

ABSTRACT
We have recently tested the transforming potential of 484 ‘long-tail’ genes, which are recurrently albeit infrequently mutated in head and neck cancers (HNSCC). We identified 15 novel tumor suppressors and our top hits converge on regulating the NOTCH signaling pathway. Therapeutic approaches activating NOTCH signaling could be a promising strategy to treat two-thirds of human HNSCC patients.
KEYWORDS: HNSCC, Notch, mouse models of cancer, long-tail genes, in vivo CRISPR
Cancer is a complex disease in which somatic cells progressively accumulate mutations that eventually disrupt their cellular functions and increase tumorigenic potential. Comprehensive sequencing efforts such as The Caner Genome Atlas (TCGA) and more recently Pan-Cancer Analysis of Whole Genomes (PCAWG) have mapped the genomic landscape of several cancer types with the promise to identify nodes with cancer networks, that can be targeted therapeutically.1,2 A big challenge, however, is the extensive mutational heterogeneity found in most cancer types, where typically only a few genes are mutated at high frequency, while the vast majority of genes are found mutated at low frequency (<5%), commonly referred to as the ‘long-tail’ distribution1 (Figure 1a).
Figure 1.
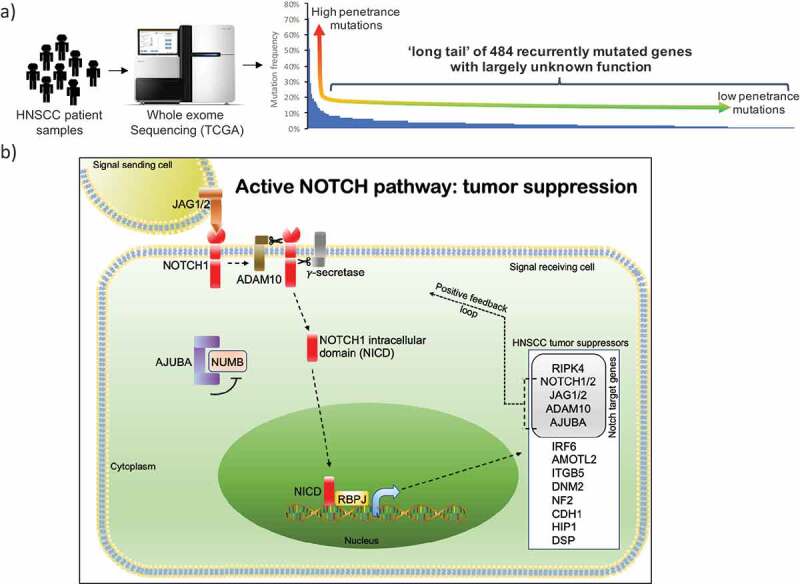
NOTCH signaling is essential for HNSCC suppression. (a) Schematic representing the long-tail genes mutated in HNSCC patients (adapted from Loganathan et al.) (b) Ligands Jagged1/2 (JAG1/2) binding results in ADAM10 (Disintegrin and metalloproteinase domain-containing protein 10) and gamma secretase-mediated cleavage of NOTCH1/2 leading to release of NOTCH intracellular domain (NICD). AJUBA sequesters NUMB, thereby blocking ubiquitination and degradation of NICD. NICD translocates to the nucleus, binds to transcription factor RBPJ, and turns on the expression of Notch target genes that include HNSCC tumor suppressors identified in our in vivo CRISPR screen.
The extensive mutational heterogeneity also inadvertently impedes the hunt for effective therapies that would be applicable to a broad set of patients. Most research has thus focused on the handful of genes that are mutated frequently, with the idea to develop therapeutic strategies for as many patients as possible. However, most of the frequently mutated genes are not druggable or targeted therapies showed limited long-term efficacy in the clinic.
The long-tail of infrequently mutated genes has attracted much less attention and the reasons for this are manifold. Tumors accumulate mutations as they grow, which means that most long-tail mutations likely do not affect tumor development – they are just random bystanders. The difficulty is to weed out these bystanders and identify the long-tail mutations that actually drive cancerous growth.2 Even if identified, those findings might only have limited clinical and therapeutic relevance due to the small number of patients carrying those mutations.
We reasoned that a small percentage of a very common disease can still constitute a lot of patients. In addition, certain long-tail driver mutations have been recently shown to be the genetic basis of extraordinary responses to cancer therapy.3,4 We thus set out to systematically characterize the transforming potential of long-tail genes in Head and Neck Squamous Cell Carcinoma (HNSCC).5
HNSCC is the sixth most common cancer worldwide with high mortality of 40–50%.5 HNSCC arises in mucosal linings of the upper airway or oral cavity and is associated with tobacco and alcohol consumption or human papillomavirus (HPV) infection. The most common genetic alterations in HNSCC affect phosphorylate tumor protein p53 (TP53, best known as p53, 71%), Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA, 35%), NOTCH1 (17%), and HRAS (6%), followed by a long-tail of mutations, the majority of which lack biological or clinical validation.6 To probe this long-tail, we developed a direct in vivo CRISPR (clustered regularly interspaced short palindromic repeats) screening methodology that employs ultrasound-guided injections7-9 of lentiviruses carrying CRISPR components into the amniotic cavity of living mouse embryos at embryonic day E9.5. At this stage, the injected virus will transduce the single-layered surface ectoderm, which will later give rise to the epithelium of the skin and mouth.7-9
Using this technique, we introduced a lentiviral sgRNA library targeting 484 long-tail HNSCC genes into the surface epithelium of four mouse strains that are sensitized for SCC development by conditional expression of oncogenic Pik3caH1047R, HRasG12V or p53R270H or expression of the HPV16 E6/7 oncogenes and followed mice for tumor development. The top hits of this screen are Adam10, Ripk4 and Ajuba followed by Notch 2, Notch 3, Dsp, Cdh1, Znf750, Nf2, Cul3, Stk19, Rasa1, Dnm2, Nudt11, and Irf6. CRISPR/Cas9-inactivation or conditional deletion of Adam10, Ajuba, and Ripk4 triggered invasive HNSCC formation on tumor-susceptible backgrounds within a matter of weeks, indicating potent tumor suppressor function.10
Mechanistically, we showed that Adam10 mediates its tumor-suppressive activity by activating the Notch pathway. Loss of Adam10 and subsequent formation of HNSCC could be rescued by forced expression of the Notch1 intracellular domain (NICD), the cleaved form of Notch1, that translocates to the nucleus to activate gene expression. In addition, we showed that Notch1 and Notch2, the Notch ligands Jag1 and Jag2, and the Notch transcriptional mediator Rpbj are required for HNSCC suppression, while other Notch pathway components such as Dll1 and Adam17 are dispensable, indicating a tightly controlled regulation. Furthermore, we demonstrated that Ajuba also activates the Notch pathway by directly interacting with Notch1/2 and Numb, a known negative regulator of Notch. Upon Notch activation Ajuba sequesters Numb, thereby permitting NICD translocation to nucleus. We also showed that Notch activation leads to recruitment of NICD and Rpbj to target genes, several of which are regulators of the Notch pathway themselves, indicating a feed-forward loop (Figure 1b). We further identified critical downstream targets of the Notch pathway with tumor-suppressive capabilities such as Ripk4, Itgb5, and Amotl2.10
Intriguingly, we found that heterozygous loss of Adam10 or Ajuba is sufficient to trigger tumor development, identifying novel, highly penetrant haploinsufficiencies. Clinically, we show that ADAM10 and AJUBA are mutated or heterozygously lost in 13% and 17% of human HNSCCs, respectively, in a pattern that is mutually exclusive with NOTCH receptor mutations. Importantly, due to the haploinsufficiency, oncogenic alterations of ADAM10 and AJUBA are actually much more frequent than initially assumed. Together, ADAM10, AJUBA, and NOTCH receptor alterations are found in at least 67% of human HNSCC cases, establishing the NOTCH pathway as a major tumor suppressor with therapeutic relevance in HNSCCs.10
Our work highlights two achievements: (1) the technological advancement of direct in vivo CRIPSR to integrate cancer genomics and mouse modeling for rapid discovery of novel tumor suppressors and (2) the conceptual advancement that potent long-tail cancer driver mutations are often haploinsufficient and can converge on specific pathway to drive tumorigenesis in the majority of a given cancer type.
Funding Statement
This work was supported by the Krembil Foundation; Canadian Institutes of Health Research (CIHR) [CIHR 365252].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interests were disclosed.
References
- 1.Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, Campbell PJ.. Universal patterns of selection in cancer and somatic tissues. Cell. 2017;171:1–3. doi: 10.1016/j.cell.2017.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell PJ, Getz G, Stuart JM, Korbel JO, Stein LD. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Ahmadie H, Iyer G, Hohl M, Asthana S, Inagaki A, Schultz N, Hanrahan AJ, Scott SN, Brannon AR, McDermott GC. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discovery. 2014;4:1014 LP– 1021. doi: 10.1158/2159-8290.CD-14-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I. Genome sequencing identifies a basis for everolimus sensitivity. Science (80-). 2012;338(6104):221 LP–221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. 2018;18:269. doi: 10.1038/nrc.2018.11. [DOI] [PubMed] [Google Scholar]
- 6.Lawrence MS, Sougnez C, Lichtenstein L, Cibulskis K, Lander E, Gabriel SB, Getz G, Ally A, Balasundaram M, Birol I , et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beronja S, Livshits G, Williams S, Fuchs E. Rapid functional dissection of genetic networks via tissue-specific transduction and RNAi in mouse embryos. Nat Med. 2010;16:821–827. doi: 10.1038/nm.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beronja S, Janki P, Heller E, Lien W-H, Keyes BE, Oshimori N, Fuchs E. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature. 2013;501(7466):185–190. doi: 10.1038/nature12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D, Reva B, Fuchs E. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science (80-). 2014;343(6168):309 LP–313. doi: 10.1126/science.1248627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loganathan SK, Schleicher K, Malik A, Quevedo R, Langille E, Teng K, Oh RH, Rathod B, Tsai R, Samavarchi-Tehrani P. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science (80-). 2020;367:1264 LP–1269. doi: 10.1126/science.aax0902. [DOI] [PubMed] [Google Scholar]