

ABSTRACT
The p53 family transcriptional factor p73 plays a pivotal role in development. Ablation of p73 results in severe neurodevelopmental defects, chronic infections, inflammation and infertility. In addition to this, Trp73−\- mice display severe alteration in the ciliated epithelial lining and the full-length N-terminal isoform TAp73 has been implicated in the control of multiciliogenesis transcriptional program. With our recently generated Trp73Δ13/Δ13 mouse model, we interrogate the physiological role of p73 C-terminal isoforms in vivo. Trp73Δ13/Δ13 mice lack exon 13 in Trp73 gene, producing an ectopic switch from the C-terminal isoforms p73α to p73β. Trp73Δ13/Δ13 mice show a pattern of expression of TAp73 comparable to the wild-type littermates, indicating that the α to β switch does not significantly alter the expression of the gene in this cell type. Moreover, Trp73Δ13/Δ13 do not display any significant alteration in the airway ciliated epithelium, suggesting that in this context p73β can fully substitute the function of the longer isoform p73α. Similarly, Trp73Δ13/Δ13 ciliated epithelium of the brain ependyma also does appear defective. In this district however expression of TAp73 is not detectable, indicating that expression of the gene might be compensated by alternative mechanisms. Overall our work indicates that C-terminus p73 is dispensable for the multiciliogenesis program and suggests a possible tissue-specific effect of p73 alternative splicing.
KEYWORDS: P53 family, ciliogenesis, development
Introduction
The p53 family proteins, including p53, p63 and p73 [1–6], share a functional and molecular interconnection in the cascade of events triggered by the DNA damage response [7–12]: genotoxicity triggers activation of these transcriptional factors leading to activation of a shared transcriptional signature, including p21, Bax, PUMA and NOXA, which is responsible for the p53 family effects on cell cycle progression and apoptosis [11,13–17]. However, while the best characterized family member, p53, plays major role in tumor suppression [18–27], the homologue p73, also exerts fundamental independent functions [3,28–30]. Structurally, Trp73 gene locus displays a complex architecture. At the 5ʹ alternative promoters can generate transactivation domain including (TAp73) and N-terminal truncated (DNp73) proteins; at the 3ʹ a wide alternative splicing provides a variety of C-terminal isoforms, which range from the full-length protein alfa to the shorter isoforms beta, gamma, delta, epsilon and zeta [31,32]. Mice carrying genetic deletion of all p73 isoforms (Trp73−\- mice) are characterized by a complex phenotype, still not fully dissected [33]. This includes chronic infections, inflammation, infertility and a severe neurodevelopmental impairment [4,34,35], which entails defective dentate gyrus architecture, ventriculomegaly, and a reduced proliferative capacity of neural stem cells [36–38].
A potentially common mechanism for the diverse abnormalities of the Trp73−\- mice involves multiciliogenesis, a process dependent on a functional TAp73 protein [39,40]. The process of multiciliogenesis is driven by a post-mitotic endocycle whereby modified centrioles or basal bodies (BBs), are amplified in large numbers through the activity of the master centriolar regulators Plk4 and Myb [41,42]. They subsequently migrate to the apical surface and dock at the plasma membrane where they nucleate the formation of microtubule-based structures [39,40]. TAp73 induces the transcription of a suite of important ciliogenesis genes by binding to promoter and/or enhancer regions of essential regulators including FOXJ1, Rfx2, Rfx3 and miR34bc [3]. Hence, the lack of TAp73 impairs the development of multiciliated epithelia, resulting in ablation of cilia structures, in upper airway tracks. However, despite the evidence for the TAp73 N-terminal isoforms in this role, little is known about the relative contribution of C-terminal isoforms to this process.
While the relevance of p73 transcriptional isoforms has been studied through a TAp73 [36] and DNp73 [43] knockout mouse models, the contribution of the p73 3ʹ-end splice-variants to its developmental functions remains to be determined. Alternative splicing of the C-terminal exons yields additional isoforms, which may or may not contain the TA domain [31]. Exons 11–13 of p73 encode a sterile alpha motif (SAM) domain that is only present in p73a, the most abundant isoform in most tissues, see Figure S1(a). This is of significance, as the SAM domain facilitates protein–protein interactions potentially affecting p73 stability, localization and transcriptional activity [44,45]. Nonetheless, the mechanistic importance of the SAM domain for the unique functions of p73 remains poorly understood.
To address the relevance of the alternative splicing isoforms, we have recently generated mice with a deleted exon 13 in the Trp73 gene, herein defined as Trp73Δ13/Δ13 mice, see Figure S1(a) (Amelio et al. PNAS 2020 in press). Deletion of this exon results in an ectopic switch of the C-terminal isoforms from p73α to p73β. Trp73Δ13/Δ13 mice display severe neurodevelopmental defects ascribed to deficiency of Cajal-Retzius (CR) cells, the pool of neurons necessary for correct hippocampal architecture (Amelio et al. PNAS 2020 in press). Here, by employing the Trp73Δ13/Δ13 mice we assess the importance of p73α in the multiciliogenesis process. Our data indicate that the ectopic p73α to p73β switch does not affect the process of multiciliogenesis in mice. p73β appears to compensate for the loss of p73α in the tracheal-multiciliated epithelium, while despite no p73 was detected in the brain ependymal cells, no phenotype was observed in the multiciliogenesis process occurring in this brain district. Taken together these findings indicate that, contrary to the hippocampal phenotype, the longest C-terminal p73 isoform is dispensable for the multiciliogenesis, suggesting a possible tissue-specific effect of p73 alternative splicing.
Results
Airway multiciliogenesis is preserved in mice lacking p73α
Deletion of exon 13 by CMV-driven recombinase generated a mouse strain (Trp73Δ13/Δ13) lacking p73α isoforms (Amelio et al. PNAS, 2020 in press). Trp73Δ13/Δ13 mice display significant neurodevelopmental defects, associated to morphological abnormalities of hippocampus and functional alterations in memory and learning (Figure 1(e), Supplementary Figure 1(b) and Amelio et al. PNAS, 2020 in press). We first verified that in the absence of exon 13, the predicted switch from p73α to p73β was induced in tracheal epithelium of Trp73Δ13/Δ13 mice. RT-PCR amplifying the 3ʹ regions of p73 mRNA (primers on exons 10 and 14) indicated that deletion of exon13 produced transcription of p73β instead than p73α (Supp. Figure S1(a)). Hence, we subsequently analyzed the cilia phenotype in our Trp73Δ13/Δ13 mouse model to assess the importance of p73α and the capacity for alternative isoforms to participate in the p73-mediated control of multiciliogenesis. Immunofluorescence staining for the cilia marker acetylated alpha-tubulin (Ac-α-tub) in tracheal epithelium indicated that the development of structurally normal cilia was maintained in Trp73Δ13/Δ13 mouse tracheal epithelium (Figure 1(a)). Furthermore, the area of Ac-α-tub staining along epithelia was not reduced relative to the wild-type and the BBs mostly displayed the correct localization (Figure 1(b)). In contrast, tracheal ciliated epithelium in Trp73−\- mice displayed the profound defects previously reported (Figure 1(a,b)). We also corroborated our data with transmission electron microscopy (TEM) and scanning electron microscopy (SEM) analyses which also displayed an abundance of full-length extensions in Trp73Δ13/Δ13 cilia and severe defects in Trp73−\- trachea epithelium (Figure 1(c,d)). These data, therefore, show that the deletion of exon 13 of the p73 gene does not impair upper airway track ciliated epithelium.
Figure 1.
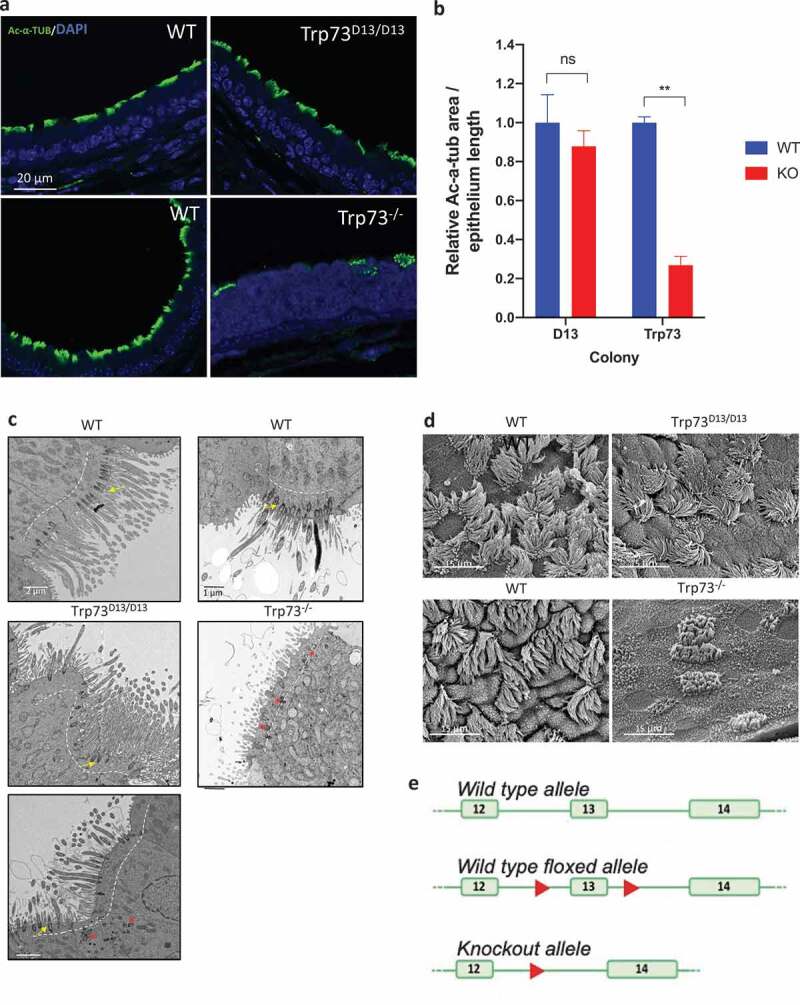
Airway ciliogenesis is preserved in the absence of p73α in Trp73Δ13/Δ13 mice. (a) Representative immunofluorescence images of Ac-α-tub expression (green) in tracheal epithelial cells of Trp73Δ13/Δ13 (top), p73 KO (bottom) mice and the corresponding wild-type shown adjacent. Nuclei were labeled with DAPI (blue). (b) Quantification of Ac-α-tub IF signal area normalized to the length of epithelia (Δ13 colony; WT & KO (Trp73Δ13/Δ13) n = 3 images from 3 mice. Trp73 colony; WT n = 3 images from 2 mice, p73 KO n = 3 images from 2 mice). (c) Representative TEM photomicrographs of tracheal epithelial cells in Trp73Δ13/Δ13 (left) and p73 KO (right) mice and the corresponding wild-type shown above. Dotted lines indicate the apical surface. Basal bodies were correctly docked at the surface of WT and most Trp73Δ13/Δ13 cells (yellow arrows) and aberrantly located in Trp73Δ13/Δ13, as in the p73 KO (red asterisks). (d) Representative SEM images of Trp73Δ13/Δ13 and p73 KO mice. Adjacent is the corresponding wild-type. All data are presented as mean values relative to WT ± SEM and ** = P < 0.01, ns = not significant. (e) Schematic representation of the targeting strategy employed to generate the Trp73Δ13/Δ13 mice. Deletion of exon 13 results in generation of an ectopic splicing junction exon 12–14, which results in p73beta. The diagram is readapted from Amelio et al, PNAS 2020.
Functional overlap of p73 C-terminal isoforms might underlie the preserved multiciliogenesis of Trp73Δ13/Δ13 mice
We next sought to investigate the p73 expression level in Trp73Δ13/Δ13 mouse tracheal epithelial cells. Despite a reduction in total TAp73 and DNp73 mRNA level (Figure 2(a)), there was no detectable alterations in the distribution of TAp73 protein in trachea epithelium (Figure 2(b,c)). This might suggest that the lack of phenotype in the Trp73Δ13/Δ13 mice might be ascribed to the functional overlap of C-terminal isoforms p73α and p73β. Consistently with this hypothesis, Trp73Δ13/Δ13 tracheal epithelia had no alterations in the pattern of expression of Foxj1 (Figure 2(d,e)), a p73 downstream target, which plays essential roles in multiciliogenesis [3,40]. Taken together, these data are suggestive of a potential tissue-specific functional overlap in p73 C-terminal isoforms, that are capable of orchestrating the ciliogenesis gene network.
Figure 2.
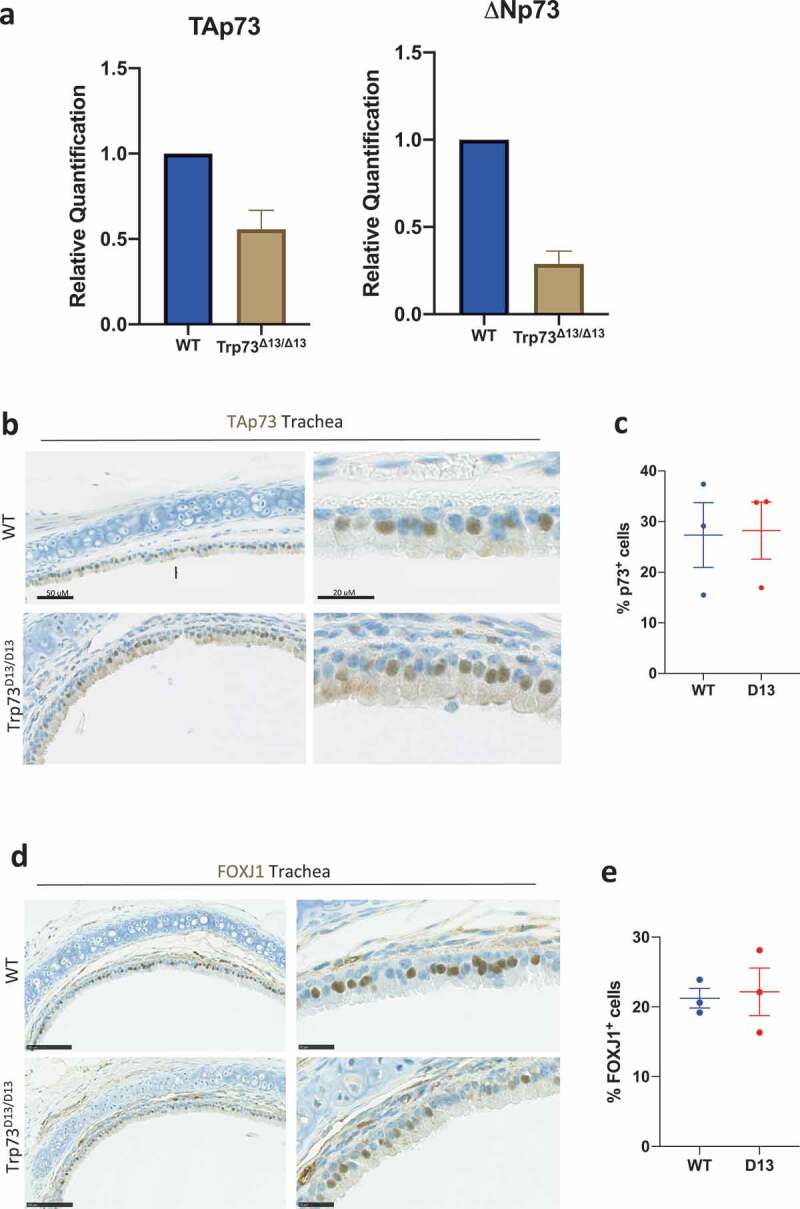
Trp73δ13/Δ13 tracheal epithelia maintain expression of TAp73 and Foxj1. (a) RT-qPCR analysis of relative TAp73 and DNp73 transcript expression in WT, Trp73Δ13/+ and Trp73Δ13/Δ13 mice. n = 3 (b) Representative IHC images showing TAp73 expression in tracheal epithelia of WT and Trp73Δ13/Δ13 mice. (c) Quantification of the percentage of p73 positive cells from staining shown in (a) (WT & Trp73Δ13/Δ13 n = 3 images from 2 mice). Data plotted as individual points with means ± SEM (d) Representative IHC images showing Foxj1 expression in tracheal epithelia of WT and Trp73Δ13/Δ13 mice. (e) Quantification of the percentage of Foxj1 positive cells from staining shown in (d) (WT & Trp73Δ13/Δ13 n = 3 images from 2 mice). Data plotted as individual points with means ± SEM.
Multiciliogenesis of brain ependymal cells is preserved in Trp73Δ13/Δ13 mice
Ependymal cells are multiciliated cells forming the neuroepithelium of the ventricular system of the brain. These cells functionally contribute to the production and regulation of cerebrospinal fluid [46,47]. p73 knock-out ependymal cells display defective maturation and ciliogenesis [48]. Consistently, Trp73−\- mice have defective functionality of cerebrospinal fluid which manifests as hydrocephalus [33]. Hence, we investigated the ependymal neuroepithelium lining in Trp73Δ13/Δ13 mice. Consistent with tracheal epithelia, ependymal ciliogenesis appears to be largely unperturbed in the absence of p73α. Ependymal cells display a similar coverage and length of cilia in Trp73Δ13/Δ13 mice and the corresponding wild-type, as shown by Ac-α-tub staining (Figure 3(a)) and electron microscopy (Figure 3(b)). To further assess the functionality of ependymal neuroepithelium, we analyzed coronal sections from Trp73Δ13/Δ13 mouse brains, to evaluate expansion of the ventricles. No evidence of development of hydrocephalus appeared (Figure S1(b,c)). This contrasted with the Trp73−\- mice which showed a striking reduction in ependymal cilia (Figure 3(a)). Unexpectedly, the Trp73Δ13/Δ13 ependymal ciliogenesis was preserved despite the immunohistochemical staining for TAp73 indicated an apparent absence of any TAp73 isoforms (Figure 3(c,e)).
Figure 3.
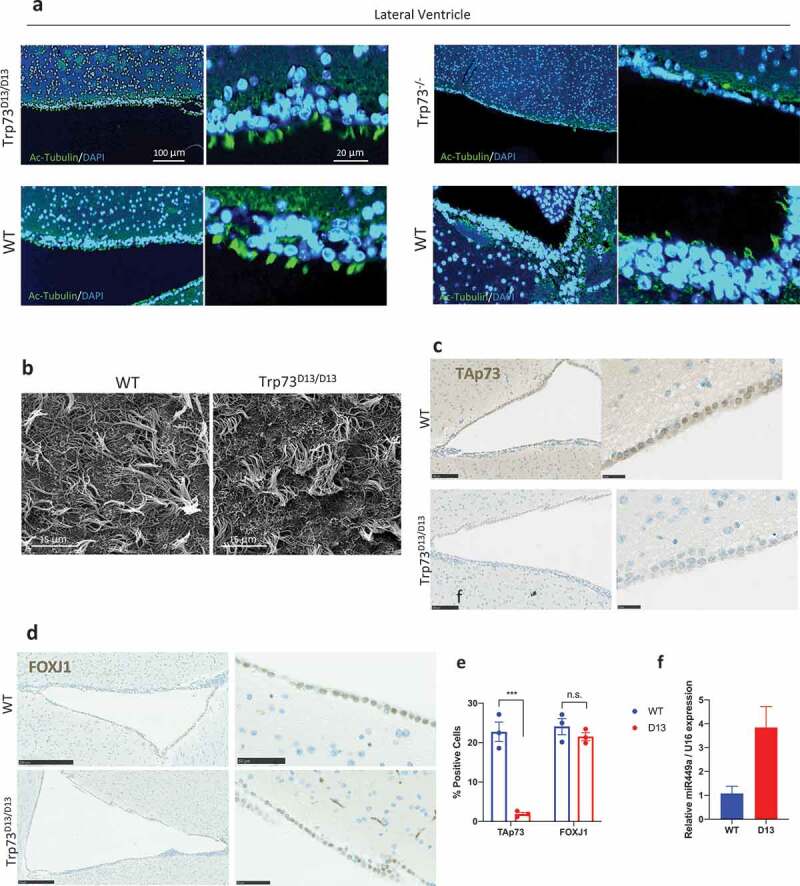
Ciliation of Trp73Δ13/Δ13 ependymal cells is not affected despite the absence of TAp73. (a) Representative immunofluorescence images of Ac-a-tub expression (green) in ependymal cells of the lateral ventricle from Trp73Δ13/Δ13 (left) and p73 KO (right) mice shown above the corresponding WT. Nuclei were labeled with DAPI (blue). (b) Representative SEM micrographs showing the apical surface of ependymal cells of the lateral ventricles of Trp73Δ13/Δ13 and WT animals. (c) and (d) Representative IHC images showing TAp73 (c) and Foxj1 (d) expression in cells lining the lateral ventricle of WT and Trp73Δ13/Δ13 mice. (e) Quantification of the percentage of p73+ and Foxj1+ cells lining the ventricle lumen from IHC images as shown in (c, d) (WT & Trp73Δ13/Δ13 n = 6 images from 2 mice). (f) RT-qPCR analysis of miR449a expression in Trp73Δ13/Δ13 mouse cortex relative to WT. n = 2, data plotted as individual points and means relative to WT ± SEM and *** = P < 0.001, n.s. = not significant.
To better understand the basis of Trp73Δ13/Δ13 ependymal cells phenotype, we analyzed the levels of FOXJ1 expression. Consistently with a lack of any evident phenotype no alteration was observed in the pattern of expression of this master regulator of the ciliogenesis program (Figure 3(d,e)). It is therefore plausible that TAp73 may be bypassed in the ciliogenesis pathway to activate downstream effectors such as FOXJ1. Indeed, recently published data has suggested that upregulation of miRNAs such as miR449 may compensate for TAp73 loss to maintain ependymal ciliogenesis [49]. Upregulation of miRNA-449a was indeed observed also in Trp73Δ13/Δ13 brain cortex (Figure 3(f)), supporting the hypothesis of a compensatory mechanism leading to proper multiciliogenesis process.
TAp73-dependent regulation of mitochondrial axis: a potential role in multiciliogenesis
Recent evidence indicates that ciliogenesis is associated to abundance of functional mitochondria [50]. Mitochondria are often localized in the proximity of the base of the cilia structure, and the motile cilia are major users of mitochondrial ATP. We and other groups have previously identified a direct effect of p53 and p73 on cellular metabolism, mitochondrial function and authophagy [51–54]. In vivo and in vitro deletion of p73 reduces ATP levels, oxygen consumption and mitochondrial complex IV activity with a subsequent enhancement of the production of ROS [55]. Indeed, p73 binds and regulates the promoter of the mitochondrial complex 4 cytochrome C oxidase subunit 4, Cox4i1 [55]. Additional molecular mechanisms for p73 to regulate mitochondria and in general metabolism operate through the direct transcriptional control of glutaminase type 2, GLS2 [56], and its metabolism on glutamine [57], the synthesis of serine [58,59] as well as through the regulation of elongation during protein translation [60]. It is also conceivable that other mitochondria regulators such as mitofusin, parkin or optic atrophy type 1 (OPA1) may also be involved. For example, the latter is an ubiquitously expressed protein with a prominent role in regulating mitochondrial fusion, and its deletion leads to fragmented mitochondria due to the inability of mitochondria to form extended networks [61]. Intriguingly, its mRNA level was massively upregulated following TAp73a or TAp73b overexpression (Figure S2(a)) and significantly reduced following the depletion of p73 by siRNA (Figure S2(b,c)) or in trachea epithelial cells from Trp73−\- mice (Figure S2(d)). Consistently, overexpressed TAp73 shows the ability to physically bind its promoter region (Figure S2(e,f)). However, the detailed mechanism of action and the biological significance of this regulation remain to be clarified.
Finally, we sought to functionally correlate mitochondrial function and p73 to multiciliogenesis by investigating mitochondrial morphology in Trp73−\- multiciliated epithelial cells. To this end, we analyzed TEM images of Trp73−\- mouse tracheal epithelium and evaluate the mitochondrial morphology. In agreement with a possible physiological role of TAp73/mitochondrial axis, we observed an increase in cristae width in epithelial cells of mice lacking p73 (Figure 4(a,b)). From TEM images it was also evident a much more fragmented mitochondrial network in the Trp73−\- cells compared to wild-type (Figure 4(b)). These data are suggestive of a potential role of TAp73/mitochondrial-dependent control of energy biology during ciliogenesis. However, further work will be needed to elucidate the functional significance of this mitochondria phenotype and the implications for this biological process.
Figure 4.
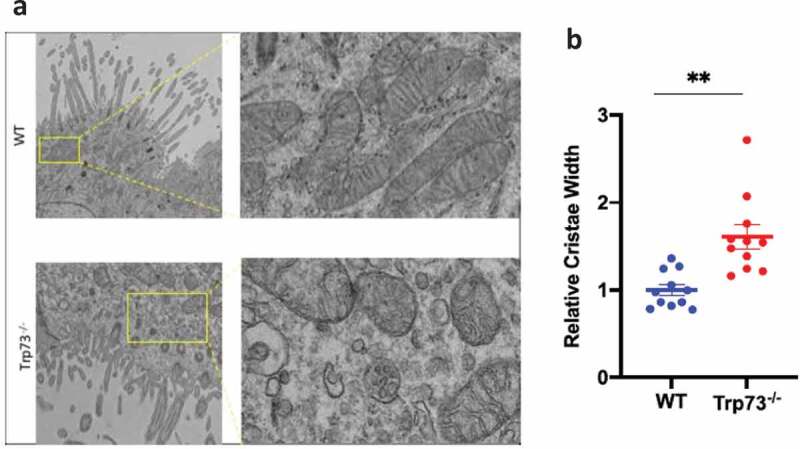
Trp73−\- mice diplay defective mitochondria. (a) Representative EM images of Trp73−\- mice display fragmented morphology in the mouse tracheal epithelium. (b) Quantification of mitochondria cristae width from EM analysis demonstrates alterations in Trp73−\- mice tracheal epithelium.
Discussion
We have shown in our Trp73Δ13/Δ13 model, carrying deletion of the C-terminal SAM domain containing isoform of p73, that p73α is dispensable for airway ciliogenesis. Indeed, in the absence of p73α in tracheal epithelia, the cilia layer is morphologically preserved and FOXJ1 remains expressed. FOXJ1 is an essential gene target of p73 in the ciliogenesis network responsible for coordinating the assembly of functional cilia. The capacity of p73β to preserve airway ciliogenesis in our model therefore might suggest that a possible functional overlap between p73 C-terminal isoforms occurs in a cell-type specific manner. It has been hypothesized that post-translational modifications of C-terminal regions of p73 that are unique to p73α modulate the stability, transcriptional activity and localization of this isoform to promote the regulation of developmental networks [29,31]. However, if the functional overlap between p73α and p73β is a correct interpretation, these regulatory networks should be cell-type specific. Questions remain on where or when p73β is physiologically expressed, as no tissue appears to express p73β as the predominant isoform [32].
We have also shown the redundancy of p73α for brain ependymal ciliogenesis. Our Trp73Δ13/Δ13 model does not show detectable defects in the ependymal neuroepithelium. However, the lack of detectable TAp73 isoforms in brain neuroepithelium leaves to inconclusive interpretations on this data, which are however in agreement with the lack of alterations in the ependymal ciliogenesis of TAp73−/- mice [49]. Reasons might lie in compensatory mechanisms that might involve microRNAs networks, such as the previously reported miR449, or roles for the N-terminal truncated isoforms, ΔNp73. However, any possible compensatory mechanism is not effective or present in tracheal epithelium as TAp73−/- showed severe impairment in airway ciliogenesis [40]. Taken together, this therefore indicates highly complex and tissue-specific roles of both C-terminal and TAp73/ΔNp73 isoforms in the context of multiciliogenesis.
Furthermore, our recent characterization of the hippocampal phenotype of Trp73Δ13/Δ13 mice sheds further light on the complexity of the roles of C-terminal isoforms of p73. We have shown that Trp73Δ13/Δ13 mice show none of the striking consequences of perturbed ependymal biology such as severe hydrocephalus and reduced cortical thickness, which are present in the Trp73−\-[62]. Nonetheless, they still display defective dentate gyrus architecture also present in both p73−/- and TAp73−/- knockouts. This therefore demonstrates the necessity of p73α for directing hippocampal development, as when p73α is replaced with p73β in the developing mouse brain, the lower blade of the dentate gyrus is truncated and malformed. These studies of the Trp73Δ13/Δ13 knockout therefore shed light on the differential requirement for p73α in different cell types of the same tissue.
We suggest a possible correlation between mitochondria biology and p73 the role in ciliogenesis. We have suggestive data of a direct metabolic control mediated by TAp73 on the dynamics of mitochondrial function. Disruption of mitochondrial function or, more specifically, deletion of selective genes such as, for example of a crucial mitochondrial gene, leads to fragmented mitochondria due to the inability of mitochondria to form extended networks, a phenomenon that frequently occurs in disease, such as ADOA where cell death is rapidly induced when OPA1 is mutated [61]. It is therefore suggestive that a role in mitochondrial fission by p73 or a synergism with that pathway may be crucial for ciliogenesis, but this has been less well defined [63]. Trp73−\- ciliated epithelium displays defective mitochondria with increased in cristae width and fragmented mitochondria network. Hence, mitochondria function might link p73 function in ciliogenesis to mitochondrial biology, opening to potentially relevant connections between ciliopathy and mitochondrial functionality. Further studies however will be first needed to clearly implicate TAp73/mitochondria axis in the ciliogenesis and determine how this molecular pathway impinge on this biological process.
Material & methods
Cell culture and transfection
NCI-H1299 cells (ATCC) were cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin (Gibco) at 37°C and 5% CO2. Cells were cultured in T175 flasks and detached prior to passage using 1% trypsin (Gibco). NCI-H1299 were transfected using Lipofectamine 2000 (Invitrogen) with overexpressing plasmid constructs for TAp73α or TAp73β. siRNA transfection was carried out using Lipofectamine RNA iMAX (Invitrogen) using 5ug predesigned TP73 siRNA (Invitrogen; AM51331) or scrambled negative control (Invitrogen).
Immunofluorescence and immunohistochemistry
Dissected trachea or whole brains were fixed in 4% paraformaldehyde (PFA) for 48 h and embedded in paraffin. Traverse sections of trachea and coronal brain sections containing the lateral ventricles were obtained for the analysis of ciliated cell populations. Sections were deparaffinized by immersion in Xylene (Sigma) followed by a gradient of 100–70% ethanol (Sigma). Antigen retrieval was performed by heating in a microwave at 292 W for 15 min in 0.01 M sodium citrate. Tissue was blocked with 5% normal goat serum for 1 h at room temperature (RT) and incubated with anti-Ac-a-tubulin (Thermofisher; T7451) diluted 1:1000 in PBS overnight at 4°C in a humidified atmosphere. Samples were washed 3 times in PBS and incubated for 1 h with Alexa Fluor 488 secondary antibodies (Molecular Probes; Invitrogen) at RT. Samples were washed 3x in PBS and mounted using Prolong Gold Antifade mountant (Thermofisher; P36930). DAPI was added to the secondary antibody solution. Images were acquired through confocal microscopy (LSM 510, Zeiss), fitted with an argon laser (488 nm excitation), and UV excitation at 405 nm. For immunohistochemistry, tissue sections were fixed and embedded as described above. IHC was carried out using the Ventana Discovery Ultra platform (Roche). Deparaffinization was performed by immersion in Ventana Discovery Wash buffer (Roche; 950–510) for 3 × 8 minute washes at 69°C. Antigen Retrieval was performed by incubation in Ventana CC1 buffer at 95°C for 32 min. The slides were blocked by incubation with Discovery goat IgG block (Roche; 760–6008) for 12 min at 37°C. Primary antibody incubations were carried out using Anti-p73 (Bethyl #A300-126A) diluted 1:200 for 3 h at 37°C or anti-Foxj1 (eBioscience #14-9965-82) diluted 1:200 for 1 h 37°C. Antibodies were diluted in EnVision Flex Antibody Diluent (Dako; DM830). The detection reaction was performed using UltraMap DAB anti-Rb Detection Kit (Roche; 760–151) according to the manufacturer’s instructions.
Electron microscopy
Conventional Scanning Electron Microscopy: Mouse trachea and ependyma were carefully dissected and chemically fixed with 2.5% glutaraldehyde (GA) and 2% PFA in 0.1 M cacodylate buffer (pH7.4) for 3 h at RT. The specimens were cut to expose to the surface of cilia organization. After several washing with 0.1 M cacodylate buffer, they were proceeded to the post-fixation with 1% osmium tetroxide/1% potassium ferrocyanide in 0.1 M cacodylate buffer for 1 h. After several washes in distilled water, a second post-fixation was performed with 5% uranyl acetate solution for overnight at 4°C in the dark fridge and then the dehydration with a series of ethanol (from 70% to absolute 100%) were completed. Finally, after special treatment with hexamethyldisilazane (HMDS, Sigma-Aldrich, St. Louis USA), the dried specimens were sputter coated with pure gold (EMITECH k950 K, Quorum Technologies, Kent UK). Fine observation was performed using FEI Quanta FEG250 (Thermo Fischer Scientific, Oregon USA). Ultrathin Section Transmission Electron Microscopy: The mouse trachea and brain ependyma were fixed with 2.5% GA and 2% PFA in the NaHCa buffer (100 mM NaCl, 30 mM HEPES, 2 mM CaCl2, adjusted at pH 7.4 with NaOH) for more than a few hours at RT. The specimens were then proceeded to the conventional post-fixation with 0.25% osmium tetroxide/0.25% potassium ferrocyanide and 1% tannic acid. After stained en bloc with 5% aqueous uranyl acetate, dehydration with a series of ethanol and infiltration were completed for the plastic embedding in TER (TAAB Epoxy Resin). After the polymerization at 65°C for a few days, the ultrathin-sections (∼60 nm) obtained by Ultramicrotome (Leica Ultracut UCT, Vienna Austria) were mounted in EM grids, stained with lead citrate, and then observed by FEI Talos F200 C 200kV transmission electron microscope (Thermo Fischer Scientific, Oregon USA) with Ceta-16 M CMOS-based camera (4 k x 4 k pixels under 16 bit dynamic range) in terms of cilia, mitochondrial, and neuronal morphogenesis.
Western blot
Cellular protein extract was obtained by incubating cell pellets with RIPA buffer for 30 minutes on ice. Protein concentrations were determined using the BCA assay (Thermofisher). 4x LDS sample buffer (Invitrogen) was added to protein samples, followed by denaturation by heating at 95°C for 10 minutes. 40 µg protein was run in 12% resolving and 4% stacking polyacrylamide gels (National Diagnostics) at 175 V for 1 h. Protein was transferred to PVDF membranes (Thermofisher) in 1x transfer buffer (Thermofisher) containing 10% (v/v) methanol by applying 0.8 mA per cm2 of membrane for 2 h on ice. Membranes were blocked in 5% milk (Marvel) in TBS/0.1% Tween 20 (TBST) for 1 h at RT before incubation with primary antibody overnight at 4°C (Anti-OPA1; BD #612,606) diluted 1:1000 in 5% milk TBST. Membranes were washed 5x in TBST and incubated with goat anti-mouse/HRP conjugated secondary antibody (Thermofisher #31,430) diluted 1:10,000 in TBST for 1 h at RT. Membranes were washed 5x in TBST and incubated with ECL substrate (GE Healthcare) for 5 min before visualization using radiographic films.
Chromatin immunoprecipitation
The MAGnify™ chromatin immunoprecipitation kit (Invitrogen) was used according to the manufacturer’s instructions. In brief, H1299 cells were transfected with TAp73α overexpression construct as described previously. Following trypsinization, cells were pelleted at 300 x g for 5 min and resuspended in PBS. 3 x 10^6 cells were suspended in 500 uL PBS and fixed by adding PFA at a final concentration of 1% and incubating for 10 min at RT. The reaction was stopped by adding glycine and cells washed twice in PBS. Cells were lysed using 150 ul lysis buffer prepared according to the manufacturer’s instructions and stored at −80°C. Chromatin was sheared using the Covaris S220 set at a duty factor of 2%, intensity 4, and 200 cycles/burst for 6 mins to obtain fragments approximately 500 bp in size for qPCR analysis. 2.5 ug anti-HA antibody or mouse immunoglobulins were bound to Dynabeads® by rotating end-over-end at 4°C for 1 h. Chromatin was diluted to 200,000 per IP and immunoprecipitated according to the manufacturer’s instructions and eluted in 150 ul elution buffer.
Reverse transcription quantitative PCR (RT-qPCR)
RNA was extracted using the RNeasy Plus mini kit (Qiagen #74136) according to the manufacturer’s instructions. The concentration and purity of RNA was measured using a nanodrop spectrophotometer (Thermofisher). 1 ug of RNA was used per reverse transcription reaction using RevertAid minus first-strand cDNA synthesis kit (Thermofisher) according to the manufacturer’s instructions. Transcribed cDNA was diluted 2x before use in qPCR. Primer sequences (Sigma) are listed in Table 1. Reactions were carried out in triplicate using Fast SYBR Green PCR Master Mix (Thermofisher #4385612). The relative quantification was obtained using the Applied Biosystems 7500 thermocycler and quantitative comparative (ΔΔCt) method normalized to TBP, which was used as an endogenous control. Quantification of miR449a was performed using the TaqMan miRNA Assay kit (Thermofisher; 001030) according to the manufacturer’s instructions. The RT reaction was carried out using the TaqMan™ MicroRNA Reverse Transcription Kit (Thermofisher; 4366596) using the specific probe for mir449a and U16 as an endogenous control.
Table 1.
Primer sequences used for PCR experiments.
| Gene | Forward Primer (5ʹ-3ʹ) Sequence | Reverse Primer (5ʹ-3ʹ) Sequence |
|---|---|---|
| hTAp73 | CCAGACCTCTTCTTCCTC | GTCAAAGTAGGTGCTGTC |
| hDNp73 | ATGCTTTACGTCGGTGAC | CTGCCCATCTGGTCCATG |
| mTAp73 | GCACCTACTTTGACCTCCCC | GCACTGCTGAGCAAATTGAAC |
| mDNp73 | ATGCTTTACGTCGGTGACCC | GCACTGCTGAGCAAATTGAAC |
| hp21 | CCTGTCACTGTCTTGTACCT | GCGTTTGGAGTGGTAGAAATCT |
| hOPA1 | TCAAGAAAAACTTGATGCTTTCA | GCAGAGCTGATTATGAGTACGATT |
| mOPA1 | CAGTTTAGCTCCCGACCTGG | TGATGACACCAGGCAAGTCC |
| hTBP | TCAACCCAGAATTGTTCTCCTTAT | CCTGAATCCCTTTAGAATAGGGTAGA |
| mTBP | TTGGCTAGGTTTCTGCGGTC | TGGAAGGCTGTTGTTCTGGT |
| hOPA1 promoter 1 | TCCATGCGCCATTGGGAG | CCTGCACTTACCAGGCCACA |
| hOPA1 promoter 2 | ATGTAAGCCTCCCTCCCACT | TGTTACATGCCTAACCCACGAA |
| hMDM2 promoter | GGTTGACTCAGCTTTTCCTCTTG | GGAAAATGCATGGTTTAAATAGCC |
Funding Statement
This work has been supported by the Medical Research Council (to GM), Associazione Italiana per la Ricerca contro il Cancro (AIRC) to GM [IG#20473; 2018-2022] and to IA [AIRC Start-Up ID 23219; 2020-2024]; Ministry of Health & MAECI Italy-China Science and Technology Cooperation [#PGR00961] to G.M. M.N. was the recipient of The Uehara Memorial Foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Chillemi G, Kehrloesser S, Bernassola F, et al. Structural evolution and dynamics of the p53 proteins. Cold Spring Harb Perspect Med. 2017;7:a028308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nemajerova A, Moll UM.. Tissue-specific roles of p73 in development and homeostasis. J Cell Sci. 2019;132:jcs233338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nemajerova A, Amelio I, Gebel J, et al. Non-oncogenic roles of TAp73: from multiciliogenesis to metabolism. Cell Death Differ. 2018;25:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vanbokhoven H, Melino G, Candi E, et al. p63, a story of mice and men. J Invest Dermatol. 2011;131:1196–1207. [DOI] [PubMed] [Google Scholar]
- [5].Levine AJ, Tomasini R, McKeon FD, et al. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12:259–265. [DOI] [PubMed] [Google Scholar]
- [6].Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113(Pt 10):1661–1670. [DOI] [PubMed] [Google Scholar]
- [7].Hafner A, Bulyk ML, Jambhekar A, et al. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199–210. [DOI] [PubMed] [Google Scholar]
- [8].Sullivan KD, Galbraith MD, Andrysik Z, et al. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018;25:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wu D, Prives C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018;25:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li X, Guo M, Cai L, et al. Competitive ubiquitination activates the tumor suppressor p53. Cell Death Differ. 2020 Jun;27(6):1807–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rada M, Barlev N, Macip S. BTK modulates p73 activity to induce apoptosis independently of p53. Cell Death Discov. 2018;4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ho CJ, Lin RW, Zhu WH, et al. Transcription-independent and -dependent p53-mediated apoptosis in response to genotoxic and non-genotoxic stress. Cell Death Discov. 2019;5:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. [DOI] [PubMed] [Google Scholar]
- [14].Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. [DOI] [PubMed] [Google Scholar]
- [15].Aubrey BJ, Kelly GL, Janic A, et al. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25:114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sankunny M, Eng C. KLLN-mediated DNA damage-induced apoptosis is associated with regulation of p53 phosphorylation and acetylation in breast cancer cells. Cell Death Discov. 2018;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pitolli C, Wang Y, Candi E, et al. p53-mediated tumor suppression: DNA-damage response and alternative mechanisms. Cancers (Basel). 2019;11:1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pitolli C, Wang Y, Mancini M, et al. Do mutations turn p53 into an oncogene? Int J Mol Sci. 2019 Dec 11;20(24):6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ. 2018;25:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26:199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018;25:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Di Gennaro A, Damiano V, Brisotto G, et al. A p53/miR-30a/ZEB2 axis controls triple negative breast cancer aggressiveness. Cell Death Differ. 2018;25:2165–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hao XL, Han F, Zhang N, et al. TC2N, a novel oncogene, accelerates tumor progression by suppressing p53 signaling pathway in lung cancer. Cell Death Differ. 2019;26:1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Amelio I. How mutant p53 empowers Foxh1 fostering leukaemogenesis? Cell Death Discov. 2019;5:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Amelio I, Mancini M, Petrova V, et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci U S A. 2018;115:E10869–E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Amelio I, Melino G. The p53 family and the hypoxia-inducible factors (HIFs): determinants of cancer progression. Trends Biochem Sci. 2015;40:425–434. [DOI] [PubMed] [Google Scholar]
- [28].Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–386. [PubMed] [Google Scholar]
- [29].Gong H, Zhang Y, Jiang K, et al. p73 coordinates with Delta133p53 to promote DNA double-strand break repair. Cell Death Differ. 2018;25:1063–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Amelio I, Inoue S, Markert EK, et al. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1alpha degradation. Proc Natl Acad Sci U S A. 2015;112:226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vikhreva P, Melino G, Amelio I. p73 alternative splicing: exploring a biological role for the C-terminal isoforms. J Mol Biol. 2018;430:1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Grespi F, Amelio I, Tucci P, et al. Tissue-specific expression of p73 C-terminal isoforms in mice. Cell Cycle. 2012;11:4474–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. [DOI] [PubMed] [Google Scholar]
- [34].Killick R, Niklison-Chirou M, Tomasini R, et al. p73: a multifunctional protein in neurobiology. Mol Neurobiol. 2011;43:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Agostini M, Tucci P, Killick R, et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc Natl Acad Sci U S A. 2011;108:21093–21098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tomasini R, Tsuchihara K, Wilhelm M, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Inoue S, Tomasini R, Rufini A, et al. TAp73 is required for spermatogenesis and the maintenance of male fertility. Proc Natl Acad Sci U S A. 2014;111:1843–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Alexandrova EM, Talos F, Moll UM. p73 is dispensable for commitment to neural stem cell fate, but is essential for neural stem cell maintenance and for blocking premature differentiation. Cell Death Differ. 2013;20:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Marshall CB, Mays DJ, Beeler JS, et al. p73 is required for multiciliogenesis and regulates the Foxj1-associated gene network. Cell Rep. 2016;14:2289–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nemajerova A, Kramer D, Siller SS, et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 2016;30:1300–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Choksi SP, Lauter G, Swoboda P, et al. Switching on cilia: transcriptional networks regulating ciliogenesis. Development. 2014;141:1427–1441. [DOI] [PubMed] [Google Scholar]
- [42].Choksi SP, Babu D, Lau D, et al. Systematic discovery of novel ciliary genes through functional genomics in the zebrafish. Development. 2014;141:3410–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wilhelm MT, Rufini A, Wetzel MK, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 2010;24:549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chi SW, Ayed A, Arrowsmith CH. Solution structure of a conserved C-terminal domain of p73 with structural homology to the SAM domain. Embo J. 1999;18:4438–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chi SG, Chang SG, Lee SJ, et al. Elevated and biallelic expression of p73 is associated with progression of human bladder cancer. Cancer Res. 1999;59:2791–2793. [PubMed] [Google Scholar]
- [46].Ohata S, Alvarez-Buylla A. Planar organization of multiciliated ependymal (E1) cells in the brain ventricular epithelium. Trends Neurosci. 2016;39:543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Paredes MF, Sorrells SF, Garcia-Verdugo JM, et al. Brain size and limits to adult neurogenesis. J Comp Neurol. 2016;524:646–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gonzalez-Cano L, Fuertes-Alvarez S, Robledinos-Anton N, et al. p73 is required for ependymal cell maturation and neurogenic SVZ cytoarchitecture. Dev Neurobiol. 2016;76:730–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wildung M, Esser TU, Grausam KB, et al. Transcription factor TAp73 and microRNA-449 complement each other to support multiciliogenesis. Cell Death Differ. 2019;26:2740–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Burkhalter MD, Sridhar A, Sampaio P, et al. Competence network for congenital heart defects. Imbalanced mitochondrial function provokes heterotaxy via aberrant ciliogenesis. J Clin Invest. 2019;129:2841–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parrales A, Thoenen E, Iwakuma T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018;25:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lonetto G, Koifman G, Silberman A, et al. Mutant p53-dependent mitochondrial metabolic alterations in a mesenchymal stem cell-based model of progressive malignancy. Cell Death Differ. 2019;26:1566–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen Y, Liu K, Shi Y, et al. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. 2018;25:639–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Goiran T, Duplan E, Rouland L, et al. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 2018;25:873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rufini A, Niklison-Chirou MV, Inoue S, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012;26:2009–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Velletri T, Romeo F, Tucci P, et al. GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell Cycle. 2013;12:3564–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Niklison-Chirou MV, Erngren I, Engskog M, et al. TAp73 is a marker of glutamine addiction in medulloblastoma. Genes Dev. 2017;31:1738–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Amelio I, Markert EK, Rufini A, et al. p73 regulates serine biosynthesis in cancer. Oncogene. 2014;33:5039–5046. [DOI] [PubMed] [Google Scholar]
- [59].Amelio I, Cutruzzola F, Antonov A, et al. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Marini A, Rotblat B, Sbarrato T, et al. TAp73 contributes to the oxidative stress response by regulating protein synthesis. Proc Natl Acad Sci U S A. 2018;115:6219–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bertholet AM, Delerue T, Millet AM, et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 2016;90:3–19. [DOI] [PubMed] [Google Scholar]
- [62].Medina-Bolivar C, Gonzalez-Arnay E, Talos F, et al. Cortical hypoplasia and ventriculomegaly of p73-deficient mice: developmental and adult analysis. J Comp Neurol. 2014;522:2663–2679. [DOI] [PubMed] [Google Scholar]
- [63].MacVicar T, Langer T. OPA1 processing in cell death and disease - the long and short of it. J Cell Sci. 2016;129:2297–2306. [DOI] [PubMed] [Google Scholar]