

ABSTRACT
Oxidative stress and Th17 cytokines are important mediators of inflammation. Treatment with beta-adrenoceptor (ADRB) antagonists (beta-blockers) is associated with induction or aggravation of psoriasis-like skin inflammation, yet the underlying mechanisms are poorly understood. Herein, we identify lysosomotropic beta-blockers as critical inducers of IL23A in human monocyte-derived Langerhans-like cells under sterile-inflammatory conditions. Cytokine release was not mediated by cAMP, suggesting the involvement of ADRB-independent pathways. NFKB/NF-κB and MAPK14/p38 activation was required for propranolol-induced IL23A secretion whereas the NLRP3 inflammasome was dispensable. MAPK14 regulated recruitment of RELB to IL23A promoter regions. Without affecting the ubiquitin-proteasome pathway, propranolol increased lysosomal pH and induced a late-stage block in macroautophagy/autophagy. Propranolol specifically induced reactive oxygen species production, which was critical for IL23A secretion, in Langerhans-like cells. Our findings provide insight into a potentially crucial immunoregulatory mechanism in cutaneous dendritic cells that may explain how lysosomotropic drugs regulate inflammatory responses.
Abbreviations
ATF: activating transcription factor; DC: dendritic cell; ChIP: chromatin immunoprecipitation; gDNA: genomic DNA; IL: interleukin; LAMP1: lysosomal associated membrane protein 1; LC: Langerhans cell; LPS: lipopolysaccharide; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MAPK: mitogen-activated protein kinase; MoDC: monocyte-derived DC; MoLC: monocyte-derived Langerhans-like cell; mtDNA: mitochondrial DNA; NAC: N-acetyl-L-cysteine; NLRP3: NLR family pyrin domain containing 3; PBMC: peripheral blood mononuclear cell; PI: propidium iodide; PYCARD/ASC: PYD and CARD domain containing; qRT-PCR: quantitative real-time PCR; ROS: reactive oxygen species; SQSTM1/p62: sequestosome 1; TLR: Toll-like receptor; TRAF6: TNF receptor associated factor 6; TNF: tumor necrosis factor; Ub: ubiquitin.
KEYWORDS: Beta blockers, dendritic cells, inflammation, skin, IL1, IL23A, MAPK14/p38, reactive oxygen species
Introduction
Macroautophagy/autophagy is a ubiquitous cellular process controlling cellular homeostasis and cytosolic content degradation [1]. Single membrane-enclosed autophagosomes, containing ubiquitin (Ub)-labeled cargo, undergo maturation and consecutively merge with lysosomes to form autolysosomes [2], a structure essential for the digestion of subcellular components [3,4]. Since lysosomal maintenance is pH sensitive, an acidic lysosomal lumen is crucial for autophagic clearance. An acidic interior contributes to ion trapping and subsequent accumulation of basic, highly lipophilic small molecules. Following lysosomal sequestration, compounds that successively increase lysosomal pH, and concomitantly the number of autophagosomes, are known as lysosomotropic compounds [5]. Lysosomotropism-induced loss-of-function of lysosomal enzymes may eventually contribute to phospholipidosis, a phospholipid storage disorder [6].
However, lysosomal dysfunctions and excessive aggregation of autophagic cargo is also associated with IL1-mediated autoinflammatory diseases [7–10]. Inhibition of autophagy was shown to regulate IL23A secretion of innate immune cells, including dendritic cells (DCs) [11,12]. Moreover, impairment of autophagy-dependent removal of dysfunctional mitochondria is associated with the formation of reactive oxygen species (ROS). Free mitochondrial DNA (mtDNA) and ROS are potent inductors of pro-inflammatory signaling, including the expression of IL1 family cytokines whose processing and expression is tightly regulated by autophagosomes [13,14]. Furthermore, activation of IL1R/IL1 receptor- and Toll-like receptor (TLR)-dependent signaling modulates chromatin and/or nucleosome remodeling thereby regulating the access of transcriptionally active NFKB/NF-κB proteins RELA, RELB and REL/c-Rel to IL12A, IL12B and IL23A promoter regions [15,16]. Thus, homeostatic feedback between autophagic activity and intra- and extracellular signaling pathways regulates pro-inflammatory cytokines [17,18].
The pathogenesis of sterile, chronic autoinflammatory diseases such as psoriasis relies strongly on Th17 cell activity [19]. A cardinal hallmark is the positive regulation of the IL23A-IL17A axis that bridges innate and adaptive immune response. Crosstalk between T cells and activated DCs is responsible for potent T cell activation and subsequent polarization. IL1A or IL1B and IL6 induce differentiation while IL23A-dependent downstream signaling stabilizes a pathogenic Th17 phenotype. Convergent data suggest a critical contribution of monocyte-derived Langerhans-like cells (MoLCs) and monocyte-derived dermal dendritic cells (MoDCs) to Th17 development in psoriasis [20]. Recent mouse studies further suggest that in particular LCs are critical regulators of IL23A production contributing to psoriasis pathogenesis [21]. We identified previously an alteration to autophagic flux induced by the anti-malarial drug chloroquine, which led to increased Th17 development, triggered by IL23A secreted from cutaneous DCs [22].
Interestingly, during ADRB (adrenoceptor beta) antagonist (hereafter referred to as “beta-blocker”) therapy, indicated primarily in cardiovascular diseases [23], numerous patients exhibit cutaneous adverse effects, so called “drug eruptions” [24,25]. Administration of the nonselective beta-blocker propranolol is linked with the emergence, maintenance and/or exacerbation of psoriasis-like skin inflammations. At present, the underlying mechanism is poorly understood. Assuming a lysosomotropic character of certain beta-blockers [26,27], we hypothesized a crucial role for autophagy in drug-provoked inflammatory reactions by epidermal LCs and dermal DCs by an enhanced secretion of IL23A under sterile-inflammatory conditions.
Results
Propranolol induces IL23A release from activated cutaneous DC subtypes
In agreement with our previous findings for inhibitors of late stage autophagy [22], the nonselective beta-blocker propranolol significantly induced IL23A release in a concentration-dependent manner, between 25 and 100 µM, under sterile-inflammatory conditions (Figure 1A). Notably, MoLCs were more sensitive to propranolol resulting in substantial cytokine secretion compared to MoDCs. Although, cytokine levels were not triggered by enhanced cell death, propranolol concentrations above 100 μM significantly increased the number of ANXA5 (annexin A5) and propidium iodide (PI) double-positive cells (Figure 1B). Thus, 75 µM was selected for further assays. Moreover, propranolol stimulation together with IL36G/IL-1F9, a new member of the IL1 family and recently implicated in the pathogenesis of psoriasis [28], upregulated Th17-related cytokine production in both DC subtypes (Fig. S1). Sharing distinct IL1R downstream signaling pathways, propranolol-induced a marginal upregulation of cytokines exclusively in LPS-activated MoLCs. Additionally, we used the (1,3)-β-glucan curdlan, a known IL23A inducer, that is recognized by CLEC7A/dectin-1 leading to subsequent activation of CARD9-dependent signaling [29]. However, propranolol significantly downregulated cytokine release in MoLCs and MoDCs, demonstrating a critical involvement of IL1R signaling for propranolol-enhanced cytokine expression. Since MoLCs appeared more prone to propranolol induced IL23A release, increasing concentrations of IL1B/IL-1β were added to MoDCs to ensure full IL1R saturation in the presence of propranolol. While this produced comparable surface expression of DC activation markers CD83 and CD86 (Fig. S2A), further IL23A production was not observed in MoDCs (Fig. S2B).
Figure 1.
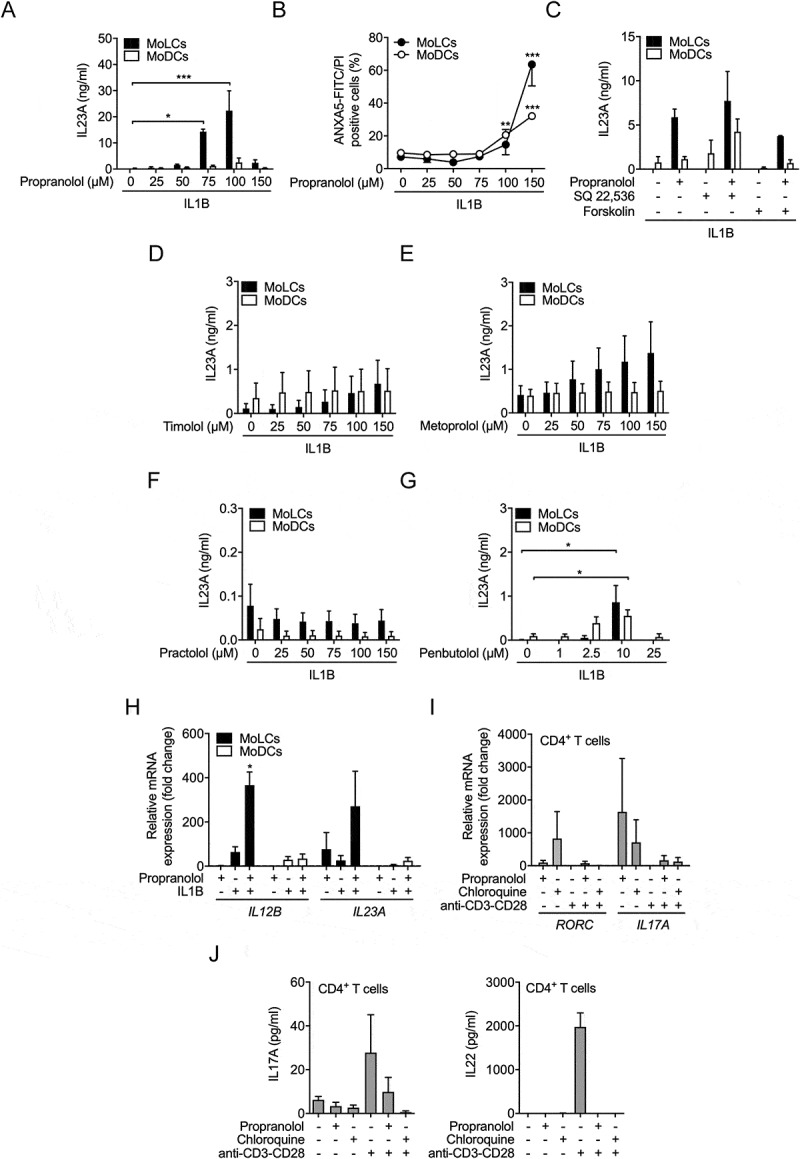
Lysosomotropic beta-blocker induce IL23A release by activated MoLCs independent of beta adrenoceptors. (A) IL23A secretion was analyzed by ELISA in the supernatant of MoLCs and MoDCs stimulated with IL1B (20 ng/ml) for 24 h in the presence or absence of propranolol at different concentrations (25–150 µM). (B) Cell viability was assessed by ANXA5-FITC/PI-double-staining using flow cytometry. (C) IL23A release by MoLCs and MoDCs, quantified after stimulation with SQ 22,536 (100 µM) or forskolin (20 µM) and followed by activation with IL1B (20 ng/ml) for 24 h in the presence or absence of propranolol (75 µM). (D-G) IL23A production by MoLCs and MoDCs after stimulation with IL1B (20 ng/ml) for 24 h in the presence or absence of timolol, metoprolol, practolol or penbutolol at different concentrations (1–150 µM) was assayed by ELISA. (H) mRNA expression of IL12B and IL23A in MoLCs and MoDCs, respectively, was assessed after 24 h of stimulation with IL1B (20 ng/ml) in the presence or absence of propranolol (75 µM). Gene expression results were normalized to GAPDH and depicted relative to unstimulated MoLCs and MoDCs (set as 1.0). Propranolol does not directly polarize naïve CD4+ T cells toward Th17 development. (I) Gene expression levels of RORC and IL17A were examined in naïve CD4+ T cells, polyclonally stimulated with anti-CD3-CD28 antibodies for 5 d with or without propranolol (75 µM) or chloroquine (20 µM). Levels of mRNA were normalized to GAPDH and presented relative to untreated CD4+ T cells (set as 1.0). (J) Naïve CD4+ T cells were stimulated with anti-CD3-CD28 antibodies for 7 d in presence or absence of propranolol (75 µM) or chloroquine (20 µM), respectively. Th17 signature cytokines IL17A and IL22 in cell culture supernatants were quantified by ELISA. (A-G) *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA test followed by Bonferroni posttest, (H and I) one-sample t-test. Data are representative of (A) n = 4–5, (B) n = 3–6, (C) n = 3–5, (D-J) n = 3 independent experiments and display mean values ± SEM.
Lysosomotropic beta-blockers elevate IL23A release independent of ADRB (adrenoceptor) blockade
To address whether IL23A secretion results from reduced cAMP levels by beta-adrenergic inhibition, we used the ADCY (adenylate cyclase) inhibitor SQ 22,536. IL23A levels were not significantly different in the presence of the inhibitor (Figure 1C). Likewise, the ADCY activator forskolin did not modulate IL23A production. Both cAMP modulators did not affect cell viability (Fig. S3A) and regulated IL6 levels in the absence of propranolol (Fig. S3B) confirming that IL6 is regulated by intracellular cAMP levels [30].
We next used beta-blockers with closely related physicochemical properties to chloroquine and propranolol to explore whether lysosomotropism induces IL23A secretion (Table 1). The nonselective beta-blocker timolol (Figure 1D) and the beta 1 selective beta-blocker metoprolol (Figure 1E) increased IL23A release in MoLCs in a concentration dependent manner but to a lesser extent than propranolol. Practolol, a specific beta 1 selective beta-blocker, which is no longer used due to the induction of inflammatory skin reactions [31], failed to promote cytokine release (Figure 1F). Given that practolol is the least lipophilic of the tested compounds, we determined the IL23A response by one of the most lipophilic clinically used nonselective beta-blocker penbutolol. Accordingly, penbutolol consequently enhanced IL23A release (Figure 1G), indicating a positive correlation between increasing clogP values and drug-induced IL23A secretion. Thus, beta-blockers with specific chemical properties orchestrate cutaneous DCs to initiate IL23A production independent of ADRB selectivity.
Table 1.
The clogP and basic pKa values of beta blockers and chloroquine used in this study.
| Compound | pKa | clogP |
|---|---|---|
| Chloroquine | 10.1 | 5.28 |
| Penbutolol | 9.76 | 3.84 |
| Propranolol | 9.42 | 3.03 |
| Metoprolol | 9.21 | 1.80 |
| Timolol | 9.67 | 1.44 |
| Practolol | 9.67 | 0.53 |
Values were obtained from www.drugbank.ca.
Propranolol alters gene expression of IL12 family members without affecting IL12 release
To investigate the regulatory effect of propranolol at the transcriptional level, we assessed mRNA expression of IL12A, IL12B and IL23A. IL12A mRNA levels were hardly detectable in MoDCs and MoLCs and not regulated (data not shown). Consistently, propranolol did not trigger IL12 release in both DC subsets under inflammatory conditions (data not shown). Propranolol alone significantly induced IL23A but had little effect on IL12B gene expression in MoLCs (Figure 1H). Activation with IL1B resulted in elevated IL12B mRNA levels while IL23A remained constant. When applied together with propranolol the transcriptional activity of both IL23A-composing subunits significantly increased. In activated MoDCs, enhanced IL12B as well as slightly upregulated IL23A transcription were observed. In the presence of propranolol, IL23A but not IL12B gene expression was further increased.
Propranolol initiates th17 differentiation in naïve CD4+ T cells
We previously confirmed that chloroquine-stimulated MoLCs promote Th17 differentiation in co-cultivation with allogeneic naïve CD4+ T cells [22]. To evaluate whether lysosomotropic drugs skew adaptive immune responses by directly affecting T cells, we determined the effect of propranolol and chloroquine on naïve CD4+ T cells. Lysosomotropic drugs increased gene expression of Th17 lineage marker RORC and Th17 signature cytokine IL17A in naïve CD4+ T cells (Figure 1I). However, anti-CD3-CD28-induced activation reduced mRNA levels of RORC and IL17A promoted by propranolol and chloroquine. We next assessed T cell cytokine response of IL17A and IL22, an accessory cytokine released by the IL17A+ IL22+ Th17 cell population [32]. Lysosomotropic compounds additionally failed to induce IL17A and IL22 release of T cells activated by anti-CD3-CD28 antibodies (Figure 1J). Thus, while propranolol and chloroquine alone triggered Th17-related gene transcription in naïve CD4+ T cells, CD3-CD28 activation resulted in inhibition of Th17 responses.
Propranolol differently regulates pro-inflammatory cytokines in cutaneous DCs
Aside from IL23A, IL6 and TNF levels are also increased in psoriatic lesions [33]. In propranolol-stimulated MoLCs, we found significantly increased IL6 and TNF secretion (Figure 2). By contrast, in MoDCs, IL6 production was reduced while TNF levels remained unaffected. In immune cell trafficking, CXCL8/IL8 plays an essential role in the large-scale infiltration of acute inflammatory cells. Concordantly, CXCL8 production was substantially increased in MoLCs and MoDCs in the presence of IL1B and propranolol (Figure 2). Similar results were observed for mRNA levels (Fig. S4).
Figure 2.
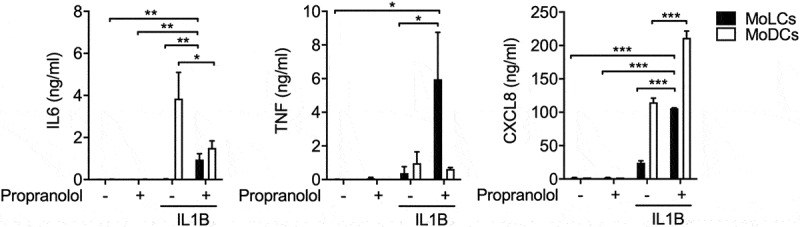
Propranolol induces production of psoriasis-like inflammation associated mediators under sterile inflammatory conditions. Indicated cytokine secretion by immature and IL1B-activated (20 ng/ml) DC subsets after 24 h in the presence or absence of propranolol (75 µM) was assessed using ELISA. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA test followed by Bonferroni posttest. Data are representative of 3–4 independent experiments and display mean values + SEM.
Propranolol alters expression of autophagy-related proteins
Lysosomotropism is strongly associated to disturbances in the autophagic machinery [5]. During phagophore formation, the mammalian autophagy marker MAP1LC3/LC3 is consecutively converted from its cytosolic, passive form LC3-I to its active LC3-II form. Protein expression demonstrated reduced amounts of LC3-I and conversely, enhanced levels of LC3-II in MoLCs after stimulation with propranolol indicating an enhanced autophagic flux (Figure 3A). Besides an induction of autophagy mediated by IL1B signaling, stimulation with IL1B and propranolol simultaneously led to barely detectable amounts of LC3-I and significantly upregulated LC3-II levels in MoLCs and MoDCs. Consequently, propranolol significantly increased LC3-II:LC3-I ratio in both subsets, indicating a late-stage block of autophagy.
Figure 3.
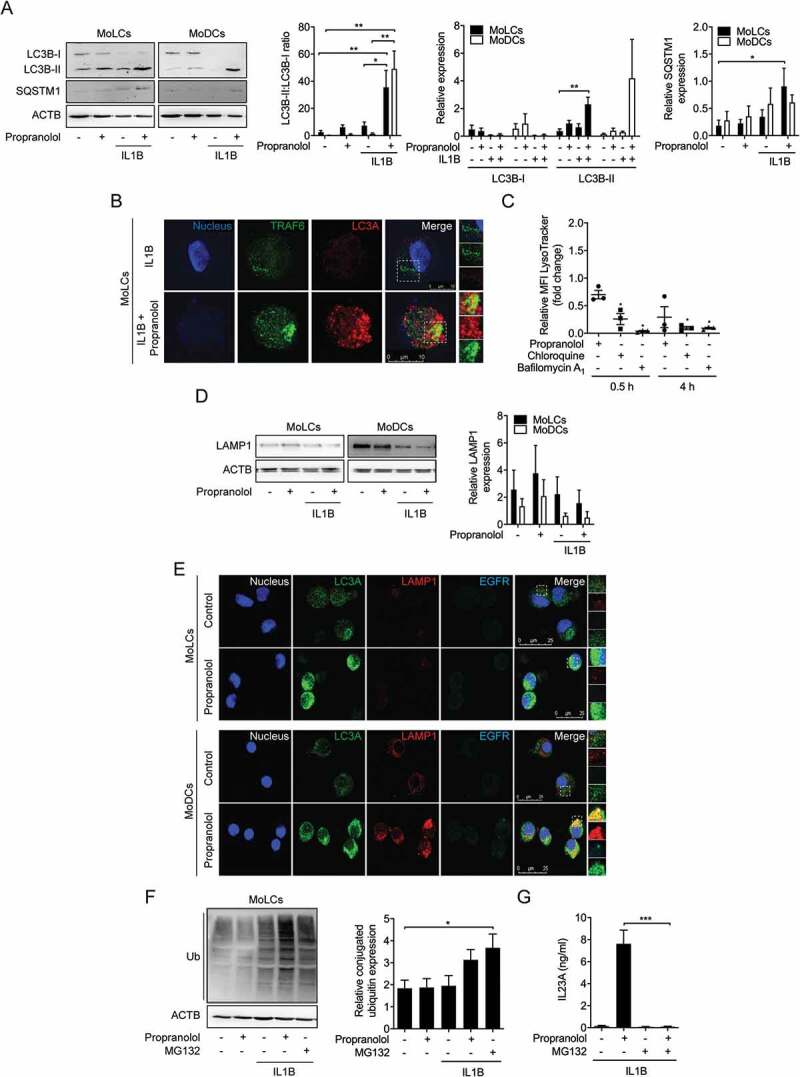
Propranolol inhibits autophagic flux and impairs endosomal maturation in MoLCs and MoDCs. (A and D) Immunoblot analysis of total LC3B-I, LC3B-II, SQSTM1, and LAMP1 expression in whole-cell lysates of MoLCs and MoDCs, stimulated with IL1B (20 ng/ml) for 24 h with or without propranolol (75 µM). (B) Immunostaining of IL1B-activated MoLCs for TRAF6 and LC3A after 24 h of stimulation with propranolol (75 µM). Scale bar represents 10 µm. (C) Acidification of intracellular compartments was examined by flow cytometry of MoLCs, incubated with propranolol (75 µM), chloroquine (20 µM) or bafilomycin A1 (1 µM) for 0.5 h or 4 h, respectively. Cells were pre-incubated for 0.5 h in medium supplemented with acidotropic LysoTracker Red DND-99 (100 nM). pH indicator-specific detection of mean fluorescence intensity (MFI) was quantified and depicted relative to untreated controls (assigned as 1.0). (E) Co-localization of LC3A and EGFR with LAMP1 was analyzed by immunofluorescence microscopy in MoLCs and MoDCs after 24 h of stimulation with propranolol (75 µM). Scale bar represents 25 µm. (F) Immunoblot analysis of whole-cell lysates from MoLCs probed with anti-Ub for total ubiquitin-conjugated constituents, stimulated with IL1B (20 ng/ml) for 24 h with or without propranolol (75 µM) or MG132 (10 µM). (G) IL23A release was analyzed by ELISA in the supernatant of IL1B-stimulated MoLCs (20 ng/ml) for 24 h in the presence or absence of propranolol (75 µM) or MG132 (10 µM). (A, D and F) Protein expression was quantified by densitometric analysis with ACTB/β-actin serving as control. (A, F and G) *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA test followed by Bonferroni posttest, (C) unpaired two-tailed t-test. Data are representative of n = 3–4 independent experiments and display mean values ± SEM.
We next quantified expression of the cargo receptor protein SQSTM1/p62, a protein known to be degraded during autophagy. Concordantly, propranolol markedly increased SQSTM1 expression in IL1B-activated MoLCs but not in MoDCs (Figure 3A). Previous data have indicated a complex interaction between the IL1R downstream signaling molecule TRAF6 and SQSTM1 [34]. Having previously reported the TRAF6-upregulating effect of chloroquine [22], we next assessed the intracellular co-staining of LC3 and TRAF6 in MoLCs with or without propranolol to determine whether TRAF6 is degraded via LC3-mediated autophagy. Indeed, propranolol led to colocalization of LC3 and TRAF6 in IL1B-stimulated MoLCs, further highlighting a block of late-stage autophagy (Figure 3B).
Propranolol attenuates lysosomal acidification and decreases LAMP1 expression
Lysosomotropic compounds are characterized by their ability to accumulate in acidic subcellular compartments and alter their pH [27]. To evaluate the influence of propranolol on the internal pH of subcellular compartments, we stained MoLCs with an acidotropic dye (LysoTracker). Propranolol-stimulated cells revealed a time-dependent decrease in positively labeled intracellular acidic compartments compared to control (Figure 3C). Similar results were obtained for chloroquine and bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase. To assess whether propranolol impairs lysosomal function, we quantified protein expression of LAMP1 (lysosomal associated membrane protein 1), representing lysosomal maintenance. In the presence of propranolol, increased LAMP1 expression was detected in both DC subtypes (Figure 3D). Under sterile-inflammatory conditions, LAMP1 levels were reduced and even further decreased when simultaneously stimulated with propranolol. We subsequently examined whether LC3 co-localizes with LAMP1, indicating the formation of autolysosomes. Besides increased numbers of LC3+ autophagosomes, immunofluorescence staining revealed no detectable co-localization with LAMP1 after stimulation with propranolol (Figure 3E). Collectively, propranolol presumably inhibits the autophagic process at a late stage characterized by impaired fusion between autophagosomes and lysosomes.
Propranolol-impeded endosomal trafficking appears dispensable
To investigate the contribution of propranolol to other intracellular vesicular trafficking, we monitored clathrin-mediated endocytosis of internalized EGFR (epidermal growth factor receptor). Addressing the fusion of lysosomes with late endosomes during endosomal maturation, we simultaneously stained LC3, LAMP1 and EGFR in MoLCs and MoDCs, respectively. Besides increased LC3 levels, propranolol hardly altered the marginal basal expression of LAMP1 and EGFR in MoLCs. However, stimulation of MoDCs with propranolol increased the amount of endo-lysosomal proteins (Figure 3E). Additionally, co-localization of LAMP1 and EGFR was solely observed in MoDCs, indicating no significant regulation of endosomal trafficking pathways by propranolol.
Propranolol contributes to the accumulation of ubiquitinated proteins
To further elucidate the interference of propranolol with the autophagy-lysosomal pathway, we determined the ubiquitination status of cytosolic proteins and organelles in MoLCs. Propranolol and IL1B alone were insufficient to enhance the amount of Ub-labeled intracellular content (Figure 3F). Elevated ubiquitination of cytosolic constituents was detected under sterile inflammatory conditions when simultaneously challenged by propranolol or the proteasome inhibitor MG132. Surprisingly, the addition of MG132 completely abolished propranolol-induced IL23A release in IL1B-activated MoLCs, whereas MG132 alone did not affect respective cytokine production (Figure 3G). Although these data indicate that propranolol contributes to the intracellular accumulation of Ub-conjugated content, we cannot fully rule out an involvement of the ubiquitin-proteasome pathway in the propranolol-promoted IL23A secretion.
Propranolol regulates IL23A secretion in a NLRP3 inflammasome-independent pathway
Next, we investigated the expression of NLRP3 (NLR family pyrin domain-containing 3) to exclude a putative regulatory effect of propranolol in the proteasomal system. NLRP3 is negatively regulated by ubiquitination and subsequently degraded in an autophagy-dependent pathway [35]. Indeed, in MoLCs, propranolol significantly increased NLRP3 expression but only in the pellet fraction (Figure 4A). IL1B-induced activation as well as co-stimulation with propranolol almost abolished NLRP3 levels. However, the addition of MG132 reinstated NRLP3 expression suggesting enhanced protein levels due to an interference of propranolol in autophagic machinery. NLRP3 expression is involved with inflammasome activity, resulting in processing and secretion of pro-inflammatory cytokines including IL1B and IL18. Propranolol alone was insufficient to increase IL1B expression in both DC subtypes (Figure 4B). By contrast, IL1B-induced IL1B upregulation was further amplified by propranolol, while in MoDCs we observed that propranolol negatively affected IL1B.
Figure 4.
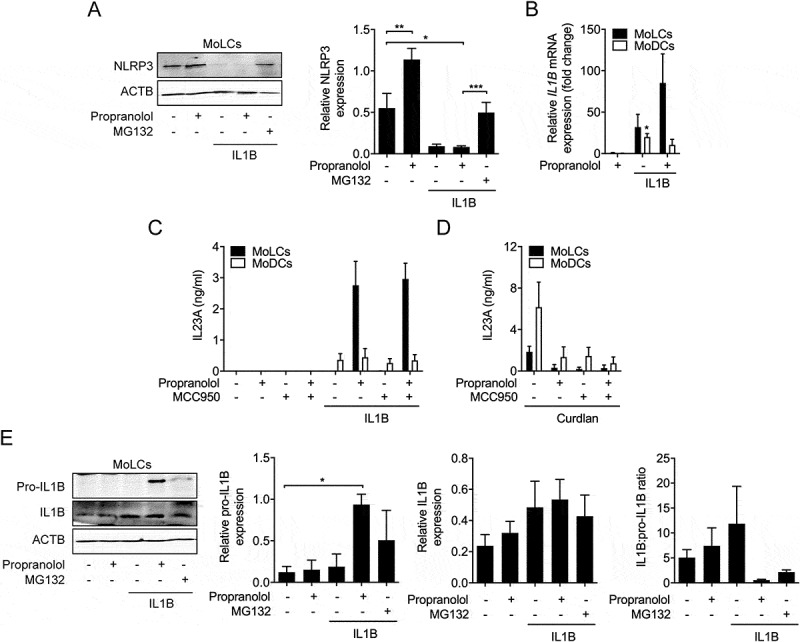
NLRP3 inflammasome activity is dispensable for propranolol-mediated induction of IL23. (A) Immunoblot analysis of cell pellets from MoLCs probed with anti-NLRP3, after 24 h activation with IL1B (20 ng/ml) in the presence or absence of propranolol (75 µM) or MG132 (10 µM). (B) Quantification of IL1B mRNA in immature and IL1B-activated (20 ng/ml) DC subsets stimulated for 3 h with or without propranolol (75 µM). Gene transcripts were normalized to GAPDH and presented relative to unstimulated controls (set as 1.0). (C and D) ELISA of IL23A released by MoLCs and MoDCs activated with IL1B (20 ng/ml) or curdlan (20 µg/ml), respectively, and stimulated with or without propranolol (75 µM) and NLRP3 inflammasome inhibitor MCC950 (5 µM) for 24 h. (E) Immunoblot analysis of IL1B in whole-cell lysates from MoLCs obtained after 24 h of stimulation with IL1B (20 ng/ml) with or without propranolol (75 µM) or MG132 (10 µM). (A and E) Protein expression was normalized to ACTB and quantified by densitometry. (A and E) *P < 0.05, **P < 0.01, one-way ANOVA test followed by Bonferroni posttest, (B) one-sample t-test. Data are representative of (A) n = 4, (B-E) n = 3 independent experiments and display mean values + SEM.
In order, to assess whether propranolol-induced IL23A secretion was also regulated by NLRP3 inflammasome activity, we used MCC950, a specific NLRP3 inflammasome inhibitor. MCC950 inhibited curdlan-induced IL23A secretion (Figure 4D) but did not affect IL23A secretion in propranolol- and IL1B-stimulated MoLCs (Figure 4C). Following detection of cellular insults, the NLRP3 inflammasome activates CASP1 (caspase 1)-dependent conversion of pro-IL1B into IL1B. Thus, to precisely investigate inflammasome activity, we quantified levels of IL1B and its precursor. In MoLCs, under sterile-inflammatory conditions, a significant increase in pro-IL1B expression promoted by propranolol was seen, while IL1B remained rather unaffected (Figure 4E). The IL1B-induced upregulated ratio of IL1B:pro-IL1B was completely abrogated in the presence of propranolol. In line, we detected no regulation of IL18 secretion by IL1B-stimulated MoLCs with or without propranolol (data not shown). Taken together, these results suggested that the NLRP3 inflammasome is dispensable for propranolol-mediated induction of IL23A.
Propranolol induces oxidative stress and leads to an abundance of ROS-producing mitochondria
Recent studies suggest a strong association between ROS levels and autophagic activity [13]. Propranolol induced a significant increase in ATF3 mRNA levels in MoLCs, indicating oxidative cellular stress (Figure 5A). No propranolol-induced alteration to ATF6 [36] was found in MoLCs (Figure 5B). Conversely, in MoDCs, no regulation of ATF3 but significant upregulation of ATF6 was observed. Subsequently, we analyzed total intracellular ROS generation. In MoLCs, propranolol increased ROS generation in a time-dependent manner, while MoDCs stayed unaffected (Figure 5C). The ROS inhibitor N-acetyl-L-cysteine (NAC) completely abrogated ROS formation in propranolol-stimulated MoLCs and completely blocked IL23A secretion, indicating a pivotal role of ROS for IL23A release (Figure 5D). Since propranolol as well as chloroquine triggered Th17-priming in naïve CD4+ T cells, we additionally assessed total intracellular ROS generation in T cells. Propranolol stimulation yielded no apparent alterations to cytosolic ROS formation (Figure 5E). Thus, propranolol and presumably other lysosomotropic compounds primarily induce oxidative cell stress in MoLCs that is crucial for the contribution to the IL23A-IL17A axis.
Figure 5.
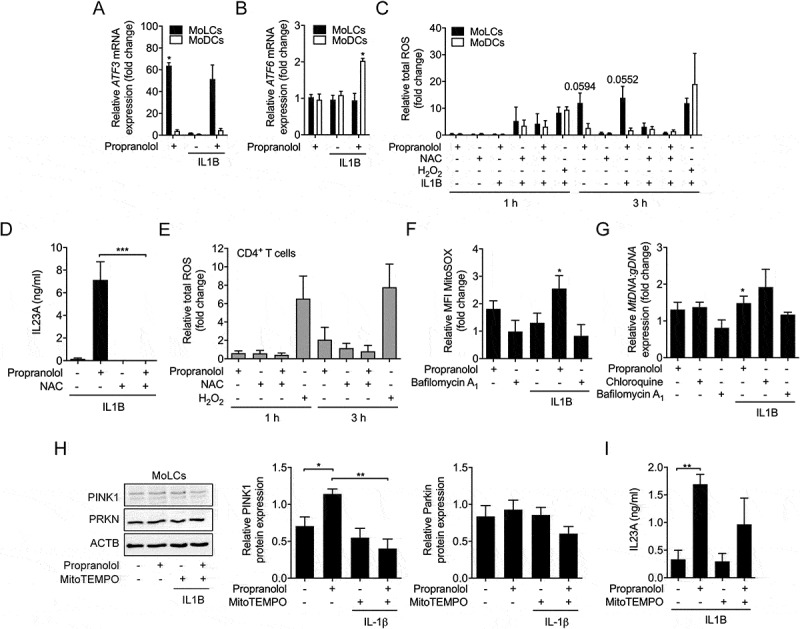
Propranolol-induced inhibition of autophagic flux is accompanied by oxidative stress and abundance of ROS-producing mitochondria in MoLCs. (A and B) qRT-PCR quantification of ATF3 and ATF6 copy numbers in immature and IL1B-activated (20 ng/ml) DC subsets stimulated for 3 h with or without propranolol (75 µM). Levels of mRNA were normalized to GAPDH and presented relative to untreated DCs (set as 1.0). (C) Flow cytometry analysis of total intracellular ROS formation in IL1B-activated DCs, pre-incubated with ROS assay stain for 1 h and subsequently stimulated with or without propranolol (75 µM) and N-acetyl-L-cysteine (20 mM) for 1 h and 3 h, respectively. H2O2 (200 µM) was used as a positive control. ROS quantification is presented relative to unstimulated controls (set as 1.0). (D) MoLCs stimulated with IL1B (20 ng/ml) were stimulated with or without propranolol (75 µM) and NAC (20 mM). Levels of IL23A were detected by ELISA. (E) Flow cytometry analysis of total intracellular ROS formation in naïve CD4+ T cells, pre-incubated with ROS assay stain for 1 h and followed by stimulation with or without propranolol (75 µM) and N-acetyl-L-cysteine (20 mM) for 1 h and 3 h, respectively. H2O2 (200 µM) served as positive control. ROS levels were displayed relative to untreated controls (set as 1.0). (F) Detection of mitochondrial-generated ROS was examined by flow cytometry and mean fluorescence intensity (MFI) was quantified in immature and IL1B-stimulated MoLCs, pre-loaded with MitoSOX (5 µM) for 10 min and subsequently cultivated for 24 h in the presence or absence of propranolol (75 µM) or bafilomycin A1 (1 µM). Detected ROS were depicted relative to unstimulated MoLCs (set as 1.0). (G) Copy numbers of mitochondrial DNA (hmito3) in unstimulated and IL1B-stimulated (20 ng/ml) with or without propranolol (75 µM), chloroquine (20 µM) or bafilomycin A1 (1 µM) were assayed after 48 h. mtDNA was normalized to ALDOA, used as a loading control for genomic DNA and displayed relative to IL1B-activated cells (set as 1.0). (H) Immunoblot analysis of PINK1 and PRKN in whole-cell lysates from MoLCs obtained after 24 h of stimulation with IL1B (20 ng/ml) with or without propranolol (75 µM) or MitoTEMPO (20 µM). Protein expression was evaluated by densitometric analysis with ACTB/β-actin assisting as control. (I) ELISA of IL23A levels collected by MoLCs activated with IL1B (20 ng/ml) and stimulated with or without propranolol (75 µM) and MitoTEMPO (20 µM) for 24 h. (A-C, F and G) *P < 0.05, one-sample t-test. (D, H and I) *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA test followed by Bonferroni posttest. Data are representative of (A, D and I) n = 3, (B, C, E, F and H) n = 4, (G) n = 3–6 independent experiments and display mean values + SEM.
It is widely known that the main source of cell stress promoted ROS production are depolarized mitochondria and/or the endoplasmatic reticulum. Thus, we stained MoLCs with a specific oxidation-sensitive fluorescent dye that accumulates in mitochondria. Indeed, in the presence of propranolol, increased fluorescent signals were detected relative to control (Figure 5F). By contrast, bafilomycin A1 failed to regulate mitochondria-derived ROS production. Immunofluorescent staining confirmed our results, showing an increase in positively labeled, ROS-producing mitochondria (Fig. S5). Collectively, propranolol led to enhanced total ROS levels and concomitantly mitochondrial-derived ROS formation in MoLCs alone that is critically involved in the secretion of IL23A. To evaluate whether late-stage block of autophagy interferes with degradation of mitochondria whereby ROS formation is promoted, mitochondrial DNA (mtDNA) was quantified. Chloroquine and propranolol but not bafilomyin A1 induced mitochondria accumulation in IL1B-activated MoLCs (Figure 5G). The selective removal of mitochondria by mitophagy is triggered by a PINK1-dependent recruitment of PRKN/parkin to dysfunctional mitochondria. Corresponding to an altered autophagic flux, IL1B-activated MoLCs stimulated with propranolol showed a significant elevation of PINK1 levels, potentially linking mitophagy to increased mtDNA deposits (Figure 5H). However, propranolol failed to sufficiently regulate PRKN. The co-administration of the mitochondria-targeted ROS scavenger MitoTEMPO strongly reduced propranolol-mediated regulation of PINK1. Moreover, MitoTEMPO decreased propranolol-provoked IL23A levels, highlighting a putative contribution of impaired mitophagy and subsequent ROS production to IL23A, induced by propranolol (Figure 5I).
MAPK14 regulates IL23A release by enhancing IL12B transcriptional activity
We previously demonstrated that MAPK14/p38 activity is essentially involved in chloroquine-induced IL23A secretion [22]. As expected, inhibition of the MAPKs MAPK1/ERK2-MAPK3/ERK1 and MAPK8/JNK by U0126 and SP600,125, respectively, did not significantly regulate IL23A release (Figure 6A). However, the MAPK14 inhibitor SB202,190 almost completely blocked IL23A secretion. To assess whether propranolol differentially regulates IL23A subunits via MAPK14, we determined mRNA expression of IL12B and IL23A. Blocking MAPK14 activity resulted in complete inhibition of IL12B but not IL23A transcription in activated MoLCs, indicating MAPK14 is required for IL12B transcription (Figure 6B).
Figure 6.
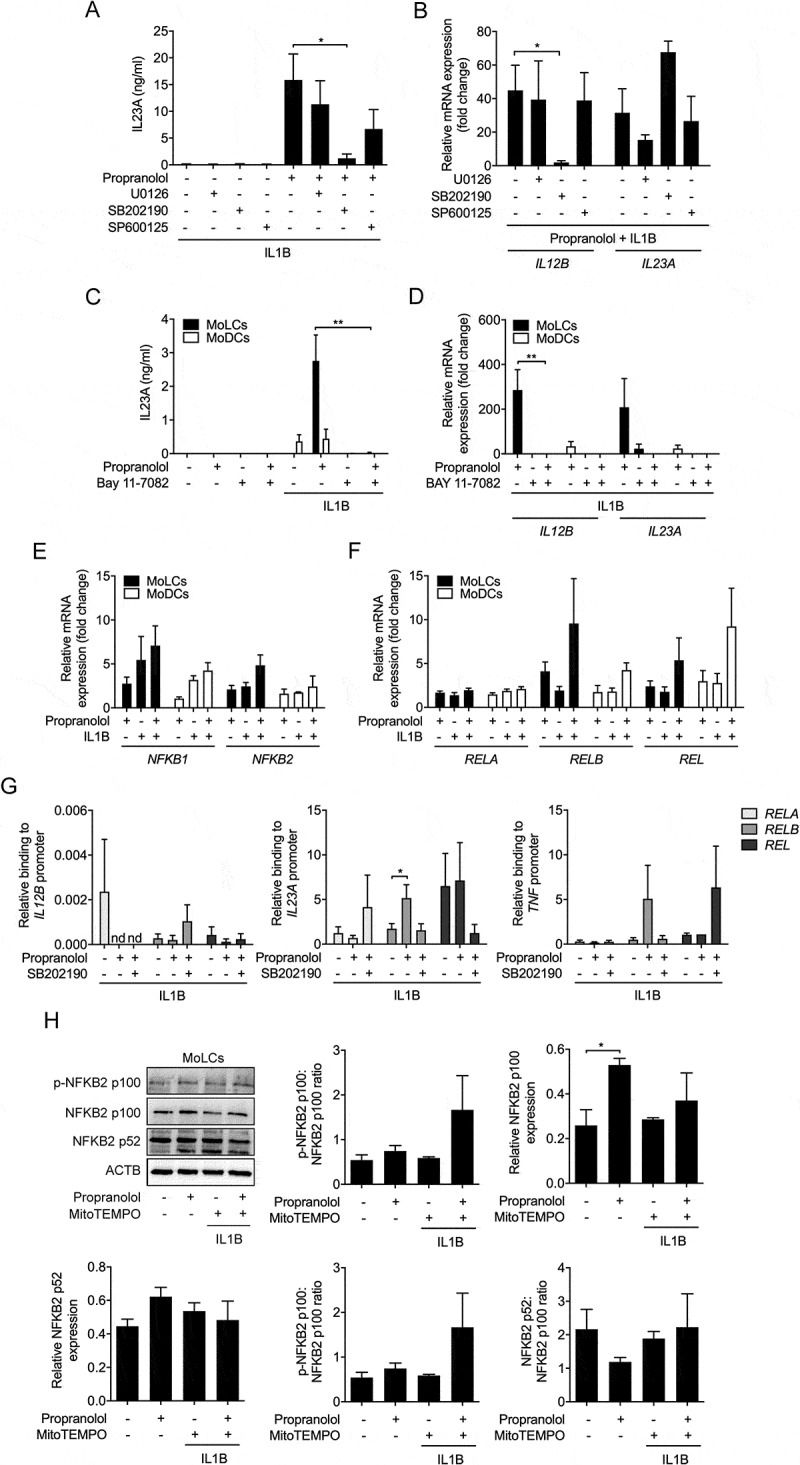
MAPK14 activity crucially regulates IL23A release. (A) IL23A secretion was analyzed by ELISA of IL1B-stimulated (20 ng/ml) MoLCs, stimulated for 24 h with or without propranolol (75 µM) in the presence or absence of a selective MAPK1-MAPK3 inhibitor (U0126, 10 µM), inhibitor of MAPK14 (SB 202,190, 10 µM) and MAPK8 inhibitor (SP 600,125, 10 µM). (B) Analysis of IL12B and IL23A mRNA expression in IL1B-activated (20 ng/ml) and propranolol-stimulated (75 µM) MoLCs with or without U0126 (10 µM), SB 202,190 (10 µM) or SP 600,125 (10 µM), respectively after 24 h. Gene transcripts were normalized to GAPDH and depicted relative to IL1B-activated MoLCs concomitantly treated with propranolol (set as 1.0). (C) IL23A release by immature and IL1B-activated (20 ng/ml) DC subsets after 24 h in presence or absence of propranolol (75 µM) and NFKB inhibitor Bay 11–7082 (10 µM) was determined by ELISA. (D) Levels of IL12B and IL23A mRNA expression in MoLCs and MoDCs, stimulated with IL1B (20 ng/ml) for 24 h with or without propranolol (75 µM) and Bay 11–7082 (10 µM). Gene copy numbers were normalized to GAPDH and presented relative to respective IL1B-stimulated DCs (set as 1.0). (E and F) Analysis of NFKB1 and NFKB2 as well as RELA, RELB and REL mRNA expression in MoLCs and MoDCs, activated with IL1B (20 ng/ml) alone or with propranolol (75 µM) for 3 h. Levels of mRNA were normalized to GAPDH and displayed relative to untreated controls (set as 1.0). (G) Binding of RELA, RELB and REL to promoters of IL12B, IL23A and TNF in MoLCs stimulated for 4 h with IL1B (20 ng/ml) in presence or absence of propranolol (75 µM) and inhibitor of MAPK14 (SB 202,190, 10 µM), respectively, assayed by ChIP and subsequently quantified by qRT-PCR. Data are presented relative to total input DNA (GAPDH). (H) Immunoblot analysis of total NFKB2 p100, NFKB2 p-NFKB2 p100, and NFKB2 p52 expression in whole-cell lysates of MoLCs, stimulated with IL1B (20 ng/ml) for 24 h with or without propranolol (75 µM) and MitoTEMPO (20 µM). (H) Protein expression was assessed by densitometric analysis with ACTB as control. (A-D, G and H) *P < 0.05, **P < 0.01, one-way ANOVA test followed by Bonferroni posttest. Data are representative of (A) n = 5, (B and H) n = 4, (C) n = 3, (D-F) n = 3–4, (G) n = 5–7 independent experiments and display mean values + SEM.
NFKB signaling plays an essential role in IL23A production
Our data indicate that IL23A secretion is crucially dependent on the activation of IL1R downstream signaling. To decipher the role of the IL1R signaling cascade mediating NFKB activity, we used BAY 11–7082 [37]. IL1B-induced IL23A secretion was abolished by BAY 11–7082 in propranolol-stimulated cells (Figure 6C). Similarly, BAY 11–7082 inhibited gene transcription of IL12B and IL23A (Figure 6D). To investigate whether propranolol regulates NFKB downstream transcription factors, we analyzed mRNA expression of NFKB-family class 1 and 2 genes. Since propranolol increased RELB and REL mRNA levels in IL1B-stimulated cells (Figure 6E,F), we next analyzed the role of propranolol in modulating IL1B-mediated recruitment of RELB, REL and RELA to IL23A promoters in MoLCs by chromatin immunoprecipitation (ChIP). Stimulation with propranolol significantly increased the recruitment of RELB to the IL23A but not IL12B promoter whereas RELA and REL were unaffected (Figure 6G). Consistent with cytokine expression, in the presence of the MAPK14 inhibitor SB202190, propranolol-induced RELB binding to the IL23A promoter region was blocked. Moreover, enhanced recruitment of RELB to TNF encoding gene binding sites was inhibited by MAPK14 blocking. Collectively, propranolol upregulated RELB recruitment to IL23A and TNF promoter regions in a manner dependent on MAPK14 activity.
In the cytosol, inactive RELB forms a heterodimeric complex with NFKB2 p100. Activating upstream signaling pathways induce post-translational processing of NFKB2 p100 via phosphorylation, consecutively leading to the formation of transcriptionally competent NFKB2 p52/RELB complexes [38]. Under sterile-inflammatory conditions, propranolol significantly increased expression of NFKB2 p100 and p-NFKB2 p100, yet in the presence of MitoTEMPO only NFKB2 p100 expression was reduced (Figure 6H).
Furthermore, the stimulation with IL1B together with propranolol led to no significant alterations of proteasomal converted NFKB2 p52. Since, neither p-NFKB2 p100:NFKB2 p100 nor NFKB2 p52:NFKB2 p100 ratios were affected, propranolol potentially increases NFKB2 p52/RELB signaling by elevating NFKB2 p100 levels without modulating post-translational modifications.
Discussion
The complex interplay between lysosomotropism, autophagy and cellular secretory machinery remains poorly understood. Here, we present a potentially critical immune modulatory effect induced by lysosomotropic beta-blockers and possibly other lysosomotropic drugs (Fig. S6). Propranolol elevated ROS formation and inhibited autophagic flux in cutaneous DCs leading to IL23A production, which is well in accordance with previous studies using chloroquine [22]. Small molecule-induced lysosomotropism, as demonstrated for chloroquine [39], depends on lipophilicity (clogP > 2) and the presence of a basic moiety (pKa > 6.5) [27]. Since propranolol induces lysosomal accumulation, we evaluated the immune regulatory effects of propranolol and other potentially lysosomotropic beta-blockers.
In IL1B-activated cutaneous DCs, that mimic sterile-inflammatory conditions, propranolol significantly promoted upregulation of IL23, independent of ADRB blockade. Correlating with decreasing clogP values, penbutolol (clogP = 3.84), metoprolol (clogP = 1.80) and timolol (clogP = 1.44) gradually increased IL23A release, while practolol (clogP = 0.53) failed to facilitate cytokine secretion. Thus, IL23A regulation and increased IL6, TNF and CXCL8/IL8 levels appear to be associated with lipophilicity of basic amines in agreement with the well-accepted criteria for lysosomotropic drugs. Although practolol provoked adverse cutaneous reactions in clinical trials, pursuant to low clogP, IL23A levels remained unchanged, indicating the involvement of other molecular mechanisms. Indeed, practolol-induced skin inflammation histologically differs from psoriatic lesions [40].
Compared to MoLCs, IL1B-stimulated MoDCs showed a less pronounced IL23A response but secreted higher amounts of CXCL8 in the presence of propranolol. The former is in line with recent data that reported a marginal upregulation of cell surface maturation markers CD83 and CD86 for MoDCs, whereas MoLCs evolved a semi-mature state under sterile-inflammatory conditions [41]. The underlying mechanisms for the latter effects are unknown, however, propranolol-induced CXCL8 mRNA levels were much lower in IL1B-activated MoDCs compared to MoLCs.
Moreover, our data indicated that specific receptor-dependent downstream signaling pathways involving MYD88- and subsequent TRAF6-mediated NFKB activation, are crucially affected by propranolol. The secretory machinery is tightly linked to NFKB activation, suggesting an immune regulatory effect of propranolol that modulates activity of closely shared IL1R and TLR4 downstream signaling molecules. Curdlan is known to potently trigger IL23A release via CLEC7A/dectin-1-mediated SYK-CARD9 signaling and polarize Th17 development [29]. Surprisingly, propranolol dampened secretion of IL23A and IL1B in both DC subsets while stimulated with curdlan. Consistent with no further evidence for its involvement in autoimmune disorders mediated by a pathogenic Th17 phenotype, CLEC7A downstream signaling is inhibited by lysosomotropic compounds.
Propranolol and chloroquine directed Th17 priming in naïve CD4+ T cells, but in the presence of anti-CD3-CD28 antibodies, mimicking antigen-presenting cell binding and thereby providing primary and co-stimulatory signals for T cell activation, lysosomotropic compounds reduced Th17 effector responses. The lack of specific polarizing and stabilizing antigen-presenting cell-derived cytokines is likely responsible for the absence of adaptive Th17-dependent signals. Supported by previously published data [22], lysosomotropism orchestrates a MoLC-dependent cytokine profile that presumably induce a Th17 phenotype under sterile-inflammatory conditions. Its role in naïve T cells interacting with DCs pre-stimulated with lysosomotropic compounds needs further exploration. Also, our work does not preclude the possibility that propranolol modulates CD8+ T cell function and T cell proliferation, which play a critical role in the pathogenesis of psoriasis. It is well established that autophagy inhibitors such as chloroquine inhibit T cell proliferation, which may be a mechanism of action in the treatment of autoimmune disease [42].
It was recently shown that IL1R downstream-dependent recruitment of TRAF6 induces autophagy [17], playing a pivotal role for the limitation of an inflammatory response [43].
Additional studies reported that autophagy terminates excessive inflammation in TLR signaling [44,45]. Mechanistically, propranolol increased MAP1LC3/LC3-II conversion, expression of autophagy marker SQSTM1/p62 and colocalization of TRAF6 and LC3, indicating propranolol as a late-stage inhibitor of autophagy. Lysosomotropic compounds are generally characterized by an accumulation within acidic organelles and alteration to their respective pH [46]. We confirmed this for both propranolol and chloroquine, which is in line with previous findings [27]. Cationic amphiphilic compounds, possessing similar physicochemical properties, have also been shown to introduce phospholipidosis, an excessive accumulation of phospholipids enclosed in lysosomal-related lamellar. However, LAMP1, representing the physical presence of lysosomal lamellar bodies, was downregulated by propranolol under sterile inflammatory conditions indicating no further evidence for phospholipidosis.
Due to functional inhibition of autophagy, elevated expression of signaling molecules downstream of IL1R might further increase autophagic flux, thereby inducing an amplification loop [47]. However, recent studies provided evidence that several lysosomotropic compounds directly induce endolysosomal LC3 lipidation in a non-canonical fashion [48,49]. Since EGFR regulation is limited to MoDCs, endocytic trafficking likely plays a secondary role. Nonetheless, an impaired IL1R internalization provoked by propranolol might contribute to an enhanced expression of IL1B-triggered signaling components.
In addition, we could not detect co-localization of LC3-II with lysosome-marker LAMP1 further confirming an impaired maturation of autolysosomes. Indeed, we observed elevated ubiquitin-conjugated intracellular content promoted by propranolol. Notably, inhibition of ubiquitin-proteasome pathway resulted in similar ubiquitination and however, abolished propranolol-induced IL23A secretion. This effect is presumably due to an indirect inhibition of IL1R downstream transduced NFKB activity, blocking proteasomal degradation of NFKBIA/IκB [50,51]. However, propranolol exclusively provoked an increased expression of NLRP3 that is known to be degraded in an autophagy-dependent pathway. In discordance to IL1B regulation, IL1B-induced conditions with or without propranolol diminished NLRP3 expression. The assembly of NLRP3 with PYCARD and CASP1, known as the NLRP3 inflammasome, is positively regulated by TRAF6 activation [52]. Thus, sterile inflammatory conditions may mask antibody binding. The NLRP3 inflammasome complex is crucial in orchestrating the production and secretion of IL1 family cytokines including IL1B and IL18. However, inhibition of NLRP3 inflammasome activity did not alter propranolol-induced IL23A production. In addition, propranolol led to increased pro-IL1B expression but neither modulated CASP1 activity by inducing IL1B maturation, nor orchestrated IL18 secretion in MoLCs. In line with previous studies, reporting a selective regulation of TRAF6 by SQSTM1 as well as based on our results, increased pro-IL1B expression is likely due to an accumulation of IL1R downstream-dependent signaling molecules that are presumably degraded by the autophagy-lysosomal pathway [18,34]. Taken together, these results potentially rule out the possibility of lysosomotropism-induced IL23A secretion triggered by the NLRP3 inflammasome.
Although MG132 is preferably used as proteasomal inhibitor, it may affect lysosomal hydrolases and thus, the involvement of the autophagy-lysosomal system cannot be excluded. Lysosomal hydrolases require acidic pH for adequate function. Propranolol, chloroquine and bafilomycin A1, alkalized lysosomal lumen, therefore we suggest an exclusive block of the proteasome by MG132.
Propranolol rapidly provoked intracellular ROS production in MoLCs. The ROS scavenger NAC completely abolished propranolol-mediated IL23A release. Intriguingly, in MoDCs, no increase of ROS was detected, possibly explaining the less pronounced increase in IL23A production. In naïve CD4+ T cells, we additionally observed no emergence of ROS intrinsically driving a Th17 phenotype. Therefore, we suggest an essential role by the specific DC-derived cytokine pattern provoked by lysosomotropism to induce a psoriasis-like skin inflammation.
Previous reports indicate a crucial role for ROS, elevating IL1B and/or IL23A secretion in autophagy-deficient and functionally autophagy-inhibited mouse BMDCs [18,53]. Although several ROS sources are known, depolarized mitochondria are most likely involved in ROS formation [54]. Indeed, ROS are profoundly derived from dysregulated mitochondria in MoLCs. The precise mechanism by which ROS are generated by propranolol remains to be identified. Furthermore, propranolol markedly induced PINK1 expression and similarly increased mtDNA deposits while PRKN levels are not effectively regulated. PINK1 is preferentially recruited to depolarized mitochondria leaking mtDNA, thereby facilitating PRKN recruitment for subsequent elimination of mitochondria [55]. Indicating a consecutive recruitment, this may account for a decelerated upregulation of PRKN. However, inhibition of propranolol-induced mitochondrial ROS overproduction downregulated PINK1 and PRKN levels and concomitantly reduced IL23A release. In line with previous studies reporting that selective mitochondria depletion is tightly linked to the PINK1- and PRKN-directed autophagic machinery, lysosomotropism induced an accumulation of mitochondria linking an impaired autophagy-lysosomal clearance to ROS formation and IL23A secretion [56].
While IL1R downstream signaling is mandatory to activate the secretory machinery, propranolol alone substantially enhanced IL12B and IL23A gene transcription. Besides IL1R engagement, TLR-induced NFKB activation leads to histone modification and chromatin remodeling, fostering accessibility of IL12B and IL23A promoter regions for transcription factors in DCs [57,58]. Concordantly, inhibition of NFKB signaling resulted in a complete abrogated gene transcription of IL23A subunits. Thus, the mandatory NFKB recruitment to specific IL12B and IL23A promoter is regulated by propranolol.
Among MAPK signaling, MAPK14/p38 has been linked to phosphorylation and phosphoacetylation of histone H3, allowing access of NFKB transcription factors to IL12B and IL23A promoter regions [59]. According to current reports, functional inhibition of MAPK signaling unraveled MAPK14 as a crucial IL1R downstream signaling molecule of IL23A production [60,61]. Importantly, MAPK14 signaling stimulates IL23A production in LCs in vivo leading to IL17A-producing T cells and psoriasiform skin inflammation in mice [21]. Consistently, we demonstrated that IL23A secretion and, in particular, IL12B gene transcription is strictly dependent on MAPK14 activity in MoLCs. We previously detected that chloroquine-stimulated MoLCs elevated MAPK expression, suggesting that IL23A induction might be dependent on amplified MAPK activity mediated by increased signaling molecules downstream of IL1R, presumably due to inhibition of autophagic flux by lysosomotropic compounds.
Several pieces of evidence indicate a requirement of REL activity for the upregulation of the Il12b promoter in mouse DCs as well as macrophages and in addition, supported by currently published data, MAPK14-regulated histone modifications possibly control REL binding to Il12b promoter [59,62]. Conversely, our data indicated an enhanced propranolol-mediated RELB recruitment to IL23A promoter regions in IL1B-activated MoLCs that was completely abolished by specific block of MAPK14. Although IL23A secretion remained almost unaffected by inhibition of MAPK1-MAPK3, we noticed a slight reduction of IL23A. Thus, besides an apparent mandatory MAPK14 activity for IL23A transcription, MAPK1-MAPK3 may additionally be involved in histone modifications, that enable access to IL23A promoter regions [16], but further studies are required. Moreover, we observed a conspicuously propranolol-induced TNF promoter binding by RELB. Again, potentially MAPK14-induced histone modifications appeared essential for enhanced TNF transcription. Expression of the RELB upstream signaling component NFKB2 p100 is profoundly increased by propranolol, whereas concurrent scavenging of mitochondria-produced ROS maintained NFKB2 p100 levels. Indicating a pivotal function for mitochondria-derived ROS. Besides a prominent regulation of NFKB2 p100, propranolol failed to alter subsequent post-translational activation. Previous studies indicate that endogenous-derived ROS and supplemented H2O2 alter the TRAF6-mediated non-canonical upstream MAP3K14/NIK (mitogen-activated protein kinase kinase kinase 14) pathway, thereby promoting RELB-NFKB2 p52 transcriptional activities [63,64]. With respect to propranolol-induced NFKB2 gene transcription as well as upregulated NFKB2 p100 levels, and in concordance that Nfkb2-deficient mouse macrophages elevated Il23a expression [16], we suggest a putative role for ROS in transducing a non-canonical NFKB activation.
Collectively, our data suggest a pharmacological off-target effect for lysosomotropic beta-blocker and possible other drugs that may critically affect late-stages of autophagic flux and induce cytokine release under inflammatory conditions. A limitation of our study is that we did not use knockout cells to confirm the role of the underlying pathways. Although recent progress has been made in genome editing of primary human immune cells such as T cells, generation of human knockout cells remains technically challenging [65]. Presumably due to a crosstalk of propranolol-induced ROS formation and enhanced non-canonical NFKB signaling, cutaneous DCs respond with IL23A secretion. Propranolol might exert its immune-regulatory functions via inhibition of autophagy-dependent negative feedback loops with subsequent increase in IL1R downstream signaling and dysregulation of mitochondria. Enhanced MAPK activity upregulates the accessibility of promoter regions for transcriptionally active NFKB proteins, and thus varies affinities to IL23A κB sites in distinct DNA sequences. ROS acting as a signal transducer may contribute to non-canonical NFKB signaling converging innate and adaptive immune responses. Thus, we propose lysosomotropic compounds are a crucial trigger to dendritic cells, exacerbating Th17-related psoriasis-like skin inflammation. Importantly, these findings may hold implications for other IL23-IL17A axis mediated auto-immune disorders.
Materials and methods
Generation of MoLCs and modcs from human monocytes
Isolated human monocytes were isolated and differentiated into MoLCs and MoDCs, respectively [22,41]. In brief, peripheral blood mononuclear cells (PBMCs) were obtained from buffy-coat donations from anonymous healthy volunteers (DRK-Blutspendedienst Ost) after informed consent. MoLCs and MoDCs were generated from adherent human monocytes cultured in RPMI 1640 (Sigma-Aldrich, R0883) supplemented with 2 mM L-glutamine (Sigma-Aldrich, G7513), 100 U/ml penicillin, 100 µg/ml streptomycin (Sigma-Aldrich, P4333), 10% heat inactivated FCS (Biochrom Ltd, S0615) and additionally with rh-CSF2/GM-CSF (100 ng/ml; Miltenyi Biotec, 130-093-866), rh-IL4 (20 ng/ml; Miltenyi Biotec, 130-093-922) and with or without rh-TGFB1/TGF-β1 (20 ng/ml; Miltenyi Biotec, 130-095-067). After 6 d, suspension cells were collected and sorted by CD1a MicroBeads (clone HI149; Miltenyi Biotech, 130-051-001).
Isolation and in vitro activation of naïve CD4+ T cells from human pbmcs
Naïve human CD4+ T cells were isolated from PBMCs by negative selection using magnetic-activated cell sorting beads according to the manufacturer’s instructions (MACS; Miltenyi-Biotec, 130-096-533) [22,66]. Purified naïve CD4+ T cells were washed with PBS (Sigma-Aldrich, D8537), seeded in a 24-well cell culture plate (VWR, 734–0056) at a density of 106 cells per ml and further cultured in RPMI 1640 (Sigma-Aldrich, R0883) supplemented with 2 mM L-glutamine (Sigma-Aldrich, 59202C), 10% heat inactivated FCS (Biochrom, S0615) for 5 and 7 d respectively. To induce activation, ImmunoCult Human CD3-CD28 T cell Activator (STEMCELL Technologies, 10971) was added to complete medium.
Functional assays of DCs in vitro
After in vitro differentiation, MoLCs and MoDCs were washed with PBS (Sigma-Aldrich, D8537) and seeded into 24-well cell culture plates (VWR, 734–0056) in complete medium without supplemented cytokines at a density of 106 cells/ml. For subsequent stimulation, the following agonists were applied to the cell culture medium: rh-IL1B/IL-1β (20 ng/ml; eBioscience, 14–8018), rh-IL36G (100 ng/ml; PeproTech, 200-36G), ultrapure LPS from Escherichia coli serotype 0111:B4 (1 µg/ml; InvivoGen, tlrl-3pelps), curdlan, a beta-1,3-glucan extracted from Alcaligenes faecalis (20 µg/ml; InvivoGen, tlrl-curd).
DCs or naïve CD4+ T cells were pre-incubated with propranolol (25–150 µM; Sigma-Aldrich, P0884), penbutolol (1–25 µM; P0307000), timolol (25–150 µM; Tocris Bioscience, 0649), metoprolol (25–150 µM; Sigma-Aldrich, A9165), practolol (25–150 µM; Tocris Bioscience, 0831), and for autophagy blocking experiments, chloroquine (20 µM; Sigma-Aldrich, C6628), bafilomycin A1 (1 µM; Tocris Bioscience, 1334) for 1 h. Afterward, MoLCs and MoDCs were activated with different agonists as mentioned above.
For functional assays, MoLCs and MoDCs were pre-incubated with SQ 22,536 (100 µM; Cayman Chemical, 13,339), forskolin (20 µM; Cayman Chemical, 11,018), MG132 (10 µM; Sigma-Aldrich, M7449), N-acetyl-L-cysteine (NAC; 20 mM; Sigma-Aldrich, A9165), MCC950 (5 µM; Sigma-Aldrich, 5.38120), MitoTEMPO (20 µM; Sigma-Aldrich, SML0737) in complete medium for 1 h with or without propranolol (75 µM). Afterward, MoLCs and MoDCs were additionally stimulated with rh-IL1B (20 ng/ml) with or without propranolol (75 µM).
To analyze the regulatory effect of propranolol on IL1R downstream signaling, experiments were performed analog to functional assays in the presence of the selective MAPK inhibitors U0126 (MAPK1-MAPK3 inhibitor; Tocris Bioscience, 1144), SB 202190 (MAPK14/p38 MAPK inhibitor; Sigma-Aldrich, S7067) and SP 600125 (MAPK8/JNK inhibitor; Sigma-Aldrich, S5567) at 10 µM or Bay 11–7082 (10 µM; Sigma-Aldrich, B5556), a functional inhibitor of NFKB/NF-κB mediated signaling.
Cell viability
Cell viability was determined by trypan blue exclusion assay (Sigma-Aldrich, T6146). Experiments were performed in quadruplicates. Additionally, cell viability was evaluated by ANXA5-FITC/PI assay (eBioscience, 88-8005-72). Single-cell suspensions were subjected to flow cytometry (CytoFlex, Beckman Coulter, Krefeld, Germany) with a total of 20 × 103 events.
Elisa
After 24 h of stimulation, cell culture supernatants were collected and assayed for cytokine secretion using commercially available ELISA kits: IL1B (ELISA-Ready Set Go; Invitrogen, 501125197), IL6, CXCL8/IL8 (ELISA-Ready Set Go; eBioscience, 88-7066-88), IL12 (ELISA-Ready Set Go; eBioscience, 88–7126), IL17A (ELISA-Ready Set Go; eBioscience, 88-7176-88), IL18 (DuoSet; R&D Systems, DY318-05), IL22 (DuoSet; R&D Systems, DY782), IL23A (DuoSet; R&D Systems, DY1290) and TNF (ELISA-Ready Set Go; eBioscience, 88–7346).
Flow cytometry
Single-cell suspensions of MoDCs were stained with PE-labeled anti-CD83 (BioLegend, 305308) and FITC-labeled anti-CD86 (Miltenyi Biotech, 130-113-571) antibodies and subsequently analyzed by flow cytometry (CytoFlex, Beckman Coulter) for 10 × 103 events.
RNA isolation, cdna synthesis and qRT-PCR
Total RNA isolation, cDNA synthesis and quantitative real-time RT-PCR (qRT-PCR) were performed as described [67]. Primers (synthesized by TIB Molbiol) with the following sequences were used: CXCL8, GAPDH, IL1B, IL6, IL12A, IL12B, IL23A, RORC and TNF as published previously [22,68,69]; ATF3, 5ʹ-CCTCGGAAGTGAGTGCTTCT-3ʹ and 5ʹ-ATGGCAAACCTCAGCTCTTC-3ʹ; ATF6, 5ʹ-AAGCCCTGATGGTGCTAACTGAA-3ʹ and 5ʹ-CATGTCTATGAACCCATCCTCGAA-3ʹ; NFKB1, 5ʹ-AGAAGTCTTACCCTCAGGTCAAA-3ʹ and 5ʹ-TCCAGCAGTTACAGTGCAGAT-3ʹ; NFKB2, 5ʹ-AAGGACATGACTGCCCAATTTAA-3ʹ and 5ʹ-ATCATAGTCCCCATCATGTTCTTCTTC-3ʹ; RELA, 5ʹ-CTGCCGGGATGGCTTCTAT-3ʹ and 5ʹ-CCGCTTCTTCACACACTGGAT-3ʹ; RELB, 5ʹ-AGCATCCTTGGGGAGAGC-3ʹ and 5ʹ-GAGGCCAGTCCTTCCACAC-3ʹ; REL/c-Rel, 5ʹ-CAACCGAACATACCCTTCTATCC-3ʹ and 5ʹ-TCTGCTTCATAGTAGCCGTCT-3ʹ; IL17A, 5ʹ-CTCATTGGTGTCACTGCTACTG-3ʹ and 5ʹ-CCTGGATTTCGTGGGATTGTG-3ʹ. Fold difference in gene expression was normalized to the housekeeping gene GAPDH, showing the most constant expression levels. The reaction mix including cDNA template, primers and SYBR Green (iTaq Universal SYBR Green Supermix; Bio-Rad, 172–5125) was run under the conditions as previously described.
DNA isolation, determination of mtdna copy number
After 48 h, total DNA was extracted using the innuPREP DNA Mini Kit (AnalytikJena, 845-KS-2040050), according to manufacturer’s instructions. For the evaluation of mitochondria DNA (mtDNA) content, the ratio of mtDNA:gDNA (mitochondrial to genomic DNA) was determined. As recently published, mtDNA copy number was assessed by using primers for a unique 129-bp fragment called hmito3 with following sequences: 5ʹ- CACTTTCCACACAGACATCA-3ʹ and 5ʹ-TGGTTAGGCTGGTGTTAGGG-3ʹ [70]. Mitochondrial DNA expression was determined by normalization to genomic DNA, represented by ALDOA expression with following sequences: 5ʹ- CGGGAAGAAGGAGAACCTG-3ʹ and 5ʹ-GACCGCTCGGAGTGTACTTT-3ʹ. The reaction mix of DNA template, primers and SYBR Green was assayed under the same conditions as described above [67].
Immunofluorescence
Immunofluorescence was performed as described previously with minor modifications [70]. Cells were fixed with ice-cold methanol (VWR, 1.06009.1000) for 10 min. Primary antibodies used, were rabbit-anti-MAP1LC3A/LC3A (1:400; Cell Signaling Technology, 4599), mouse-anti-TRAF6 (1:50; Santa Cruz Biotechnology, sc-8409), rat-anti-EGFR (1:100; Bio-Rad, MCA1784) and mouse-anti-LAMP1 (1:400; BioLegend, 328601). Secondary DyLight488-conjugated anti-rabbit (1:400; Dianova, A23220-05) or AlexaFluor488-conjugated anti-mouse antibodies (Cell Signaling Technology, 4408) as well as AlexaFluor594-conjugated anti-rabbit or anti-mouse antibodies (1:400; Cell Signaling Technology, 4412 or 8890) and AlexaFluor647-conjugated anti-rat (1:1000; Thermo Fisher Scientific, A-21247) were applied after washing for 1 h at room temperature. Cells were mounted in ImmunoSelect Antifading Mounting Medium with 4ʹ,6-diamidino-2-phenylindole (DAPI, Dianova, SCR-38448). Images were captured using a confocal laser scanning microscope Leica SP8 (Leica microsystems, Wetzlar, Germany).
Internal pH measurement
The acidotropic agent LysoTracker Red DND-99 (Invitrogen, L7528) was freshly diluted to a final concentration of 100 nM in RPMI complete in the absence of supplemented cytokines. After 0.5 h and 4 h, respectively, cells were collected, washed in PBS and subsequently loaded with LysoTracker Red DND-99 (100 nM) for 30 min at 37°C according to the manufacturer’s recommendations. Again, MoLCs were harvested, washed and LysoTracker Red-uptake was determined by flow cytometry (CytoFlex, Beckman Coulter), examining a total of 10 × 103 events.
ROS quantification
Total internal ROS formation was determined with Total Reactive Oxygen Species Assay Kit (eBioscience, 88–5930) in concordance with manufacturer’s instructions. In brief, prior to stimulation, MoLCs and MoDCs were labeled with provided ROS assay stain for 1 h at 37°C in complete medium without supplemented cytokines. Following stimulation for 1 h and additionally 3 h, cells were collected, washed and immediately analyzed by using flow cytometry (CytoFlex, Beckman Coulter) for 10 × 103 events.
Mitosox assay
MitoSOX Red (Invitrogen, M36008) was diluted in medium without supplemented cytokines to a final concentration of 5 µM. MoLCs were re-suspended in MitoSOX Red containing medium for 10 min at 37°C and subsequently cultivated. The following fluorescence-labeled cells were stimulated for 24 h. Afterward, MoLCs were harvested, washed with PBS and simultaneously examined by flow cytometry (CytoFlex, Beckman Coulter) for 10 × 103 events or mounted on poly-lysine coated slide (Thermo Fisher Scientific) using cytospin procedure as described [22]. ImmunoSelect Antifading Mounting Medium with 4ʹ,6-diamidino-2-phenylindole (DAPI, Dianova, SCR-38,448) was applied and microscopy was carried out using a BZ-8000 fluorescence microscope (Keyence, Itasca, United States).
Western blotting
Cell lysis and protein isolation was performed as previously described [70]. Protein lysates were separated by 10% SDS polyacrylamide gel containing Rotiphorese Gel 40 (Carl Roth, T802), TRIS (Carl Roth, 5429), SDS (Carl Roth, 0183), TEMED (Carl Roth, 2367), ammonium persulfate (Sigma-Aldrich, A3678) in distilled deionized water using Mini-PROTEAN electrophoresis system (Bio-Rad, Munich, Germany). Gels were blotted onto polyvinylidene difluoride membranes (Immun-Blot PVDF; Bio-Rad, 1620177). After blocking with 5% skimmed-milk powder (Sucofin, 12307) for 1 h at 37°C, membranes were incubated with primary rabbit antibodies at 1:1000: MAP1LC3B/LC3B (Cell Signaling Technology, 2775), SQSTM1/p62 (Cell Signaling Technology, 8025), Ubiquitin (Cell Signaling Technology, 3933), NLRP3 (Cell Signaling Technology,15101), IL1B (Cell Signaling Technology,12703), PRKN/parkin (Cell Signaling Technology, 2132), PINK1 (Cell Signaling Technology, 6946), phospho-NFKB2 p100 (Cell Signaling Technology, 4810), NFKB2 p100/p52 (Cell Signaling Technology, 4882) and LAMP1 (BioLegend, 328601) overnight at 4°C, and subsequently incubated with anti-rabbit HRP-conjugated secondary antibody (1:1000; Cell Signaling Technology, 7074) for 1 h. Then blots were developed with SignalFire ECL reagent (Cell Signaling Technology, 6883) and signals were visualized by PXi/PXi Touch gel imaging system (Syngene, Cambridge, UK). The membranes were stripped with restore western blot stripping buffer (Thermo Fisher Scientific, 21059) and further re-incubated with anti-ACTB/β-actin rabbit antibody (1:1000; Cell Signaling Technology, 4970). Values of protein expression were analyzed by densitometry and normalized to ACTB levels using ImageJ version 1.46r (NIH) verifying for non-saturation and subtracting background.
Chip assay
ChIP was assayed with EpiQuik ChiP kit (Epigentek, P-2002) in concordance with manufacturer’s instructions. In particular, pre-stimulated cells were cross-linked with 1% formaldehyde (Sigma-Aldrich, F8775) in complete medium for 10 min and subsequently lysed. DNA was sheared by sonication. Chromatin fraction was immuno-precipitated for 90 min with rabbit-anti-RELA (1:100; Cell Signaling Technology, 8242), rabbit-anti-RELB (1:50; Cell Signaling Technology, 10544) and rabbit-anti-REL (1:50; Cell Signaling Technology, 12659). Cross-linking was reversed at 65°C for 90 min. Input DNA and precipitated DNA were purified and subsequently analyzed with qRT-PCR by primers including IL12B, IL23A, TNF, 5ʹ-CCCAGGGACCTCTCTCTAATCA-3ʹ and 5ʹ-GCTACAGGCTTGTCACTCGG-3ʹ and normalized to GAPDH.
Statistical analysis
Data are expressed as means ± SEM. For multiple comparisons, statistically significant differences were determined by one-way ANOVA followed by a Bonferroni post-hoc test and considered significant at *P < 0.05, **P < 0.01, ***P < 0.001. Statistical differences against unstimulated control (fold change) were assessed by unpaired Student`s t-test or one-sample t-test. Statistical analysis was performed using GraphPad Prism software.
Supplementary Material
Acknowledgments
We gratefully acknowledge the technical support provided by Dr. Katharina Achazi (Core Facility BioSupraMol, FU Berlin, Germany). We thank Dr. Guy Yealland for language editing.
Funding Statement
This work was supported by the German Ministry of Education and Research under Grant [031A262A].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Klionsky DJ, Emr SD.. Cell biology - Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Klionsky DJ, Eskelinen EL, Deretic V.. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes … wait, I’m confused. Autophagy. 2014;10:549–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dunn WA Jr. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994;4:139–143. [DOI] [PubMed] [Google Scholar]
- [4].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- [5].Ashoor R, Yafawi R, Jessen B, et al. The contribution of lysosomotropism to autophagy perturbation. PloS One. 2013;8:e82481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reasor MJ, Hastings KL, Ulrich RG. Drug-induced phospholipidosis: issues and future directions. Expert Opin Drug Saf. 2006;5:567–583. [DOI] [PubMed] [Google Scholar]
- [7].Schön M, Behmenburg C, Denzer D, et al. Pathogenic function of IL-1 beta in psoriasiform skin lesions of flaky skin (fsn/fsn) mice. Clin Exp Immunol. 2001;123:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. [DOI] [PubMed] [Google Scholar]
- [9].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Deretic V, Kimura T, Timmins G, et al. Immunologic manifestations of autophagy. J Clin Invest. 2015;125:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Harris KM, Fasano A, Mann DL. Cutting edge: IL-1 controls the IL-23 response induced by gliadin, the etiologic agent in celiac disease. J Immunol. 2008;181:4457–4460. [DOI] [PubMed] [Google Scholar]
- [12].Peral de Castro C, Jones SA, Ni Cheallaigh C, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol. 2012;189:4144–4153. [DOI] [PubMed] [Google Scholar]
- [13].Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. [DOI] [PubMed] [Google Scholar]
- [14].Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Utsugi M, Dobashi K, Ishizuka T, et al. Rac1 negatively regulates lipopolysaccharide-induced IL-23 p19 expression in human macrophages and dendritic cells and NF-kappaB p65 trans activation plays a novel role. J Immunol. 2006;177:4550–4557. [DOI] [PubMed] [Google Scholar]
- [16].Mise-Omata S, Kuroda E, Niikura J, et al. A proximal kappa B site in the IL-23 p19 promoter is responsible for RelA- and c-rel-dependent transcription. J Immunol. 2007;179:6596–6603. [DOI] [PubMed] [Google Scholar]
- [17].Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Singh TP, Zhang HH, Borek I, et al. Monocyte-derived inflammatory Langerhans cells and dermal dendritic cells mediate psoriasis-like inflammation. Nat Commun. 2016;7:13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zheng T, Zhao W, Li H, et al. p38alpha signaling in Langerhans cells promotes the development of IL-17-producing T cells and psoriasiform skin inflammation. Sci Signal. 2018;11:eaao1685. [DOI] [PubMed] [Google Scholar]
- [22].Said A, Bock S, Lajqi T, et al. Chloroquine promotes IL-17 production by CD4+ T cells via p38-dependent IL-23 release by monocyte-derived Langerhans-like cells. J Immunol. 2014;193:6135–6143. [DOI] [PubMed] [Google Scholar]
- [23].Wachter SB, Gilbert EM. Beta-adrenergic receptors, from their discovery and characterization through their manipulation to beneficial clinical application. Cardiology. 2012;122:104–112. [DOI] [PubMed] [Google Scholar]
- [24].Tsankov N, Angelova I, Kazandjieva J. Drug-induced psoriasis. Recognition and management. Am J Clin Dermatol. 2000;1:159–165. [DOI] [PubMed] [Google Scholar]
- [25].Brauchli Y, Jick S, Curtin F, et al. Association between beta‐blockers, other antihypertensive drugs and psoriasis: population‐based case–control study. Brit J Dermatol. 2008;158:1299–1307. [DOI] [PubMed] [Google Scholar]
- [26].Lemieux B, Percival MD, Falgueyret JP. Quantitation of the lysosomotropic character of cationic amphiphilic drugs using the fluorescent basic amine Red DND-99. Anal Biochem. 2004;327:247–251. [DOI] [PubMed] [Google Scholar]
- [27].Nadanaciva S, Lu S, Gebhard DF, et al. A high content screening assay for identifying lysosomotropic compounds. Toxicol In Vitro. 2011;25:715–723. [DOI] [PubMed] [Google Scholar]
- [28].Towne JE, Sims JE. IL-36 in psoriasis. Curr Opin Pharmacol. 2012;12:486–490. [DOI] [PubMed] [Google Scholar]
- [29].Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang YH, Lin JX, Vilcek J. Synthesis of interleukin 6 (Interferon-Beta-2/B cell stimulatory factor-Ii) in human-fibroblasts is triggered by an increase in intracellular Cyclic-Amp. J Biol Chem. 1988;263:6177–6182. [PubMed] [Google Scholar]
- [31].Felix RH, Ive FA, Dahl MG. Cutaneous and ocular reactions to practolol. Br Med J. 1974;4:321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liang SC, Tan XY, Luxenberg DP, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Arican O, Aral M, Sasmaz S, et al. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005;2005:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sanz L, Diaz-Meco MT, Nakano H, et al. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. Embo J. 2000;19:1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chuang SY, Yang CH, Chou CC, et al. TLR-induced PAI-2 expression suppresses IL-1beta processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci U S A. 2013;110:16079–16084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shuda M, Kondoh N, Imazeki N, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38:605–614. [DOI] [PubMed] [Google Scholar]
- [37].Strickson S, Campbell DG, Emmerich CH, et al. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem J. 2013;451:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. [DOI] [PubMed] [Google Scholar]
- [39].Kazmi F, Hensley T, Pope C, et al. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab Dispos. 2013;41:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sondergaard J, Wadskov S, Jensen HA, et al. Aggravation of psoriasis and occurrence of psoriasiform cutaneous eruptions induced by practolol (Eraldin). Acta Derm Venereol. 1976;56:239–243. [PubMed] [Google Scholar]
- [41].Said A, Bock S, Müller G, et al. Inflammatory conditions distinctively alter immunological functions of Langerhans-like cells and dendritic cells in vitro. Immunology. 2015;144:218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jia W, He MX, McLeod IX, et al. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy. 2015;11:2335–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Giegerich AK, Kuchler L, Sha LK, et al. Autophagy-dependent PELI3 degradation inhibits proinflammatory IL1B expression. Autophagy. 2014;10:1937–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ishizaki J, Yokogawa K, Ichimura F, et al. Uptake of imipramine in rat liver lysosomes in vitro and its inhibition by basic drugs. J Pharmacol Exp Ther. 2000;294:1088–1098. [PubMed] [Google Scholar]
- [47].Moscat J, Diaz-Meco MT. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell. 2009;137:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Florey O, Gammoh N, Kim SE, et al. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy. 2015;11:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jacquin E, Leclerc-Mercier S, Judon C, et al. Pharmacological modulators of autophagy activate a parallel noncanonical pathway driving unconventional LC3 lipidation. Autophagy. 2017;13:854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Meiners S, Laule M, Rother W, et al. Ubiquitin-proteasome pathway as a new target for the prevention of restenosis. Circulation. 2002;105:483–489. [DOI] [PubMed] [Google Scholar]
- [51].Qin Z, Cui B, Jin J, et al. The ubiquitin-activating enzyme E1 as a novel therapeutic target for the treatment of restenosis. Atherosclerosis. 2016;247:142–153. [DOI] [PubMed] [Google Scholar]
- [52].Xing Y, Yao X, Li H, et al. Cutting edge: TRAF6 mediates TLR/IL-1R signaling-induced nontranscriptional priming of the NLRP3 inflammasome. J Immunol. 2017;199:1561–1566. [DOI] [PubMed] [Google Scholar]
- [53].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. [DOI] [PubMed] [Google Scholar]
- [54].Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. [DOI] [PubMed] [Google Scholar]
- [55].Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Weinmann AS, Plevy SE, Smale ST. Rapid and selective remodeling of a positioned nucleosome during the induction of IL-12 p40 transcription. Immunity. 1999;11:665–675. [DOI] [PubMed] [Google Scholar]
- [58].Garrett S, Fitzgerald MC, Sullivan KE. LPS and poly I: C induce chromatin modifications at a novel upstream region of the IL-23 p19 promoter. Inflammation. 2008;31:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Saccani S, Pantano S, Natoli G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol. 2002;3:69–75. [DOI] [PubMed] [Google Scholar]
- [60].Canavese MPM, Dombrowski Y, Koglin S, et al. VEGF induces IL-23 expression in keratinocytes through p38 signaling. J Clin Exp Dermatol Res. 2011;S2:002. [Google Scholar]
- [61].Zhu L, Wu Y, Wei H, et al. Up-regulation of IL-23 p19 expression in human periodontal ligament fibroblasts by IL-1beta via concurrent activation of the NF-kappaB and MAPKs/AP-1 pathways. Cytokine. 2012;60:171–178. [DOI] [PubMed] [Google Scholar]
- [62].Sanjabi S, Hoffmann A, Liou HC, et al. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci U S A. 2000;97:12705–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang C, Deng L, Hong M, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. [DOI] [PubMed] [Google Scholar]
- [64].Li Q, Engelhardt JF. Interleukin-1beta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J Biol Chem. 2006;281:1495–1505. [DOI] [PubMed] [Google Scholar]
- [65].Simeonov DR, Marson A. CRISPR-Based Tools in Immunity. Annu Rev Immunol. 2019;37:571–597. [DOI] [PubMed] [Google Scholar]
- [66].Bock S, Murgueitio MS, Wolber G, et al. Acute myeloid leukaemia-derived Langerhans-like cells enhance Th1 polarization upon TLR2 engagement. Pharmacol Res. 2016;105:44–53. [DOI] [PubMed] [Google Scholar]
- [67].Weindl G, Castello F, Schäfer-Korting M. Evaluation of anti-inflammatory and atrophogenic effects of glucocorticoids on reconstructed human skin. Altern Lab Anim. 2011;39:173–187. [DOI] [PubMed] [Google Scholar]
- [68].Weindl G, Naglik JR, Kaesler S, et al. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. The J Clin Invest. 2007;117:3664–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hamidi S, Schäfer-Korting M, Weindl G. TLR2/1 and sphingosine 1-phosphate modulate inflammation, myofibroblast differentiation and cell migration in fibroblasts. Biochim Biophys Acta. 2014;1841:484–494. [DOI] [PubMed] [Google Scholar]
- [70].Malik AN, Shahni R, Rodriguez-de-Ledesma A, et al. Mitochondrial DNA as a non-invasive biomarker: accurate quantification using real time quantitative PCR without co-amplification of pseudogenes and dilution bias. Biochem Biophys Res Commun. 2011;412:1–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.