

ABSTRACT
Centrosomes are the primary microtubule-organizing centers that are important for mitotic spindle assembly. Centrosome amplification is commonly observed in human cancer cells and contributes to genomic instability. However, it is not clear how centrosome duplication is dysregulated in cancer cells. Here, we report that ATAD5, a replisome protein that unloads PCNA from chromatin as a replication factor C-like complex (RLC), plays an important role in regulating centrosome duplication. ATAD5 is present at the centrosome, specifically at the base of the mother and daughter centrioles that undergo duplication. UAF1, which interacts with ATAD5 and regulates PCNA deubiquitination as a complex with ubiquitin-specific protease 1, is also localized at the centrosome. Depletion of ATAD5 or UAF1 increases cells with over-duplicated centrosome whereas ATAD5 overexpression reduces such cells. Consistently, the proportion of cells showing the multipolar mode of chromosome segregation is increased among ATAD5-depleted cells. The localization and function of ATAD5 at the centrosomes do not require other RLC subunits. UAF1 interacts and co-localizes with ID1, a protein that increases centrosome amplification upon overexpression. ATAD5 depletion reduces interactions between UAF1 and ID1 and increases ID1 signal at the centrosome, providing a mechanistic framework for understanding the role of ATAD5 in centrosome duplication.
KEYWORDS: Centrosome duplication, ATAD5, UAF1, ID1
Introduction
Centrosomes are the primary microtubule organizing center and important for bipolar spindle assembly during mitosis [1]. A centrosome is composed of two centrioles, a mother and a daughter centriole. The mother centriole is a mature centriole capable of forming the pericentriolar matrix which is a microtubule organizing center while the daughter centriole has no such abilities. In dividing cells, both these centrioles are disengaged, duplicated, separated, and moved to opposite poles to form a bipolar spindle in a timely fashion that is coordinated with the cell cycle [2]. This centrosome cycle consists of several structural and functional transitions that are regulated by changes in expression level and post-translational modification of participating proteins [3]. Consequently, defects in regulation lead to centrosome aberrations, usually increase centrosome numbers, and contribute to chromosome instability, which is related to diseases, especially cancers [4,6]. However, it is still not clearly understood how centrosomes are dysregulated in cancer cells.
Centrosomal localization and non-replicative roles for the DNA replication-related proteins have been reported. ORC1, a subunit of the origin recognition complex (ORC), prevents Cyclin E-dependent reduplication of both centrioles and centrosomes in a single cell cycle [7]. ORC2 also localizes to centrosomes, even though its actual role at centrosomes has not yet been investigated [8]. Cdc6, a DNA replication licensing factor, negatively regulates centrosome duplication by inhibiting spindle assembly abnormal protein 6 (Sas-6) which is a cartwheel protein [9]. The expression of a subunit of replicative MCM helicase, MCM5, but not MCM2, significantly inhibits over-duplication of centrosomes [10]. In addition, TopBP1, a protein critical for DNA replication and DNA damage response, is localized at the centrosome and is required for proper mitotic progression [11].
Inhibitor of DNA Binding 1 (ID1) is a helix-loop-helix protein that inhibits the transcriptional activation ability of basic helix-loop-helix proteins [12]. It has been previously reported that a portion of ID1 is localized at the centrosome and ectopic expression of ID1 results in supernumerary centrosomes [13]. High expression levels of ID1 consistently correlate with centrosomal abnormalities in human tumor cells [14]. A strong binding of ID1 to the proteasomal subunit S5A/Rpn10 and stabilization of Aurora A kinase through Cdh1 binding have been suggested as causes of centrosomal abnormalities [15,16]. A Previous study reported that overexpression of ubiquitin-specific protease 1 (USP1) increases ID1 expression, leading to centrosome duplication [17]. Although stabilization of ID proteins by USP1-mediated deubiquitination has been reported as a mechanism underlying the maintenance of the stem cell-like state [18], it is still not clear how ID1 is dysregulated in cancer cells.
ATAD5 forms a pentameric complex, similar to the clamp loader complex (replication factor C, RFC), that consists of ATAD5 and RFC2-5, referred to as ATAD5-RFC-like complex (RLC) [19,25]. ATAD5-RLC is important for maintaining genomic stability in eukaryotic organisms, from yeasts to humans and the importance of this function is underscored by the fact that Atad5 heterozygote mutant mice develop tumors [26]. Additionally, somatic mutations of ATAD5 were found in patients with several types of cancers and a genome-wide analysis indicated that the ATAD5 locus confers enhanced susceptibility to endometrial, breast, and ovarian cancers [26,28]. A recent study revealed that ATAD5 promotes replication fork restart under replication stress [29]. ATAD5-RLC has two major molecular functions in the regulation of the replication sliding clamp, proliferating cell nuclear antigen (PCNA), at the replication forks: (1) PCNA unloading during normal DNA replication to terminate DNA synthesis [30,33]. (2) PCNA deubiquitination to limit the DNA damage bypass pathway by recruiting the USP1/USP1-associated factor 1 (UAF1) complex [34].
UAF1 forms distinct complexes with various proteins, such as USP1, USP12, USP46, RAD51AP1 and FANCI [34,38]. UAF1 has been reported to be an important factor for the stability and catalytic activity of USP1 [37]. USP1 regulates DNA damage tolerance pathway and the Fanconi anemia pathway through its association with UAF1 and by deubiquitinating PCNA and FANCD2, respectively [37]. The USP1/UAF1 complex has also been reported to participate in homologous recombination [35,38,39]. However, not much has been studied regarding the roles of the USP1/UAF1 complex in centrosome regulation. Since USP1 affects centrosome duplication and ATAD5 interacts with the USP1/UAF1 complex [17,34], we hypothesized that ATAD5 plays a role in centrosome duplication. In this study, we found that ATAD5 and UAF1 are localized at the centrosome and play important roles in regulating centrosome duplication and bipolar segregation. UAF1 interacts with ID1 and their interaction is altered according to ATAD5 protein level. Consistent with centrosome over-duplication in ATAD5-depleted cells, ID1 signals are increased at the centrosomes. Our results strongly suggest that ATAD5 is involved in controlling centrosome duplication by modulating ID1 levels at the centrosomes.
Materials and methods
Cell lines
Human embryonic kidney (HEK) 293 T, HeLa, U2OS, and NIH3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (GE Healthcare) or 10% calf serum (GE Healthcare) for NIH3T3 cells, 100 U/mL penicillin G (Life Technologies), and 100 μg/mL streptomycin (Life Technologies) at 5% CO2, 37°C. HeLa cells expressing green fluorescence protein (GFP)-tagged Centrin1 (HeLa_Centrin1-GFP) were generated by infection of lentiviral vector pMF1454. AB2.2 mouse embryonic stem cells (mESCs) were maintained in M15 medium (high-glucose DMEM supplemented with 15% fetal bovine serum, 100 μM mercaptoethanol, 1 mM glutamine, 3 mg/ml penicillin, 5 mg/ml streptomycin) containing 1,000 U/ml ESGRO leukemia inhibitory factor. Atad5 knockout AB2.2 mESCs were generated using CRISPR-mediated gene targeting. Briefly, cells were electroporated with two guide RNAs (pX330, Addgene plasmid number 42,230) which target sequences in 5ʹ of mouse Atad5 exon2 (guide RNA #1: 5ʹ- GTTGTACGCCTTTGTCGATTG-3ʹ and guide RNA #2: 5ʹ- GACTAAACCTTCCCTAGCTG-3ʹ) coupled with the targeting vector and selected in HAT media (0.5 mM sodium hypoxanthine, 2 μM aminopterin, 80 μM thymidine) before colony picking. ATAD5 knockout U2OS cell line was generated using a commercial ATAD5 CRISPR/CAS9 knockout plasmid (Santa Cruz biotechnology; SC-405,654) with the manufacturer’s protocol. Briefly, U2OS cells were transfected with the CRISPR/CAS9 plasmid, and 48 h later GFP positive single cells were sorted using a FACSAria cell sorter (BD biosciences). After 3 wk, single-cell colonies were picked and tested for complete ATAD5 knockout using immunoblotting.
Drugs and antibodies
The following drug was used in this study: CDK1 inhibitor (RO3306; Sigma-Aldrich). The following antibodies were used: anti-UAF1, anti-Centrin2, anti-RFC5, anti-CEP164 (Santa Cruz Biotechnology); anti-FLAG, anti-HA, anti-γ-tubulin, anti-α-tubulin (Sigma-Aldrich); anti-NUF2 (Abcam); anti-histone H3 antibodies (Thermo Fisher Scientific). Polyclonal anti–human ATAD5 antibody was raised against the N-terminal fragment (residues 202–367 amino acids) in rabbits. The anti-ATAD5 antibody was purified using ATAD5 fragment (residues 1–400 amino acids) blotted to nitrocellulose membrane. Briefly, the blot was incubated with 5 mL of serum, washed three times with Tris-buffered saline containing 0.1% Tween 20. Then, the bound antibody was eluted with 0.1 M glycine buffer (pH 2.7) three times, and the eluted antibody was neutralized with 1/10 volume of 1.5 M Tris (pH 8.8).
Transfections and RNA interference
Transfections of plasmid DNA and small interfering RNAs (siRNAs, 20 nM) were performed using X-tremeGENE™ HP (Roche) and RNAiMAX (Thermo Fisher Scientific) respectively, according to the manufacturer’s instructions. Transfection reagent was removed 6 h post transfection and fresh medium was added. Cells were analyzed 48 h after transfection. The siRNA sequences used in this study were: ATAD5 3ʹ UTR (5ʹ-GUAUAUUUCUCGAUGUACA-3ʹ); UAF1 (5ʹ-AAUCAGCACAAGCAAGAUCCAUAUA-3ʹ); RFC4 (5ʹ-AAGAGAUUAGGAAGAUCUG-3ʹ).
Immunoprecipitation and immunoblot analysis
For immunoprecipitation, cells were lysed in ice-cold buffer X (100 mM Tris-HCl, 250 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1 M PMSF) with phosphatase inhibitors (Roche), protease inhibitors (Roche) and Benzonase nuclease (250 U/mL) followed by sonication and centrifugation. Target proteins within the lysate were immunoprecipitated with specific antibodies. For chromatin-bound fraction, Triton X-100 insoluble fractions were obtained according to the methods described previously [31] with slight modifications. In brief, cells were incubated with buffer A (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES (pH 6.8), 1 mM EGTA, 0.2% Triton X-100) containing phosphatase inhibitors and protease inhibitors for 5 min on ice followed by centrifugation. Then, the chromatin-bound fraction was isolated by resuspending the pellet in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate) containing 0.1 M PMSF, phosphatase inhibitors, protease inhibitors, and Benzonase nuclease for 30 min on ice followed by sonication and centrifugation. For whole cell extracts, cells were incubated in RIPA buffer with Benzonase nuclease for 10 min on ice followed by sonication and centrifugation.
For immunoblot analysis, proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were incubated in Tris-buffered saline containing 0.1% Tween 20 supplemented with 5% skim milk powder for blocking. Membranes were then incubated with primary antibodies overnight. After washing, the blot was incubated with horseradish peroxidase-conjugated secondary antibodies (Enzo Life Sciences). Signals were detected using enhanced chemiluminescence reagent (Thermo Fisher Scientific) and an automated imaging system (ChemiDoc™; Bio-Rad Laboratories).
Flow cytometry
Cells were washed with PBS, fixed with 70% ethanol for 1 h, and then incubated with 0.1 mg/mL RNase A at 37°C for 1 h. DNA was stained with 0.05 mg/mL propidium iodide. Flow cytometry was performed on a FACSVerse™ flow cytometer using BD FACSuite™ software (BD Biosciences). Data analysis was performed using FlowJo software.
Immunostaining
U2OS, HeLa, and NIH3T3 cells were plated on LabTek™ chamber slides (Thermo Fisher Scientific), and mESCs were plated on μ-Slide (ibidi). Cells were pre-extracted with CSK buffer (10 mM PIPES, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, and 0.5% Triton X-100) for 10 min at 4°C and fixed with 4% paraformaldehyde for 20 min at room temperature. The fixed cells were incubated with the primary antibodies diluted in PBS supplemented with 10% fetal bovine serum for 1 h at room temperature. After three washes with 0.05% Triton X-100 in PBS, Alexa Fluor®–conjugated secondary antibodies (Thermo Fisher Scientific) were added and incubated for 30 min. Cells were mounted using ProLong® Gold antifade reagent with DAPI (Vector Laboratories).
Image acquisition and analysis
Confocal images were acquired using LSM880 confocal microscope (Carl Zeiss) with 40x/1.2 lens objective. For all centrosome counting experiments, whole cell depth was covered by z-stack imaging method. Z-stack images were then combined by “maximum intensity projection” analysis method in “Zen 2.6 blue edition” (Carl Zeiss) software. For ID1 intensity analysis, each centriole was firstly masked and ID1 intensity within the masked regions was measured. This signal was then normalized by the size of the centriole.
Structural illumination images were acquired using a Zeiss ELYRA S.1 microscope (Carl Zeiss) with Plan-Apochromat 63x/1.4 Oil DIC. Green (488 nm) and Red (561 nm) channels were acquired by sequentially alternating the excitation whilst retaining the same setting with respect to phase, rotation, and multi-cube filterset. Images were acquired with five phases, five rotations. Green channel signal was acquired with a grating of 28.0 μm and red was 34.0 μm. Images were processed using the automatic reconstruction settings in the Zen software.
Statistical analysis
Prism 8 (GraphPad Software) was used to generate graphs and analyze data. For all data, two-tailed paired Student’s t-test was used; * p ≤ 0.05, ** p ≤ 0.01, ns, not significant. Statistical parameters are described in the figures and the figure legends.
Results
ATAD5 is localized at the centrosomes without forming an RLC complex
ATAD5 plays an important role at replication forks as a replisome protein [30,31,33]. Several replisome proteins have been reported to have additional, replication-independent roles. To investigate possible roles of ATAD5 in other cellular processes, we immunostained endogenous ATAD5 and visualized its cellular localization. ATAD5 signals were observed as small focal forms throughout the nucleus in U2OS human osteosarcoma cell line (Figure 1(a)). Interestingly, one or two large focal signals adjacent to each other have been observed at the nucleus edge or in the cytoplasmic region close to the nucleus. Since the numbers and locations of these focal signals were similar to those of centrosomes, cells were co-immunostained with antibodies to ATAD5 and γ-tubulin, which is a marker for centrosomes. We found that the large focal signals of ATAD5 were co-localized with either of the two γ-tubulin signals. The co-localization of ATAD5 and γ-tubulin was also observed in NIH3T3 cells, which is a mouse fibroblast cell line (Figure 1(b)) and in mouse embryonic stem cells (mESCs) (Figure 1(c)). These signals were completely missing in Atad5-knockout mESCs. These results indicate that the anti-ATAD5 antibody that we used specifically detects endogenous ATAD5. Additionally, when mNeonGreen-fused ATAD5 N-terminal domain was expressed in cells, mNeonGreen signals were co-localized with γ-tubulin signals (Figure 1(d)), which further supports our finding that ATAD5 is localized at the centrosomes.
Figure 1.
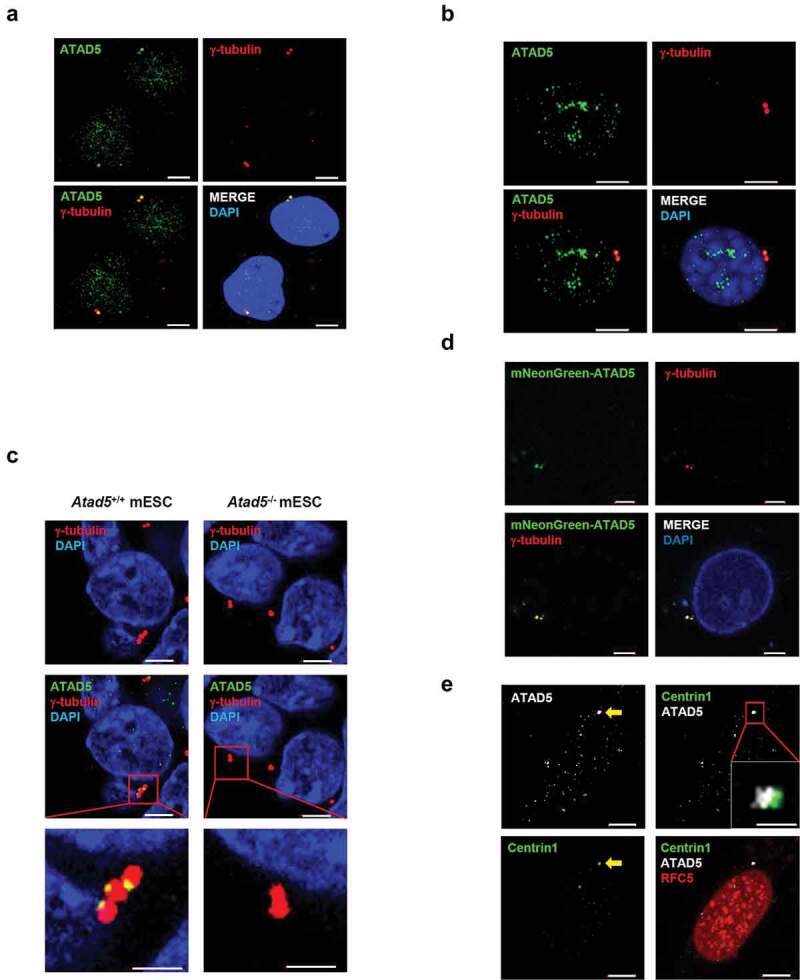
ATAD5 is localized at the centrosomes in an RFC independent manner.
(a) U2OS cells and (b) NIH3T3 cells were fixed at asynchronous condition for immunostaining. Bar, 5 μm. (c) The wild type (Atad5+/+) mouse embryonic stem cells (mESCs) and Atad5 knock out (Atad5−/-) mESCs were fixed for immunostaining. Bar, 5 μm. The inset shows high-magnification image of the centrosome. Bar, 2 μm. (d) U2OS cells were transfected with a DNA vector expressing mNeonGreen-fused ATAD5 N-terminal fragment (1–693 amino acids). After 48 h of transfection, cells were fixed for immunostaining as indicated. Bar, 5 μm. (e) HeLa cells stably expressing GFP-tagged Centrin1 (HeLa_Centrin1-GFP) were fixed at asynchronous condition for immunostaining. The arrow indicates a centrosome. Bar, 5 μm. The inset shows high-magnification image. Bar, 1 μm.
ATAD5 regulates PCNA at replication forks as a pentameric complex, ATAD5-RLC [32]. We examined whether ATAD5 localizes at the centrosome as a component of ATAD5-RLC by doing co-immunostaining of RFC5 protein, a small subunit of RLC. To visualize centrioles, we generated HeLa cells that constitutively express green fluorescence protein (GFP)-tagged Centrin1, a centriole protein (HeLa_Centrin1-GFP). RFC5 was observed throughout the nucleus as a punctate signal. However, unlike ATAD5, RFC5 did not co-localize with Centrin1 (Figure 1(e)). These results suggest that the localization of ATAD5 at the centrosome does not require the formation of the ATAD5-RLC complex.
ATAD5 is localized at the base of mother and daughter centrioles that undergo duplication
After disengagement of the mother and daughter centrioles, the pericentriolar matrix containing γ-tubulin ring complexes (γ-TuRCs) are recruited to the centrioles and surround them [1,2]. Since the large focal signals of ATAD5 were co-localized with either one or both of the γ-tubulin signals (Figure 1(a)), we further examined the co-localization of these two proteins at the centrosomes. ATAD5 signals were co-localized with two adjacent γ-tubulin signals with three different patterns. One of the two γ-tubulin signals always co-localized with ATAD5 signal, but the other showed no (i), intermediate (ii) or strong (iii) ATAD5 signal (Figure 2(a)). When we examined the co-localization of ATAD5 and Centrin2, a centriole protein, we obtained a similar pattern in general but structurally somewhat different results. ATAD5 signal was co-localized with one of the two adjacent Centrin2 signals (i, before maturation of daughter centriole) or both separated Centrin2 signals (ii, after centrosomal cohesion is lost and two matured centrosomes are separated at the G2/M transition) (Figure 2(b)). In both cases, however, the ATAD5 and Centrin2 signals were visually separated without overlapping. The conventional confocal microscopy was not enough to resolve the centriole pairs (Figure 2(b)). Therefore, we used a structured illumination microscopy to obtain high-resolution images of ATAD5 and centriole pairs. We could observe clearly discernible one or two centriole pairs (Figure 2(c)). We observed ATAD5 signals right beside a pair of Centrin1 signals. Considering the structure of a centriole pair, we concluded that ATAD5 is localized at the base of centriole pairs.
Figure 2.
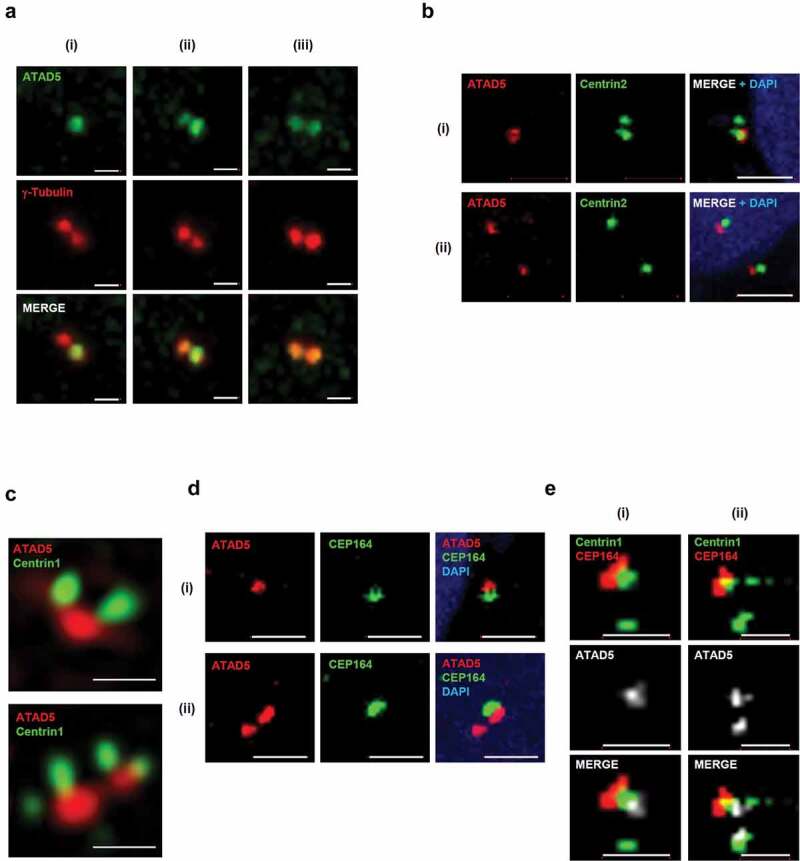
ATAD5 is localized at the base of mother and daughter centrioles that undergo duplication.
(a-e) U2OS cells (A, B, D) and HeLa_Centrin1-GFP cells (c, e) were fixed at asynchronous condition for immunostaining as indicated. Bar, 1 μm. Images were acquired using a confocal microscope (a, b, d, e) or a structured illumination microscopy (c).
Since ATAD5 signals are detected only at a single centrosome in some cells (Figure 2(a)), we hypothesized that ATAD5 is localized only at the mother centriole upon centriole disengagement and expands its localization at the daughter centriole as it matures. To test this hypothesis, cells were co-immunostained with antibodies to ATAD5 and CEP164, a marker for the appendages located at the top of the mother centriole. Until a daughter centriole matures into a mother centriole, it lacks appendages and CEP164 signal would be negative. ATAD5 signals were detected next to CEP164 (Figure 2(d)). However, ATAD5 signals were also detected without an adjacent CEP164 signal (Figure 2(d), ii). Immunostaining of HeLa_Centrin1-GFP cells with ATAD5 and CEP164 spotted ATAD5 at the centrosome in a more structurally ordered manner. ATAD5 signals were detected at the base of the mother centrioles, which is at the opposite end from the CEP164 signal (Figure 2(e)). Collectively, we conclude that ATAD5 primarily localizes to the mother centriole, but also accumulates at the daughter centriole even before it fully matures into the mother centriole.
ATAD5 depletion results in centrosome amplification
To obtain insights into the biological meaning of localization of ATAD5 at the centrosomes, we examined the effects of ATAD5 depletion on centrosome biology. Since ATAD5 begins to localize at the daughter centriole that undergoes duplication, we first enumerated the centrosomes. To more accurately examine the centrosome numbers, we used HeLa_Centrin1-GFP cells. Small interfering RNA (siRNA)-mediated ATAD5 depletion almost doubled the frequency of cells with centrosome over-duplication (3.05 ± 1.29%) compared to non-target siRNA treated cells (1.79 ± 0.95%). We then performed ATAD5 recovery experiments. Ectopic expression of ATAD5 reduced the frequency of cells with centrosome over-duplication to basal levels (1.31 ± 0.30%) (Figure 3(a)), indicating that the effect is ATAD5-specific. We obtained similar results with U2OS cells. ATAD5 depletion doubled the frequency of centrosome over-duplication (10.34 ± 2.62%) compared to non-target siRNA treated cells (4.38 ± 1.94%) (Figure 3(b)).
Figure 3.
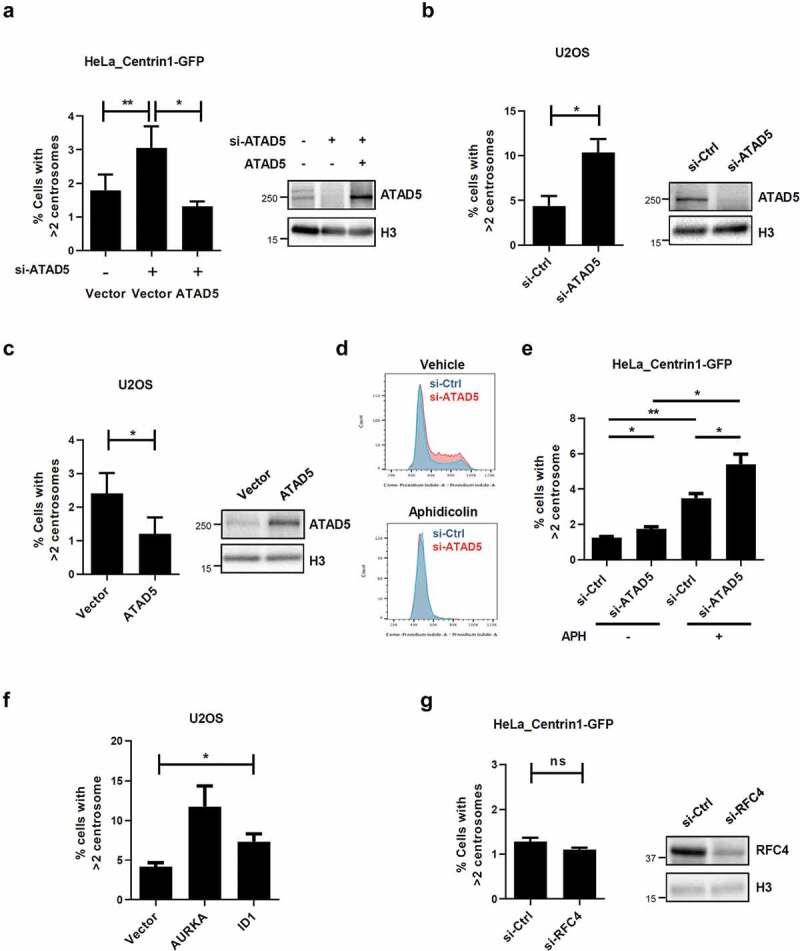
Centrosomes are over-duplicated in ATAD5-depleted cells.
(a-c, e-g) 48 h after transfection of siRNA or cDNA as indicated, cells were immunostained with an anti-γ-tubulin antibody. The percentage of cells with more than two centrosomes was displayed. Three independent experiments were performed and at least 300 cells were counted per experiment. Values represent mean ± SEM. Statistics: paired t-test. *, p ≤ 0.05, **, p ≤ 0.01, ns, not significant. (a-c, g) The right panels of graphs show immunoblot results from whole cell extracts prepared 48 h after transfection. (d, e) Cells were treated with 2 ug/mL aphidicolin (APH) for 24 h to arrest cells at the G1/S phase boundary. (d) Cells were fixed and DNA was stained with propidium iodide for cell cycle analysis.
Prolonged S phase has been reported to cause multiple rounds of centrosome duplication in a single S phase [40,41]. ATAD5 depletion delays S phase progression, [31] which could result in centrosome amplification. Therefore, we tested the effects of overexpression of ATAD5, which does not alter cell cycle profile (data not shown), on centrosome number. ATAD5 overexpression reduced the frequency of cells with centrosome amplification to 1.2 ± 0.99% compared to control cells (2.4 ± 1.23%) (Figure 3(c)), suggesting that a small delay in S phase progression by ATAD5 depletion is not the cause of centrosome amplification. To completely rule out the effect of S phase delay upon ATAD5 depletion, we arrested cells at the G1/S boundary using polymerase α inhibitor, aphidicolin, which is commonly used for centrosome duplication studies (Figure 3(d)). We found that ATAD5 depletion increased centrosome over-duplication even when cells were arrested at the G1/S boundary (Figure 3(e)). Overexpression of Aurora A kinase or ID1 has been reported to cause an increase in cells with centrosome over-duplication [13,16,42]. The level of defects caused by overexpression of Aurora A kinase or ID1 in our experimental conditions (figure 3(f)) was comparable to that caused by ATAD5 depletion, which suggests that the observed effects of ATAD5 depletion on centrosome numbers are significant.
We also tested whether small subunits of RLC were involved in centrosome duplication. In line with our previous finding that ATAD5 is localized at centrosomes without forming an RLC complex (Figure 1(e)), RFC4 depletion did not affect the frequency of cells with centrosome amplification (Figure 3(g)). Taken together, these results suggest that ATAD5 is important for the proper regulation of centrosome duplication and this regulation is independent of RLC formation.
ATAD5 depletion results in multipolar segregation
Centrosomes mediate mitotic microtubule organization. A pair of centrosomes is pulled to the opposite poles and drives stable bipolar segregation of chromosomes to facilitate correct cell division. Therefore, over-duplicated centrosomes or fragmented centrosomes often cause multipolar segregation leading to abnormal distribution of chromosomes [2]. Since ATAD5 depletion results in supernumerary centrosomes (Figure 3), we speculated that it would eventually lead to an increase in cells with multipolar segregation. Cells were co-immunostained with antibodies to α-tubulin and NUF2, a centromere protein, to clearly visualize chromosome segregation. As expected, ATAD5-depleted U2OS cells showed significantly increased events of multipolar segregation (4.48 ± 2.09%) compared to the control cells (1.89 ± 0.31%) (Figure 4(a,b)). Furthermore, this phenotype was consistently observed in HeLa cells (Figure 4(c,d); 1.56 ± 0.28% vs. 5.72 ± 0.53%) and ATAD5 knockout U2OS cells (Figure 4(e); 2.81 ± 0.31% vs. 8.01 ± 0.71%). Apart from the defects in multipolar segregation, we did not observe any significant defects in centriole or microtubule orientation in ATAD5-depleted cells (Figure 4(c)). Even in ATAD5-depleted cells that undergo multipolar segregation, there were two centrioles at each pole and were focused at microtubule minus ends (Figure 4(c)). However, our findings clearly suggest that ATAD5 is required for accurate centrosome duplication and is further responsible for correct bipolar segregation.
Figure 4.
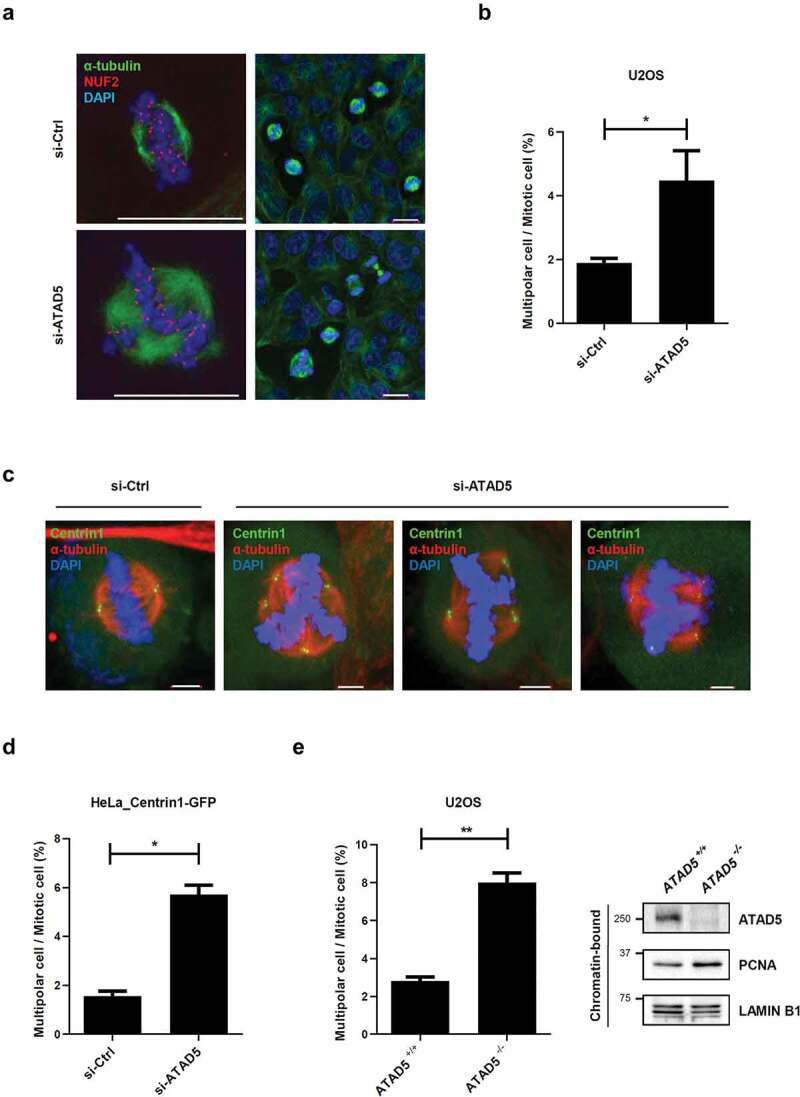
Bipolar segregation is defective in ATAD5-depleted cells.
(a-d) U2OS cells (a, b) or HeLa_Centrin1-GFP cells (c, d) were transfected with ATAD5 siRNA for 48 h before fixation for immunostaining. (e) Wild type or ATAD5 knockout U2OS cells were fixed for immunostaining. (a-e) Cells were arrested at the G2 phase by treatment of 10 μM RO3306, a CDK1 inhibitor, for 20 h and released in the fresh media for 1 h (a, b) and 1.5 h (c, d, e) before fixation. (a, c) Representative images. Bar, 20 μm (a) and 5 μm (@). (b, d, e) The percentage of cells with multipolar segregation was displayed. Three independent experiments were performed and at least 100 cells were counted per experiment. Values represent mean ± SEM. Statistics: paired t-test. *, p ≤ 0.05. (e) The right panel of graph shows immunoblot results from chromatin-bound protein fraction.
UAF1 and ID1 interact, and co-localize at the centrosomes
Since ATAD5 interacts with the USP1/UAF1 complex and USP1 has been reported to regulate centrosome duplication by increasing ID1 levels [17,34], we speculated that ATAD5 modulates ID1 to regulate centrosome duplication. We first tested whether ATAD5 physically interacts with ID1. When we immunoprecipitated ATAD5 or ID1, we could not detect either proteins in immunoprecipitates from each pull-down assay (data not shown). Instead, we found that UAF1, a strong interacting partner of ATAD5 and USP1, interacted with ID1. FLAG-tagged UAF1 was immunoprecipitated with HA-tagged ID1 (Figure 5(a)). This interaction was also confirmed by pulling down HA-tagged ID1 (Figure 5(b)). It has been previously reported that UAF1 stabilizes USP1 in vitro [37] and that USP1 increases ID1 protein levels [18]. However, the protein levels of USP1 and ID1 were not affected by UAF1 overexpression in our experimental conditions (Figure 5(a)). We also found that the protein levels of USP1 and ID1 were not affected by UAF1 depletion (Figure 5(c)). Different from the previous studies where gene knockout or small hairpin RNA was used to deplete UAF1 [35,39,43], transient depletion by siRNA does not appear to decrease USP1 levels.
Figure 5.
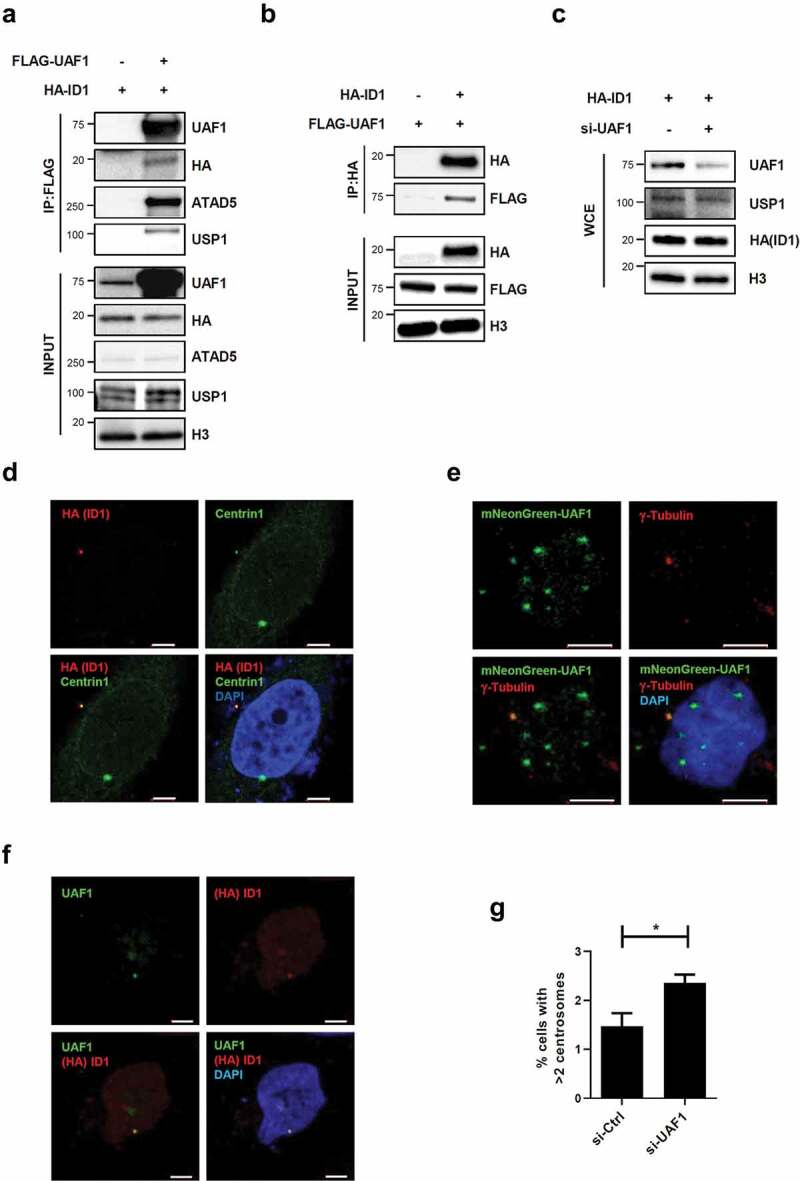
UAF1 and ID1 interact, and co-localize at the centrosome.
(a, b) HEK293 T cells were transfected with 3xFLAG-tagged UAF1 cDNA (FLAG-UAF1) and HA-tagged ID1 cDNA (HA-ID1). (a, b) After 48 h, whole cell extracts were prepared for immunoprecipitation (IP) with an anti-FLAG antibody (A) and an anti-HA antibody (b). Immunoprecipitates were eluted, resolved by SDS-PAGE, and immunoblotted using the indicated antibodies. (c) HEK293 T cells were transfected with a combination of UAF1 siRNA and HA-ID1 cDNA. After 48 h of transfection, whole cell extracts (WCE) were subjected for immunoblotting. (d-f) After 48 h of transfection, cells were fixed for immunostaining as indicated. HeLa_Centrin1-GFP cells (d) and HeLa cells (f) were transfected with HA-ID1 cDNA. (e) HeLa cells were transfected with mNeonGreen-fused UAF1 cDNA. Bar, 5 μm. (g) HeLa_Centrin1-GFP cells were transfected with UAF1 siRNA for 48 h and fixed. The percentage of cells with more than two centrosomes was displayed. Three independent experiments were performed. Values represent mean ± SEM. Statistics: paired t-test. *, p ≤ 0.05.
We also examined the centrosomal localization of ID1 and UAF1 proteins. HA-tagged ID1 was co-localized with Centrin1 (Figure 5(d)), as previously reported [13]. Immunostaining of UAF1 showed punctuation signals and one of signals was co-localized with γ-tubulin (Figure 5(e)). However, we could not detect USP1 signals at the centrosomes. As expected, co-immunostaining of UAF1 and ID1 showed co-localization of the two proteins (figure 5(f)). Interestingly, UAF1 depletion increased the frequency of cells with centrosome amplification (2.36 ± 0.29%) compared to non-target siRNA treated cells (1.47 ± 0.46%) (Figure 5(g)). These data suggest that the interrelationship between ATAD5, UAF1, and ID1 is important for centrosome regulation and that the effect of ATAD5 depletion on centrosome duplication is not mediated by changes in USP1 and ID1 protein levels.
ATAD5 regulates interactions between UAF1 and ID1
Based on the binding of UAF1 with both ATAD5 and ID1, we investigated whether ATAD5 is required for the interaction between UAF1 and ID1 proteins. We found that ATAD5 depletion reduced interactions between UAF1 and ID1 in whichever ways the proteins were pulled down (Figure 6(a,b)). Conversely, overexpression of ATAD5 increased interactions between UAF1 and ID1 proteins (Figure 6(c)). Neither ATAD5 depletion nor overexpression affected the total protein level of ID1 (Figure 6(a-d)). These data suggest that ATAD5 regulates interactions between UAF1 and ID1 and this regulatory mechanism for ID1 is different from USP1-mediated ID1 regulation.
Figure 6.
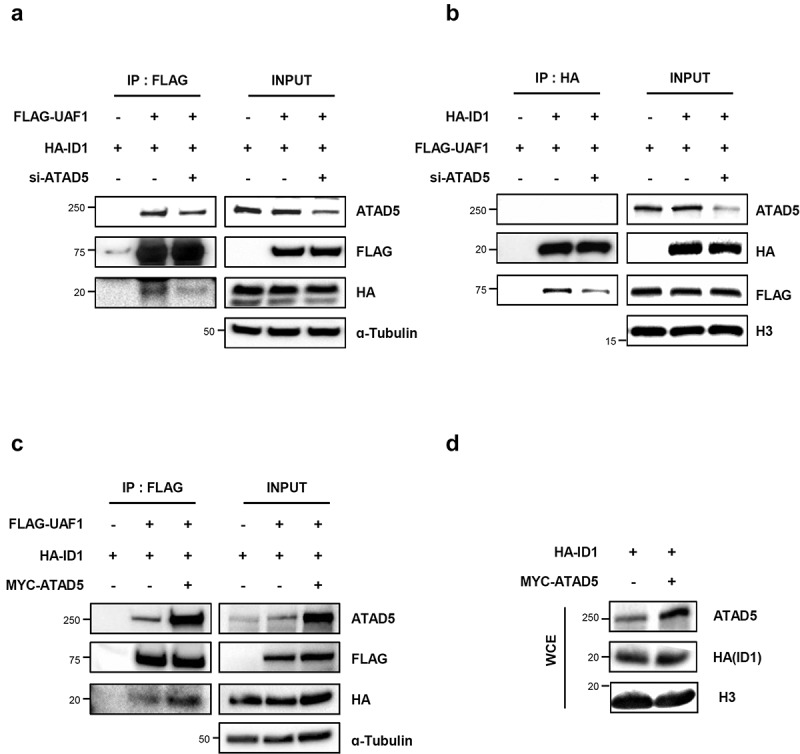
ATAD5 regulates interactions between UAF1 and ID1.
(a-c) HEK293 T cells were transfected with a combination of ATAD5 siRNA, 3xFLAG-tagged UAF1 (FLAG-UAF1) cDNA, MYC-tagged ATAD5 (MYC-ATAD5) cDNA, and HA-tagged ID1 (HA-ID1) cDNA. After 48 h, whole cell extracts were prepared for immunoprecipitation (IP) with an anti-FLAG antibody (a, c) and an anti-HA antibody (b). Immunoprecipitates were eluted, resolved by SDS-PAGE, and immunoblotted using the indicated antibodies. (d) HEK293 T cells were transfected with MYC-ATAD5 cDNA and HA-ID1 cDNA. After 48 h, whole cell extracts were resolved by SDS-PAGE and immunoblotted using the indicated antibodies.
ATAD5 depletion increases ID1 signals at the centrosomes
Next, we checked the effects of ATAD5 depletion on ID1 at the centrosomes. We measured the mean intensities of ID1 signals at the centrioles and normalized them by numbers and sizes of centrioles. We found that ID1 signals, specifically at the centrosomes, were increased in ATAD5-depleted cells compared to control cells (Figure 7(a and b)), and this was consistent in ATAD5 knockout U2OS cells (Figure 7(c and d)). These data suggest that ATAD5 modulates the localization of ID1 at the centrosomes.
Figure 7.
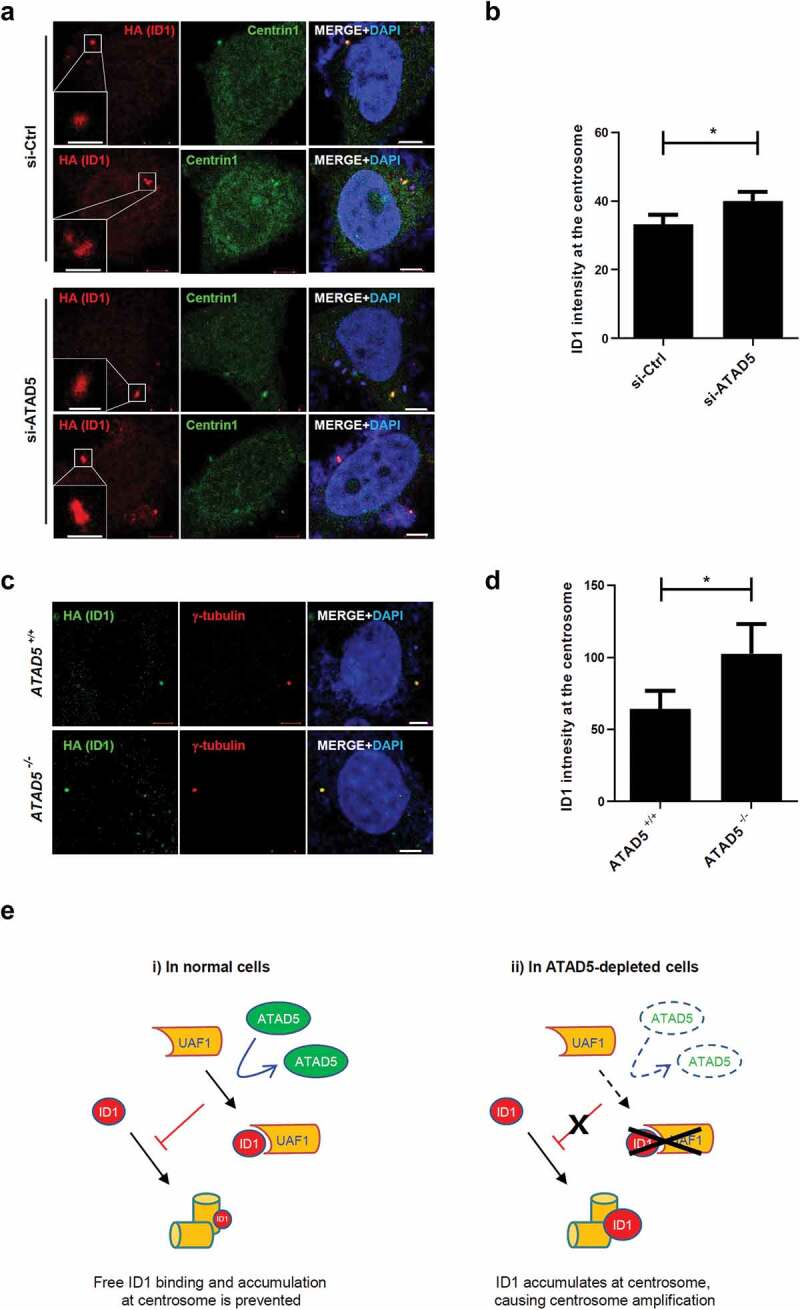
ATAD5 depletion increases ID1 signals at the centrosomes.
(a-d) HeLa_Centrin1-GFP cells (a, b) or ATAD5 knockout U2OS cells (c, d) were transfected with ATAD5 siRNA and HA-ID1 cDNA for 48 h and fixed for immunostaining. Bar, 5 μm. (a, c) Representative images. The inset shows high-magnification image. Bar, 2 μm. (b, d) Mean HA signal intensities at the centrosome were measured and displayed. Three independent experiments were performed. Values represent mean ± SEM. Statistics: paired t-test. *, p ≤ 0.05. (e) A graphical model of how ID1 and UAF1 fit in the regulation of centrosome by ATAD5. In normal cells, ATAD5 facilitates interactions between UAF1 andID1 and therefore inhibits free ID1 from accumulating at the centrosome. However, in the absence of ATAD5, ID1 is released from UAF1, thereby free ID1 accumulates at centrosome, leading to centrosome defects.
Discussion
We have demonstrated that ATAD5 and UAF1 are localized at the centrosomes and that both these proteins play important roles in centrosome regulation. How ATAD5 controls centrosome duplication and how its dysregulation causes centrosome amplification is not yet fully defined, but several possibilities are suggested based on previous works and our studies. ATAD5 depletion increases ID1 signals at the centrosomes (Figure 7(a-d)). Since ID1 overexpression has been previously reported to lead to centrosome amplification [13], increased ID1 expression at the centrosomes can result in centrosome amplification in ATAD5-depleted cells. Deubiquitination activity of USP1 has been reported to increase ID1 protein levels [17,18]. Although ATAD5 depletion does not alter protein levels of USP1 [34], since UAF1 increases USP1 activity [37], it is still possible that ATAD5 regulates deubiquitination activity of USP1 through UAF1. However, depletion or overexpression of ATAD5 or UAF1 did not alter the protein levels of ID1 (Figures 5 and 6). Collectively, these results indicate that in ID1 regulation, ATAD5 appears to use a different regulatory mechanism than USP1. Instead, we found that ID1 interacts with UAF1 and this interaction is mediated by ATAD5 (Figure 6(a-c)). However, we could not detect ATAD5 in ID1 immunoprecipitates (Figure 6(b)). Therefore, it is speculated that UAF1 physically interacts with ID1 under certain conditions, and ATAD5 can promote their interactions in a temporary manner (Figure 7(e, i)). Although relative contributions of nuclear UAF1 and centrosomal UAF1 is not clear, one conjecture based on the observation that ATAD5 depletion increases ID1 levels at the centrosomes is as follows; UAF1 sequesters ID1, which is promoted by ATAD5, and this consequently inhibits hyper-accumulation of free ID1 at the centrosomes (Figure 7(e, i)). Therefore, ATAD5 depletion releases ATAD5/UAF1-bound ID1, which in turn results in increase of centrosomal ID1 localization and centrosome over-duplication (Figure 7e, ii). This might explain the unaltered levels of total ID1 protein, together with altered UAF1/ID1 interactions upon ATAD5 depletion. Alternatively, ATAD5/UAF1 might control centrosomal ID1 levels by direct protein binding and its regulatory dysfunction can lead to ID1 accumulation.
It is also possible that ATAD5 regulates other centrosomal proteins that participate in centriole duplication by protein–protein interactions. After centriole disengagement, ATAD5 accumulates at the base of the daughter centriole before it fully matures into the mother centriole (Figure 2(d)). This is the period when procentrioles start to form and elongate at the base of the mother and daughter centrioles. Centrosomal proteins that localize at the proximal end of the centriole participate in centriole duplication and procentriole formation processes. Sas-6 promotes procentriole formation [44], and it also stabilizes centriole intermediates [45]. Recruitment of Sas-6 to the centrioles is promoted by Polo-like kinase 4 (Plk4) and SCL/TAL1 interrupting locus (STIL) [46]. In addition, CEP135 interacts with centrosomal P4.1-associated protein (CPAP), which regulates procentriole formation and its elongation [47,48]. Loss of any of these proteins, Plk4, STIL, Sas-6, or CEP164, blocks centriole elongation and sequentially inhibits centrosome duplication. Conversely, overexpression of these proteins results in centriole amplification. ATAD5 uses C-terminal ATPase and RFC interaction domains to regulate PCNA unloading [32] while participating in other cellular processes by interacting with other related proteins through its N-terminal domain [31,49]. Mass spectrometry-based interactome analysis showed that ATAD5 is likely to interact with centrosome proteins [50]. Our preliminary mass spectrometry analysis also spotted out several proteins related to centrosome regulation, including Plk1 and Aurora B kinase as ATAD5-interacting proteins. Based on these results, ATAD5 that localizes at the base of the centriole likely regulates centrosomal proteins by protein–protein interactions.
ATAD5 has been studied with an emphasis on its role as a replisome protein associated with DNA replication and repair. ATAD5 is primarily detected in Triton X-100 insoluble protein fractions containing chromatin-bound and nuclear matrix-bound proteins [31]. Consistently, ATAD5 has a putative nuclear localization signal sequence at the N-terminal regions [32]. In this study, we unexpectedly found that ATAD5 is present at the centrosomes. Given that RFC5 is not detected at the centrosomes (Figure 1(e)), it is possible that ATAD5 takes a different conformation at the centrosomes compared to when it is in a complex with other RLC subunits. It is still not clear how the N-terminal domain of ATAD5 is structurally located relative to the C-terminal domain. Since the N-terminal half of ATAD5 interacts with many other proteins, binding to centrosome-specific proteins might interfere with proper nuclear localization or RLC formation. ATAD5 immunostaining with purified anti-ATAD5 antibody shows clearer signals at the centrosomes than at the nucleus (Figure 1). It is likely that the polyclonal antibodies raised against the N-terminal portion of ATAD5 do not have good accessibility to ATAD5 when it forms the RLC complex, but instead, efficiently binds to ATAD5 when it exists as a non-complex at the centrosomes. The N-terminal region of ATAD5 likely plays an important role in its localization to the centrosome, independent of RLC, and further, regulates centrosome biology through its unique conformation.
Funding Statement
This work was supported by the Institute for Basic Science [IBS-R022-D1]; UNIST research fund [1.180063].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Doxsey S, McCollum D, Theurkauf W.. Centrosomes in cellular regulation. Annu Rev Cell Dev Biol. 2005;21(1):411–434. [DOI] [PubMed] [Google Scholar]
- [2].Nam HJ, Naylor RM, van Deursen JM.. Centrosome dynamics as a source of chromosomal instability. Trends Cell Biol. 2015;25:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fu J, Hagan IM, Glover DM. The centrosome and its duplication cycle. Cold Spring Harb Perspect Biol. 2015;7:a015800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gonczy P. Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer. 2015;15:639–652. [DOI] [PubMed] [Google Scholar]
- [5].Nigg EA, Cajanek L, Arquint C. The centrosome duplication cycle in health and disease. FEBS Lett. 2014;588:2366–2372. [DOI] [PubMed] [Google Scholar]
- [6].Vitre BD, Cleveland DW. Centrosomes, chromosome instability (CIN) and aneuploidy. Curr Opin Cell Biol. 2012;24:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hemerly AS, Prasanth SG, Siddiqui K, et al. Orc1 controls centriole and centrosome copy number in human cells. Science. 2009;323(5915):789–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Prasanth SG, Prasanth KV, Siddiqui K, et al. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J. 2004;23(13):2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Xu X, Huang S, Zhang B, et al. DNA replication licensing factor Cdc6 and Plk4 kinase antagonistically regulate centrosome duplication via Sas-6. Nat Commun. 2017;8(1):15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ferguson RL, Maller JL. Cyclin E-dependent localization of MCM5 regulates centrosome duplication. J Cell Sci. 2008;121(19):3224–3232. [DOI] [PubMed] [Google Scholar]
- [11].Bang SW, Ko MJ, Kang S, et al. Human TopBP1 localization to the mitotic centrosome mediates mitotic progression. Exp Cell Res. 2011;317(7):994–1004. [DOI] [PubMed] [Google Scholar]
- [12].Perk J, Iavarone A, Benezra R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5(8):603–614. [DOI] [PubMed] [Google Scholar]
- [13].Hasskarl J, Duensing S, Manuel E, et al. The helix-loop-helix protein ID1 localizes to centrosomes and rapidly induces abnormal centrosome numbers. Oncogene. 2004;23:1930–1938. [DOI] [PubMed] [Google Scholar]
- [14].Manthey C, Mern DS, Gutmann A, et al. Elevated endogenous expression of the dominant negative basic helix-loop-helix protein ID1 correlates with significant centrosome abnormalities in human tumor cells. BMC Cell Biol. 2010;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hasskarl J, Mern DS, Munger K. Interference of the dominant negative helix-loop-helix protein ID1 with the proteasomal subunit S5A causes centrosomal abnormalities. Oncogene. 2008;27:1657–1664. [DOI] [PubMed] [Google Scholar]
- [16].Man C, Rosa J, Yip YL, et al. Id1 overexpression induces tetraploidization and multiple abnormal mitotic phenotypes by modulating aurora A. Mol Biol Cell. 2008;19:2389–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jung JK, Jang SW, Kim JM. A novel role for the deubiquitinase USP1 in the control of centrosome duplication. Cell Cycle. 2016;15:584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Williams SA, Maecker HL, French DM, et al. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell. 2011;146:918–930. [DOI] [PubMed] [Google Scholar]
- [19].Kubota T, Myung K, Donaldson AD. Is PCNA unloading the central function of the Elg1/ATAD5 replication factor C-like complex? Cell Cycle. 2013;12:2570–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fox JT, Lee KY, Myung K. Dynamic regulation of PCNA ubiquitylation/deubiquitylation. FEBS Lett. 2011;585:2780–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bellaoui M, Chang M, Ou J, et al. Elg1 forms an alternative RFC complex important for DNA replication and genome integrity. EMBO J. 2003;22:4304–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ben-Aroya S, Koren A, Liefshitz B, et al. ELG1, a yeast gene required for genome stability, forms a complex related to replication factor C. Proc Natl Acad Sci U S A. 2003;100:9906–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kanellis P, Agyei R, Durocher D. Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr Biol. 2003;13:1583–1595. [DOI] [PubMed] [Google Scholar]
- [24].Banerjee S, Myung K. Increased genome instability and telomere length in the elg1-deficient Saccharomyces cerevisiae mutant are regulated by S-phase checkpoints. Eukaryot Cell. 2004;3:1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sikdar N, Banerjee S, Lee KY, et al. DNA damage responses by human ELG1 in S phase are important to maintain genomic integrity. Cell Cycle. 2009;8:3199–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bell DW, Sikdar N, Lee KY, et al. Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLoS Genet. 2011;7:e1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Maleva Kostovska I, Wang J, Bogdanova N, et al. Rare ATAD5 missense variants in breast and ovarian cancer patients. Cancer Lett. 2016;376:173–177. [DOI] [PubMed] [Google Scholar]
- [28].Kuchenbaecker KB, Ramus SJ, Tyrer J, et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet. 2015;47:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Park SH, Kang N, Song E, et al. ATAD5 promotes replication restart by regulating RAD51 and PCNA in response to replication stress. Nat Commun. 2019;10:5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kubota T, Nishimura K, Kanemaki MT, et al. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol Cell. 2013;50:273–280. [DOI] [PubMed] [Google Scholar]
- [31].Lee KY, Fu H, Aladjem MI, et al. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J Cell Biol. 2013;200:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kang MS, Ryu E, Lee SW, et al. Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat Commun. 2019;10:2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shiomi Y, Nishitani H. Alternative replication factor C protein, Elg1, maintains chromosome stability by regulating PCNA levels on chromatin. Genes Cells. 2013;18:946–959. [DOI] [PubMed] [Google Scholar]
- [34].Lee KY, Yang K, Cohn MA, et al. Human ELG1 regulates the level of ubiquitinated proliferating cell nuclear antigen (PCNA) through Its interactions with PCNA and USP1. J Biol Chem. 2010;285:10362–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cohn MA, Kee Y, Haas W, et al. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J Biol Chem. 2009;284:5343–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liang F, Longerich S, Miller AS, et al. Promotion of RAD51-mediated homologous DNA pairing by the RAD51AP1-UAF1 complex. Cell Rep. 2016;15:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cohn MA, Kowal P, Yang K, et al. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell. 2007;28:786–797. [DOI] [PubMed] [Google Scholar]
- [38].Cukras S, Lee E, Palumbo E, et al. The USP1-UAF1 complex interacts with RAD51AP1 to promote homologous recombination repair. Cell Cycle. 2016;15:2636–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Murai J, Yang K, Dejsuphong D, et al. The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Mol Cell Biol. 2011;31:2462–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Meraldi P, Lukas J, Fry AM, et al. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93. [DOI] [PubMed] [Google Scholar]
- [41].Balczon R, Bao L, Zimmer WE, et al. Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol. 1995;130:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 2002;21:483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Park E, Kim JM, Primack B, et al. Inactivation of Uaf1 causes defective homologous recombination and early embryonic lethality in mice. Mol Cell Biol. 2013;33:4360–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Strnad P, Leidel S, Vinogradova T, et al. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev Cell. 2007;13:203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yoshiba S, Tsuchiya Y, Ohta M, et al. HsSAS-6-dependent cartwheel assembly ensures stabilization of centriole intermediates. J Cell Sci. 2019;132(12). [DOI] [PubMed] [Google Scholar]
- [46].Vulprecht J, David A, Tibelius A, et al. STIL is required for centriole duplication in human cells. J Cell Sci. 2012;125:1353–1362. [DOI] [PubMed] [Google Scholar]
- [47].Chang J, Cizmecioglu O, Hoffmann I, et al. PLK2 phosphorylation is critical for CPAP function in procentriole formation during the centrosome cycle. EMBO J. 2010;29:2395–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tang CJ, Fu RH, Wu KS, et al. CPAP is a cell-cycle regulated protein that controls centriole length. Nat Cell Biol. 2009;11:825–831. [DOI] [PubMed] [Google Scholar]
- [49].Wessel SR, Mohni KN, Luzwick JW, et al. Functional analysis of the replication fork proteome identifies BET proteins as PCNA regulators. Cell Rep. 2019;28:3497–509 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hein MY, Hubner NC, Poser I, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. [DOI] [PubMed] [Google Scholar]