of mutations at the 5′ splice sites. Nucleic Acids Res. 2007;35:5995–6003. doi: 10.1093/nar/gkm647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey SE, Cheng C. Methods for characterization of alternative RNA splicing. Methods Mol Biol. 2016;1402:229–241. doi: 10.1007/978-1-4939-3378-5_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. R Foundation for Statistical Computing: R: A language and environment for statistical computing, 2018. https://www.R-project.org.
- 23. Daly MB, Pilarski R, Berry M, et al: NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Genetic/Familial High-Risk Assessment: Breast and Ovarian (Version 3.2019). Plymouth Meeting, PA, National Comprehensive Cancer Network, 2019. [Google Scholar]
- 24.Lincoln SE, Kobayashi Y, Anderson MJ, et al. A systematic comparison of traditional and multigene panel testing for hereditary breast and ovarian cancer genes in more than 1000 patients. J Mol Diagn. 2015;17:533–544. doi: 10.1016/j.jmoldx.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Mersch J, Brown N, Pirzadeh-Miller S, et al. Prevalence of variant reclassification following hereditary cancer genetic testing. JAMA. 2018;320:1266–1274. doi: 10.1001/jama.2018.13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. doi: 10.1200/PO.18.00217. Idos GE, Kurian AW, Ricker C, et al: Multicenter prospective cohort study of the diagnostic yield and patient experience of multiplex gene panel testing for hereditary cancer risk. JCO Precision Oncol 10.1200/PO.18.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benson AB, Venook AP, Al-Hawary MM, et al: NCCN Guidelines Version 4.2019 Colon Cancer. Plymouth Meeting, PA, National Comprehensive Cancer Network, 2019.