

ABSTRACT
Major histocompatibility complex class I (MHC-I) is a key molecule in anti-tumor adaptive immunity. MHC-I is essential for endogenous antigen presentation by cancer cells and subsequent recognition and clearance by CD8+ T cells. Defects in MHC-I expression occur frequently in several cancers, leading to impaired antigen presentation, immune evasion and/or resistance to immune checkpoint blockade (ICB) therapy. Pancreatic ductal adenocarcinoma (PDAC), a deadly malignancy with dismal patient prognosis, is resistant to ICB and shows frequent downregulation of MHC-I independent of genetic mutations abrogating MHC-I expression. Previously, we showed that PDAC cells exhibit elevated levels of autophagy and lysosomal biogenesis, which together support the survival and growth of PDAC tumors via both cell-autonomous and non-cell-autonomous mechanisms. In our recent study, we have identified NBR1-mediated selective macroautophagy/autophagy of MHC-I as a novel mechanism that facilitates immune evasion by PDAC cells. Importantly, autophagy or lysosome inhibition restores MHC-I expression, leading to enhanced anti-tumor T cell immunity and improved response to ICB in transplanted tumor models in syngeneic host mice. Our results highlight a previously unknown function of autophagy and the lysosome in regulation of immunogenicity in PDAC, and provide a novel therapeutic strategy for targeting this deadly disease.
KEYWORDS: Pancreatic cancer, autophagy, lysosome, MHC-I, anti-tumor immunity, immune checkpoint blockade
MHC-I is frequently dysregulated in cancer. Therefore, we investigated the expression level and localization of MHC-I in PDAC [1]. We found significant enrichment of MHC-I within intracellular punctate structures in PDAC cells that colocalize with markers of autophagosomes and lysosomes. Moreover, MHC-I is enriched in intact lysosomes purified from PDAC cells. Importantly, intracellular MHC-I localization is also observed in resected PDAC patient specimens. Moreover, inhibition of autophagy or the lysosome, but not LC3-associated phagocytosis (LAP) or endocytosis (LANDO), increases MHC-I levels and plasma membrane (PM) localization in PDAC cells, confirming the role of the autophagy-lysosomal pathway in regulation of MHC-I. Indeed, we identified the selective autophagy receptor NBR1 as an interactor of MHC-I and confirmed colocalization of NBR1 with MHC-I in PDAC cells, which requires the ubiquitin-associated (UBA) domain of NBR1. Consistent with this result, we also found that MHC-I is poly-ubiquitylated in PDAC cells. Importantly, NBR1 knockdown restores MHC-I levels and restores PM localization in PDAC cells. Together, these data indicate that MHC-I is actively degraded via NBR1-mediated selective autophagy in PDAC.
We hypothesized that restoration of PM MHC-I following autophagy-inhibition may improve anti-tumor immune responses against PDAC. Indeed, autophagy-inhibited PDAC cells exhibit increased MHC-I-restricted antigen presentation, leading to enhanced CD8+ T cell activation and tumor-cell killing in in vitro co-culture experiments. Using syngeneic transplantation experiments in mice, we further showed that autophagy-deficient PDAC cells or PDAC cells with low basal autophagy flux – isolated using a fluorescent reporter – similarly display increased MHC-I expression, more CD8+ T cell infiltration and give rise to smaller tumors in vivo. As expected, depletion of CD8+ T cells abrogates these phenotypes, confirming a role for adaptive immune responses in regulating the growth of autophagy-deficient tumors. Furthermore, autophagy inhibition, either genetically or pharmacologically with chloroquine, synergizes with ICB, an important finding given the resistance of human PDAC patients to immunotherapy.
We found that autophagy-inhibition in PDAC cells increases intratumoral CD8+ T cells and dendritic cells (DCs). Importantly, the immune response is likely a direct consequence of MHC-I restoration, as CD8+ T cell and DC infiltration is abrogated by depleting cell surface MHC-I on autophagy-deficient PDAC cells. Thus, our model proposes that: (1) autophagy inhibition restores MHC-I expression on the PM of PDAC cells; (2) improves recognition of tumor antigens by CD8+ T cells leading to activation of tumor-infiltrating DCs (possibly through release of cytokines such as IFNG/IFN-γ); and (3) activated DCs augment adaptive immune response by promoting CD8+ T cell priming and trafficking into the tumor (Figure 1). This model suggests that tumors containing preexisting CD8+ T cells or expressing strong antigens may show greater anti-tumor T cell responses upon autophagy inhibition. Accordingly, tumors completely lacking preexisting CD8+ T cells or neo-antigen expression might show minimal response to autophagy-inhibition. Indeed, these factors – lack of tumor-infiltrating CD8+ T cells and low mutational burden – have long been attributed to the lack of response to ICB of PDAC tumors. However, recent evidence has revealed that human PDAC tumors display varying amounts of infiltrating CD8+ T cells, with considerable intratumoral heterogeneity, and can harbor antigens capable of eliciting potent T cell immunity. Therefore, therapies such as chemotherapy or radiation may lead to cancer cell death and release of antigen, and promote antigen uptake and CD8+ T cell priming by DCs, further improving the efficacy of autophagy-lysosome inhibition plus ICB. Consistent with this finding, a recent study showed that combining hydroxychloroquine (HCQ) with chemotherapy can significantly increase anti-tumor responses and can lead to increased immune cell infiltration into tumors. Therefore, it is possible that using an initial combination of HCQ with chemotherapy to increase tumor cell killing, followed by HCQ combined with ICB to elicit an effective anti-tumor immune response, may lead to greater tumor inhibition.
Figure 1.
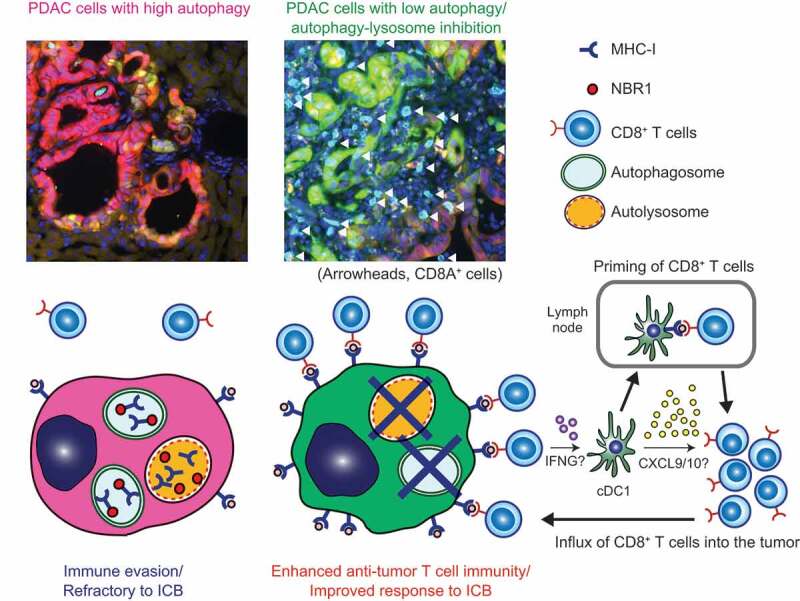
Basal autophagy flux as a determinant of immunogenicity of PDAC. (Top) Representative images of liver metastasis of mouse PDAC cells carrying the GFP-LC3-RFP autophagy reporter. CD8+ T cells (cyan) are scarce around PDAC cells with high autophagy flux (red), whereas PDAC cells with low or impaired autophagy flux (green) are surrounded by abundant CD8+ T cells (white arrowheads). (Bottom) Proposed model. (Left) Cell surface MHC-I expression is decreased by active degradation through NBR1-mediated autophagy, enabling PDAC cells to escape from immune surveillance. (Right) Cells with low autophagy flux or autophagy deficiency exhibit increased MHC-I expression, leading to enhanced recognition and killing by CD8+ T cells and improved response to immune checkpoint blockade (ICB). CD8+ T cells that recognize tumor cells then secrete molecules including IFNG/IFN-γ, which stimulate tumor-infiltrating conventional type 1 dendritic cells (cDC1), leading to further priming and recruitment of CD8+ T cells into the tumor.
Finally, there are several factors to be investigated in order to successfully translate these findings to the clinic. First, given the previously reported favorable and unfavorable effects of autophagy inhibition on immune cell types including CD4+ and CD8+ T cells, DCs and macrophages, the net effect on anti-tumor immune response requires additional investigation. Second, given the generally poor potency of HCQ, it is possible that more potent inhibitors are required to induce significant clinical responses. Third, the sequence and duration of treatment with chemotherapy, HCQ and ICB needs to be rigorously determined. Lastly, it may be important to establish whether autophagy inhibition at an early (autophagosome biogenesis) or late (lysosomal degradation) stage will induce the greatest anti-tumor response. Deeper insights into these questions may increase the likelihood of successful translation of these findings into patients.
Disclosure statement
A.C.K. has financial interests in Vescor Therapeutics, LLC. A.C.K. is an inventor on patents pertaining to KRAS regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, and the autophagic control of iron metabolism. A.C.K. is on the SAB of Rafael/Cornerstone Pharmaceuticals. A.C.K. is a consultant for Deciphera. The other authors declare no competing interests.
Reference
- [1].Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by downregulating MHC-I. Nature. 2020;581(7806):100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]