

ABSTRACT
Macroautophagy/autophagy is critical in maintaining cellular functions and homeostasis. Dynamic regulation of autophagy is associated with development of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH); however, the mechanisms involved in the regulation of autophagy in NAFLD/NASH are not well understood. Here we discuss our recent work identifying MLKL as an important nexus between autophagy and necroptosis in models of NAFLD/NASH. Mlkl, but not Ripk3, deficiency protects mice from Western diet-induced liver injury. Mlkl deficiency also prevents the accumulation of SQSTM1/p62 and LC3-II in liver in response to Western diet feeding or challenge with the protease inhibitor leupeptin. Western diet increases expression, phosphorylation and oligomerization of MLKL. In hepatocytes, palmitic acid (PA) induces the expression and translocation of MLKL to autophagosomes prior to the plasma membrane. Importantly, Mlkl, but not Ripk3, deficiency prevents the inhibition of autophagy by PA or chloroquine in hepatocytes. In contrast, overexpression of Mlkl blocks autophagic flux. Importantly, inhibition of autophagy by leupeptin or chloroquine triggers MLKL translocation to the plasma membrane, suggesting that MLKL is intimately involved in the regulation of autophagy under multiple conditions. These data indicate that MLKL contributes to Western diet-induced liver injury through inhibition of autophagy and induction of necroptosis.
KEYWORDS: Autophagic flux, LC3, MLKL, necroptosis, RIPK3, Western diet
Autophagy is an important physiological process for removing injurious intracellular components and maintaining cellular homeostasis. Dynamic regulation of autophagy is associated with development of NAFLD/NASH; however, the mechanisms involved in this regulation are not well understood. The canonical necroptotic signaling pathway involves RIPK3-mediated phosphorylation of MLKL, and then MLKL oligomerization and translocation to the plasma membrane. Limited evidence suggests that MLKL-necroptosis and autophagy may be inter-related. Previous studies identified differential contributions of RIPK3 to progression of liver injury in multiple murine models of liver diseases, including NAFLD/NASH. However, the role of MLKL in the pathogenesis of NAFLD/NASH remains unclear. We recently reported that MLKL contributes to Western diet-induced liver injury by regulating autophagy [1].
Impaired autophagy in hepatocytes is associated with liver injury in patients with NAFLD/NASH. Activation of autophagy protects mice from liver injury. Our work found that Western diets increase the accumulation of SQSTM1/p62 and LC3-II, as well as induce ER stress in the liver. Furthermore, we also found an increase in accumulation of SQSTM1/p62 and LC3-II in both PA-treated primary hepatocytes and AML12 cells, which is not further increased by bafilomycin A1, an inhibitor of autophagy. These findings indicate that PA may inhibit autophagy at the late stage of lysosomal fusion. Using an mRFP-GFP-LC3 sensor, we further demonstrated that the number of yellow-fluorescent puncta (indicative of autophagosome accumulation) sustainably increases within 16 h, suggesting that PA blocks autophagic flux by inhibiting fusion of autophagosomes with lysosomes in hepatocytes.
Accumulating evidence suggests that MLKL translocates from the cytosol both to intracellular compartments and the plasma membrane, mediating both canonical and multiple non-canonical functions, including autophagy. In mouse dermal fibroblasts, RIPK3-mediated activation of MLKL inhibits autophagy during necroptosis by translocation to autolysosomes, whereas in endothelial cells and smooth muscle cells challenged with oxidized LDL, MLKL-mediated inhibition of autophagy is associated with activation of an MTOR-dependent pathway at an early stage of autophagy. Therefore, MLKL functions are likely cell/tissue and/or stimulus specific. Interestingly, in the liver, Mlkl deficiency prevents accumulation of SQSTM1/p62 and LC3-II in response to a protease inhibitor, leupeptin. In cultured hepatocytes, Mlkl deficiency prevents inhibition of autophagic flux by chloroquine (CQ). In contrast, overexpression of Mlkl blocks autophagic flux by inhibiting the fusion of autophagosomes with lysosomes. Importantly, CQ induces expression and translocation of MLKL to the plasma membrane in hepatocytes. We hypothesized that by inhibiting lysosomal proteases, leupeptin impairs autophagic degradation of proteins, leading to an increase in MLKL protein in liver. Therefore, these data suggest co-regulatory interactions between MLKL-necroptosis and autophagy in hepatocytes.
Given the role of autophagy in NAFLD/NASH and the critical interaction between MLKL and autophagic flux, we next investigated whether MLKL contributes to Western diet-induced liver injury by inhibiting autophagy. Mlkl, but not Ripk3, deficiency protects mice from accumulation of SQSTM1/p62 and LC3-II, as well as reduces liver injury, inflammation and hepatocellular death in response to Western diet. Western diet induces the expression, phosphorylation and oligomerization of MLKL in liver, independently of Ripk3. In cultured hepatocytes, PA triggers the expression and translocation of MLKL to the plasma membrane and MLKL-dependent, caspase-independent cell death. Importantly, PA induces the translocation of MLKL to LC3-positive autophagosomes prior to the movement of MLKL to the plasma membrane. Intracellular and membrane translocation of MLKL is independent of Ripk3. Using an LC3 reporter, we found that PA induces accumulation of autophagosomes in cultured hepatocytes. This is prevented in the absence of Mlkl, but not Ripk3. Meanwhile, the number of red-fluorescent puncta (autolysosomes) is increased in primary hepatocytes from mlkl−/-, but not ripk3−/-, mice. These findings suggest that Mlkl-mediated inhibition of autophagy and induction of necroptosis likely contribute to Western diet-induced liver injury.
Of future interest, LC3-associated phagocytosis (LAP) is a non-canonical autophagy pathway that utilizes components of the canonical autophagy machinery, contributing to inflammatory responses and immune regulation. While the canonical pathway of autophagy and LAP utilize both shared and unique components, they do share the common process of lysosomal fusion. Because our data indicate that PA inhibits autophagic flux at the lysosomal fusion and degradation stage, we are unable to distinguish between canonical autophagy and LAP. This distinction will need to be the focus of future studies.
In summary, Mlkl-dependent, but Ripk3-independent, signaling contributes to Western diet-induced liver injury through inhibition of autophagy and induction of necroptosis (Figure 1). Increased expression of MLKL observed in response to dietary and pharmacological interventions that inhibit autophagy likely creates a vicious circle that exacerbates the impairment of autophagic flux. These data identify a novel co-regulatory mechanism between autophagic and necroptotic pathways in NAFLD/NASH.
Figure 1.
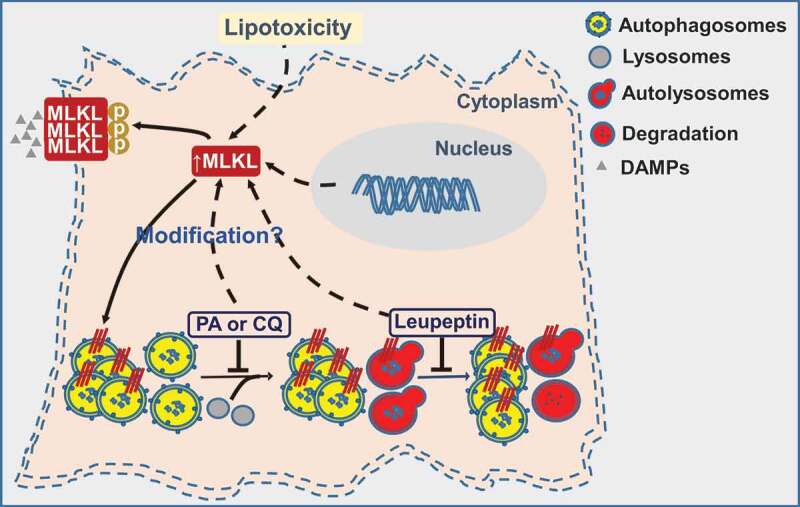
MLKL contributes to Western diet-induced liver injury through inhibition of autophagy and induction of necroptosis. Western diet or palmitic acid (PA) generates lipotoxicity, which induces the expression and translocation of MLKL to LC3-positive autophagosomes to inhibit autophagy, as well as the plasma membrane to trigger necroptosis and release damage-associated molecular patterns (DAMPs), thereby contributing to liver injury and inflammatory response. In addition, inhibition of autophagy by chloroquine (CQ) or leupeptin induces MLKL expression in hepatocytes. Increased expression of MLKL observed in response to dietary and pharmacological interventions that inhibit autophagy likely creates a vicious circle that exacerbates the impairment of autophagic flux.
Funding Statement
This work was supported by the National Institute on Alcohol Abuse and Alcoholism [P50 AA024333].
Disclosure statement
The authors have declared that no conflict of interest exists.
Reference
- [1].Wu X, Poulsen KL, Sanz-Garcia C, et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcoholic fatty liver disease. J Hepatol. 2020. S0168-8278(20)30185-9. DOI: 10.1016/j.jhep.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]