

ABSTRACT
To maintain genome stability, chromosomes must be equally distributed among daughter cells at the end of mitosis. The accuracy of chromosome segregation requires sister-kinetochores to stably attach to microtubules emanating from opposite spindle poles. However, initial kinetochore-microtubule interactions are able to turnover so that defective attachment configurations that typically arise during early mitosis may be corrected. Growing evidence supports a role for the RZZ complex in preventing the stabilization of erroneous kinetochore-microtubule attachments. This inhibitory function of RZZ toward end-on attachments is relieved by DYNEIN-mediated transport of the complex as chromosomes congress and appropriate interactions with microtubules are established. However, it remains unclear how DYNEIN is antagonized to prevent premature RZZ removal. We recently described a new mechanism that sheds new light on this matter. We found that POLO kinase phosphorylates the DYNEIN adaptor SPINDLY to promote the uncoupling between RZZ and DYNEIN. Elevated POLO activity during prometaphase ensures that RZZ is retained at kinetochores to allow the dynamic turnover of kinetochore-microtubule interactions and prevent the stabilization of erroneous attachments. Here, we discuss additional interpretations to explain a model for POLO-dependent regulation of the RZZ-SPINDLY-DYNEIN module during mitosis.
KEYWORDS: RZZ, SPINDLY, POLO/PLK1, kinetochore-microtubule attachments, dynein adaptor
Mitosis corresponds to a challenging moment in the cell cycle where multiple mechanisms need to be precisely coordinated in time and space to ensure that the genomic content is evenly partitioned between the two new nuclei. A critical event is the interaction between spindle microtubules (MTs) and kinetochores (KTs), multi-protein complexes that assemble on the centromeric region of each sister chromatid [1]. These interactions allow chromosomes to be physically positioned at the cell equator before segregation of chromatids takes place. At this point, it is critical that sister KTs are attached to the plus ends of MTs from opposite spindle poles – amphitelic attachment – so as to ensure the accurate distribution of chromatids among both daughter cells. Errors in this process lead to numerical variations in the karyotype, a genetic state known as aneuploidy and a hallmark of cancer cells [2]. Therefore, it is of utmost importance that KT-MT interactions are precisely monitored and regulated to preserve genomic integrity [3,4]. Inducing subtle changes on the rate of MT turnover at KTs was shown to be sufficient to impact segregation fidelity in human cells lines, thus arguing for a need to stringently and precisely control KT-MT attachments [5,6].
KTs first interact with spindle MTs during early mitosis in a stochastic manner. As a result, different attachment configurations can be established, including erroneous interactions like syntelic – where both sister KTs bind to MTs emanating from the same spindle pole – or merotelic – where a single KT interacts with MTs from both spindle poles. Both attachment configurations must eventually be converted and stabilized as amphitelic [7,8]. KT-MT attachments are mediated by multiple sets of a group of proteins localized at the outer KT, collectively known as the KMN (KNL1/MIS12/NDC80) network [9]. The NDC80 complex constitutes the major MT-binding component and assumes a fundamental role in tethering KTs to the plus-ends of MTs. This is achieved through the interaction of tubulin with the CH domain and the N-terminal tail of NDC80 and eventually results in the formation of end-on attachments [10–12]. However, end-on binding is frequently preceded by transient interactions between KTs and the lateral surface of MTs. Lateral attachments are thought to increase the efficiency of chromosome congression to the cell equator by rapidly incorporating KTs into the nascent spindle [13–15]. These interactions are mainly mediated by the KT-localized motor proteins DYNEIN and CENP-E, which respectively drive the transport of chromosomes toward MT minus or plus ends [16]. Once KTs reach the plus tip of MTs, lateral associations are converted to end-on interactions [17–20]. Interestingly, both motors localize to the fibrous corona, a ring-shaped structure present on the outermost region of KTs that exhibits maximal expansion in the absence of interacting MTs [21–23]. The presence of an enlarged KT is proposed to improve MT capture and contribute to the rather short time required for full chromosome alignment [21,24]. This is however achieved at the expense of accuracy, as the increased surface area exposed to MTs may also facilitate the formation of erroneous interactions [21,25].
Kinases and phosphatases operate at KTs to ensure that proper end-on interactions are formed and stabilized [8,26,27]. AURORA B kinase plays a key role in modulating KT-MT attachment status through phosphorylation of several KT proteins [8,28,29]. AURORA B-mediated phosphorylation of multiple sites on NDC80 N-terminal tail decreases the binding affinity toward MTs [11,12,30–33]. Furthermore, AURORA B phosphorylates the functionally related SKA and DAM1 complexes, decreasing their recruitment to KTs and delaying the formation of stable end-on attachments [8,28]. Therefore, AURORA B contributes to error correction by generating unattached KTs that can then attempt to establish new correct interactions with MTs. Importantly, KTs signal the lack of MT attachment by accumulating proteins that activate the Spindle Assembly Checkpoint (SAC) and consequently prevent anaphase onset until all chromosomes are attached to the mitotic spindle [34,35]. Once bioriented, sister KTs experience tension across the inter-KT axis as a result of opposite MT pulling forces. This was proposed to contribute to the stabilization of KT-MT attachments by effectively increasing the distance between centromeric AURORA B and its outer KT targets [28,36,37]. However, recent studies have challenged this view and propose that a non-centromeric pool of AURORA B is responsible for KMN phosphorylation [38–40]. Given the essential role for centromeric AURORA B in error correction, new data implies that additional substrates may exist to promote resolution of erroneous attachments [39]. Nevertheless, the establishment of tension translates into a progressive increase in KT-MT attachment stability [40], which is further reinforced by dephosphorylation of AURORA B substrates through the recruitment of PP1 and PP2A-B56 phosphatases to KTs [8,26]. As PP2A-B56 localizes to KTs already in prometaphase, it is positioned to counteract AURORA B destabilizing activity as MT interactions are converted from lateral to end-on interactions [18,41–44]. In addition to AURORA B-dependent regulation, the affinity of NDC80 to MTs is also modulated by the interaction between NDC80 CH domain and the ROD-ZW10-ZWILCH (RZZ) complex [45]. This recently uncovered regulatory pathway is proposed to be required for fine-tuning KT-MT attachments and ensure accurate chromosome segregation [45–49].
The three-subunit RZZ complex was originally identified in Drosophila and later shown to be conserved in higher eukaryotes [50]. The complex assembles with a 2:2:2 (ROD:ZW10:ZWILCH) stoichiometry with ROD subunits assuming an antiparallel configuration [51,52]. Each ZW10 subunit interacts with a central domain on ROD, while each ZWILCH subunit binds to ROD N-terminus [52]. Once at KTs, the RZZ can regulate SAC response by recruiting both SAC-activating and SAC-silencing components, such as MAD1/MAD2 and DYNEIN/DYNACTIN, respectively [49,50]. Intriguingly, how the RZZ complex is recruited to KTs in metazoans remains elusive. The ZW10-interacting protein ZWINT has been proposed to function as a KT receptor for the RZZ in human cells [53–57]. However, it should be noted that knock-down of human ZWINT leads to a significant reduction in the KT levels of KNL1, which is a major platform for the recruitment of numerous KT components and regulators [56,58]. Hence, decreased accumulation of the human RZZ at ZWINT-depleted KTs should be interpreted with reservations. Interestingly, the C. elegans ZWINT homologue was shown to be dispensable for ZW10 localization [56] and no structural homologue for ZWINT was yet identified in Drosophila [50], thus further implying that additional recruitment factors are likely involved. Recently, human BUB1 and Drosophila CAL1 were both proposed to contribute for KT accumulation of RZZ [58,59]. Moreover, KT localization of the RZZ complex is under the regulation of the mitotic kinases MPS1 and AURORA B [23,54]. MPS1-mediated phosphorylation of ROD is required for RZZ-dependent expansion of the fibrous corona [23]. AURORA B phosphorylates ZWINT but a comprehensive understanding of its role is still lacking. KT localization of the RZZ complex is only affected by the absence of AURORA B activity when KTs interact with MTs, thus suggesting a more intricate regulatory mechanism [60,61]. In human cells treated with MT-depolymerizing agents, the RZZ complex is able to associate with KTs in an AURORA B-independent manner [21,60–62]. However, inhibition of AURORA B in cells incubated with taxol, which still allows for KT-MT interactions to take place, prevents RZZ accumulation at KTs [60]. Given the role of DYNEIN in shedding KT proteins along MTs tracks, these observations suggest that AURORA B exerts a negative impact on DYNEIN-mediated removal of RZZ from KTs. Interestingly, the accumulation of RZZ at KTs of taxol-treated cells was not followed by an equivalent increase in DYNEIN localization. This suggests that inhibition of DYNEIN-mediated removal acts to break the RZZ-DYNEIN boundary [60]. DYNEIN and ZW10 do not appear to be directly targeted by AURORA B [54]. On the other hand, AURORA B phosphorylates ZWINT and this could be required to retain RZZ at KTs [54]. However, expression of ZWINT phosphomimetic mutants for AURORA B sites not only retained the RZZ at KTs, but also its binding partners MAD2 and DYNEIN [54]. Together, these results argue against a role for AURORA B at the RZZ-DYNEIN interface and, instead, support AURORA B-mediated phosphorylation of ZWINT as a mechanism to control the ZWINT-RZZ boundary. It is therefore plausible that AURORA B acts indirectly to inhibit DYNEIN-dependent removal of RZZ from KTs.
Given the pivotal role of the RZZ complex in preventing chromosome mis-segregation, it is not surprising that its function at KTs is under precise regulation [50]. Recent studies provided detailed knowledge on the role of RZZ in KT-MT attachment dynamics [45–49]. The N-terminal B-propeller domain of ROD was shown to interact with NDC80 N-terminal tail in a yeast two hybrid assay [45]. Furthermore, a partially reconstituted RZZ complex (ROD ß-propeller and ZWILCH) interacted in vitro with an NDC80 fragment in a tail-dependent manner and was able to inhibit NDC80 complex binding to MTs in co-sedimentation assays. These in vitro data together with in vivo observations in C. elegans lead to a model where the interaction of RZZ (through ROD) with the N-terminal tail of NDC80 precludes the MT-binding capacity of the adjacent CH domain [45]. Hence, RZZ-dependent negative regulation of KT-MT interactions is proposed to prevent premature stabilization of erroneous end-on attachments during early mitosis. This mechanism is relieved by DYNEIN-mediated removal of the RZZ complex from KTs [21,45–49]. By recruiting DYNEIN to KTs, RZZ sets the stage for its own removal and, consequently, for the silencing of SAC- and attachment-related functions [45,63].
DYNEIN has multiple binding partners at KTs but the RZZ complex emerges as the essential recruitment platform [64]. ZW10 can directly interact both with DYNEIN INTERMEDIATE CHAIN (DIC) [64,65] and with the p50 subunit of the DYNACTIN complex that is required for DYNEIN activation [64,66]. However, RZZ can also target DYNEIN to KTs indirectly through SPINDLY, an adaptor for the motor protein [21,48,49,63,67]. Similarly to other DYNEIN adaptors, SPINDLY encompasses several coiled coil (CC) segments along its length and contains different N-terminal motifs that have been demonstrated to be required for the interaction with DYNEIN/DYNACTIN (Figure 1(a)) [21,52,63,68]. Careful dissection of each domain revealed that the “Spindly motif” acts as a DYNACTIN interacting interface, adding new insights on the initial observations describing this motif as a DYNEIN recruiting signature [63,68]. Direct binding to the DYNEIN complex is mediated by the CC1 box, a N-terminal motif that interacts with DYNEIN LIGHT INTERMEDIATE CHAIN (DLIC) [68,69]. SPINDLY has an additional N-terminal CC2 box that is also required for DYNEIN recruitment, but its interacting partner remains elusive [21]. We have recently identified a new motif within a region associated with SPINDLY-cargo binding regulation [47]. Interestingly, all motifs appear to be shared by other DYNEIN adaptors, suggesting that SPINDLY may function in an analogous manner to promote DYNEIN linkage to specific cargos [21,68]. A general mechanism for adaptor-mediated DYNEIN recruitment proposes that the adaptor’s N-terminal domain engages with DYNEIN/DYNACTIN while the C-terminal domain is assigned to cargo recognition [68,70]. In agreement with this model, SPINDLY requires its C-terminal domain to interact with the RZZ complex, the DYNEIN cargo at KTs [68]. This interaction may involve different RZZ subunits as it has been demonstrated that human SPINDLY can interact with ROD ß-propeller [52], while the C. elegans orthologue binds both to ROD and ZWILCH [68]. More recently, we showed that Drosophila SPINDLY also interacts directly with ZWILCH [47]. In human cells, farnesylation of SPINDLY is also required for its KT localization and SPINDLY-driven corona expansion [21,52,71,72]. Addition of the farnesyl group occurs at the outmost C-terminal cysteine residue present within a CAAX motif on human SPINDLY, a sequence signature that is absent from Drosophila and C. elegans orthologues. It is possible that farnesylation of SPINDLY renders binding to ZWILCH dispensable for KT localization in human cells. Although the role of SPINDLY farnesylation is still poorly understood, recent data suggests that it contributes to strengthen the association with ROD by releasing SPINDLY from an auto-inhibitory folding [21]. This model is based on the observation that an N-terminal SPINDLY truncation no longer requires farnesylation to associate with KTs. A regulatory mechanism based on inhibitory intramolecular interactions appears to be a common feature of DYNEIN adaptors. A similar behavior has been described for BICD proteins, a well-studied family of adaptor proteins. In this case, the C-terminal cargo binding domain of BICD/BICD2 attenuates the N-terminal interaction affinity to DYNEIN and this inhibitory effect is relieved upon cargo binding [70]. It would be interesting to examine whether SPINDLY farnesylation acts in a similar manner to relieve the N-terminus and, therefore, promote binding to DYNEIN. In addition to farnesylation, human SPINDLY also requires the activity of MPS1 kinase to localize to KTs. However, whether SPINDLY is a direct MPS1 substrate remains to be demonstrated [21,23,73]. It has also been suggested that ROD is phosphorylated by MPS1 and that this contributes to the KT recruitment of RZZ-SPINDLY and expansion of the fibrous corona into crescent-like shapes [23]. Interestingly, MPS1 activity was shown to be dispensable for both SPINDLY KT targeting and corona expansion when an N-terminal SPINDLY truncation was expressed [21]. It is important to mention that in human cells both RZZ and SPINDLY are required for corona expansion and, therefore, each protein may be influencing different steps during the KT enlargement process. MPS1-dependent phosphorylation of ROD could be involved in early enrichment and/or conversion of farnesylated SPINDLY into fully uninhibited SPINDLY molecules at KTs. In fact, expression of ROD phospho-defective mutants prevents recruitment of wild type levels of both ROD and SPINDLY, but equivalent amounts of the two proteins can still localize indicating that farnesylated SPINDLY is targeted to the available ROD binding sites [23]. However, KTs fail to expand possibly due to the inability to produce uninhibited SPINDLY required to drive RZZ oligomerization. Conversely, expression of the KT high-affinity and constitutively uninhibited SPINDLY protein (N-terminal truncation) can bypass the need for SPINDLY farnesylation or MSP1-dependent phosphorylation of ROD, hence contributing to a significant rescue in corona expansion [21]. This also suggests that additional RZZ molecules that are incorporated into the growing corona do not required MPS1-dependent phosphorylation. Thus, the activities of farnesyl transferase and MPS1 enzymes could be important to induce the prompt expansion of the outermost KT domain in early mitosis. The rapid assembly of an enlarged corona could represent an important adaptation of mammalian cells with large volumes and monocentric chromosome to fine-tune MT capture. Further detailed biochemical analysis will be important to understand exactly how SPINDLY promotes RZZ oligomerization. Surprisingly, formation of RZZ-driven higher-order structures does not require SPINDLY in C. elegans [22].
Figure 1.
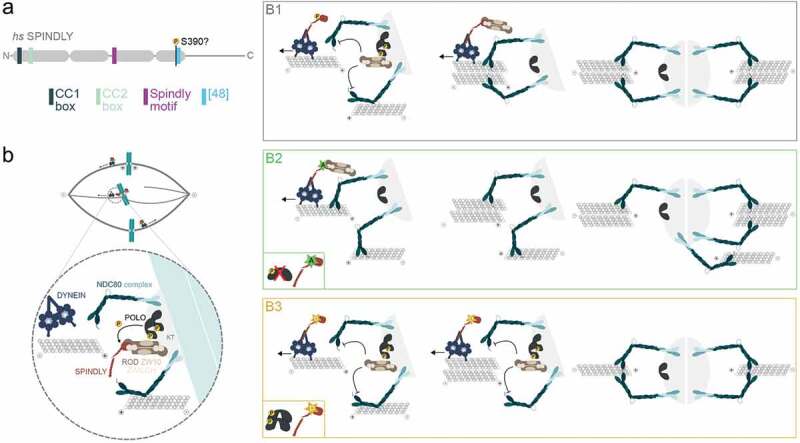
POLO regulates DYNEIN-dependent removal of RZZ-SPINDLY to fine-tune KT-MT attachments in mitosis. (a) Schematic representation of the different motifs present in human SPINDLY. Regions predicted to adopt coiled-coil conformations are represented in gray. We propose that an additional motif exists C-terminal to the Spindly motif [47]. A putative Plk1-dependent phosphorylation site on human SPINDLY is shown (S390). Our work identifies a Polo-dependent phosphorylation site within an equivalent motif on Drosophila SPINDLY (S499) [47]. (b) Proposed model for POLO-dependent regulation of the RZZ-SPINDLY-DYNEIN module at KTs: (B1) During prometaphase, POLO activity promotes RZZ disengagement from SPINDLY-DYNEIN by phosphorylating SPINDLY. The transient enrichment in RZZ at KTs opposes MT binding by NDC80, thereby, inhibiting the formation of stable attachments. This regulatory mechanism may be important to prevent premature stabilization of erroneous interactions that can form. (B2) In the absence of POLO activity or upon expression of non-phosphorylatable SPINDLY (A) mutant, DYNEIN can shed RZZ-SPINDLY of KTs. Consequently, untimely removal of RZZ can contribute to the stabilization of erroneous attachments (for instance, merotelic interaction). (B3) Conversely, in cells expressing phospho-mimetic SPINDLY (d) mutant or with unrestricted POLO kinase activity, RZZ is uncoupled from SPINDLY-DYNEIN. This allows the RZZ to inhibit NDC80 affinity toward MTs and promote MT turnover at KTs. Nevertheless, continuous destabilization of KT-MT interactions may affect the robustness of attachments.
SPINDLY is critical for the function of the fibrous corona not only by promoting RZZ-dependent expansion, but also because it sets the stage for DYNEIN-mediated compaction [21,63]. Before corona disassembly initiates, DYNEIN accumulated at enlarged KTs facilitates interactions with the lateral surface of MTs [16]. These lateral attachments allow the rapid capture of chromosomes during early prometaphase and promote biorientation by driving the rotation of the inter-KT axis to a position parallel to the spindle axis [15,24]. Sister KTs of laterally attached chromosomes become primarily exposed to MTs emanated from opposite poles, hence favoring chromosome biorientation as lateral interactions are converted to end-on attachments. However, it is important that the corona is able to compact during this conversion process. A constitutively expanded corona would be detrimental to the accuracy of chromosome segregations, as it favors long-lived lateral interactions that compromise end-on attachment formation [21]. Failure to establish end-on attachments may, in part, result from corona-enriched RZZ exerting a negative effect over stable KT-MT end-on attachments (discussed above). Thus, DYNEIN is required to silence the RZZ-dependent inhibitory function and allow stable conversion of lateral into end-on attachments. However, premature RZZ shedding from KTs must be avoided to allow some degree of KT-MT attachment plasticity and prevent stabilization of erroneous end-on attachments that might form during early mitosis [45,47]. Similarly to a recently proposed “DYNEIN brake” mechanism to maintain CENP-E at KTs [74], it has been suggested that cells must block untimely removal of RZZ from KTs. However, the molecular underpinnings of such mechanism had never been established [61]. Our recent work brings new insight into this matter [47]. We found that POLO kinase regulates the RZZ-SPINDLY-DYNEIN module at KTs by uncoupling the RZZ complex from SPINDLY-DYNEIN during early mitosis (Figure 1(b)). This causes retention of the RZZ at KTs and maintains KT-MT attachments labile enough to allow error correction. We demonstrate that SPINDLY-DYNEIN disengagement from the RZZ complex results from a decrease in the affinity of POLO-phosphorylated SPINDLY toward ZWILCH. Importantly, SPINDLY phosphorylation does not preclude its own KT localization, but instead primes SPINDLY for removal when DYNEIN arrives at KTs. Accordingly, it has been shown that RZZ-SPINDLY accumulates at KTs early in mitosis whereas DYNEIN localizes later [14]. Hence, our model predicts that during early prometaphase, when POLO kinase activity is elevated, DYNEIN can remove the loosely KT-bound phosphorylated SPINDLY adaptor but not its KT cargo (Figure 1B1). Consequently, KT-associated RZZ delays the stable conversion of lateral into end-on KT-MT attachments. This constitutes a safeguard mechanism to counteract the stabilization of early erroneous end-on attachments that may arise due to the stochastic nature of KT-MT interactions. We could recapitulate different scenarios using SPINDLY versions bearing phospho-mutations in the residue targeted by POLO kinase (S499) (Figure 1B2,B3). Drosophila S2 cells expressing a phospho-defective SPINDLY mutant exhibited increased levels of DYNEIN-dependent RZZ shedding along MTs indicating that the inability to disengage the RZZ-SPINDLY-DYNEIN axis leads to premature removal of RZZ from KTs. In agreement, these cells accumulated merotelic attachments as a result of increased stabilization of KT-MT interactions (Figure 1B2). Conversely, expression of a phospho-mimetic SPINDLY mutant caused an increase in RZZ levels at congressing KTs as a result of SPINDLY-DYNEIN disengagement from the cargo. As predicted by the model, permanent destabilization of KT-MT interactions had an impact on the robustness of end-on KT-MT attachments (Figure 1B3).
KTs harness the force generated by MT-depolymerization to power chromosome movement during anaphase and full MT occupancy is required to ensure faithful chromosome segregation [75]. Interestingly, our data shows that in Drosophila neuroblasts, chromosome migration during anaphase occurs in an asynchronous manner when POLO is constitutively active. The presence of unrestricted kinase should mimic constant SPINDLY phosphorylation and, consequently, RZZ-mediated inhibition of stable KT-MT attachments (Figure 1B3). We envisage that some chromosomes eventually lag due to insufficient MT-dependent pulling forces as a result of low MT occupancy [47]. However, this scenario does not occur when both POLO activity and SPINDLY phosphorylation levels decrease as cells progress from prometaphase to metaphase (Figure 1B1, [44,72–79];). Although future work is necessary to understand how POLO activity is antagonized in this context, we anticipate PP2A-B56 phosphatase as a good candidate to counteract SPINDLY phosphorylation and relieve RZZ inhibitory action toward stable KT-MT interactions. POLO phosphorylates BUBR1 to direct PP2A-B56 recruitment to prometaphase KTs leading to a local tug-of-war between kinase and phosphatase activities [26,80]. We reason that a suitable environment toward substrate phosphorylation may be provided by high CDK1/CYCLIN A levels during early mitosis which can prime SPINDLY for POLO-dependent phosphorylation [48]. Also, a decrease in CYCLIN A levels from prometaphase to metaphase appears to correlate with a stepwise increase in KT-MT stability [81]. Hence, as mitosis progresses and CYCLIN A levels drop, dephosphorylation of SPINDLY will prevail. Additionally, dephosphorylation of POLO T-loop by PP1 and ubiquitination-dependent POLO removal may contribute to silence POLO activity and tip the balance against a KT-MT destabilizing environment [76,78]. PP2A-B56 itself restrains POLO accumulation at KTs during prometaphase [80].
Although the involvement of POLO/PLK1 in the destabilization of KT-MT attachments had been put forward previously, the underlying mechanisms had remained elusive [41,47,76,82–86]. POLO activity is required for proper centromeric localization of AURORA B and activation, thus indirectly contributing to promote destabilization of KT-MT interactions [85,87]. In line with that, POLO depletion in Drosophila cells significantly increases the frequency of stable syntelic attachments [83]. However, depletion or inhibition of AURORA B fails to recapitulate the severity of the phenotype [83], hinting for an extra layer of KT-MT regulation. The mechanism that we describe may contribute to elucidate this matter [47]. Loss of POLO activity at KTs leads to premature relieve of RZZ-dependent inhibitory function and consequently allows the stabilization of erroneous attachments that typically occur during early mitosis (Figure 1B2). Additionally, a decrease in RZZ levels at KTs is expected to be accompanied by a decline in DYNEIN levels. The absence of minus-end-directed force production leaves plus-end-directed polar ejection forces (PEFs) unopposed which can also contribute to the stabilization of KT-MT attachments [13,88,89]. Importantly, PEFs are present in PLK1-depleted cells and could further stabilize erroneous KT-MT attachments [90]. It would be interesting to test if a decrease in PEFs could partially restore the attachment defects in POLO/PLK1-depleted cells.
The existence of a DYNEIN brake mechanism that prevents premature RZZ removal is therefore critical for mitotic fidelity. It has been previously suggested that human CENP-I is required to inhibit DYNEIN-mediated shedding of RZZ/Mad1 from KTs. However, the underlying mechanism remains unclear [61]. CENP-I is a centromeric protein that belongs to the constitutive centromere-associated network (CCAN) and its depletion dampens the recruitment of another CCAN sub-complex composed by CENP-O/P/Q/U/R [91,92]. Importantly, CENP-U (or PBIP1) was proposed to recruit PLK1 to KTs [93–95]. A plausible inference is that CENP-I depletion indirectly causes a reduction in PLK1 accumulation at KTs. The lower levels of PLK1 in CENP-I depleted KTs could account for the faster loss of RZZ from prometaphase KTs as a result of a more stable SPINDLY-DYNEIN engagement with the RZZ. However, it remains to be confirmed whether CENP-I depleted cells have decreased levels of PLK1 at KTs and if targeting PLK1 to these KTs would rescue RZZ localization.
It is important to note that POLO/PLK1 also promotes the stabilization of KT-MT attachments. This occurs mainly through BUBR1-dependent recruitment of PP2A-B56 [43,79,96]. PP2A-B56 accumulation at KTs allows the dephosphorylation of key substrates including NDC80, which subsequently can establish stable interactions with MTs as end-on attachments are formed [41]. Despite the apparent contradictory roles, it is conceivable that POLO/PLK1 phosphorylates different substrates that will direct opposite inputs at distinct mitotic time points or attachment configurations. The prevalence of one input over the other may be dictated by mitotic regulators like CDK1/CYCLIN A or may result from specific subcellular accumulation of POLO/PLK1 [97]. Recent findings bring new insights to the complex array of functions of POLO/PLK1 and propose a regulatory mechanism to control an important new role uncovered for the RZZ complex [45–49]. RZZ is required for corona expansion during early mitosis. In addition, the RZZ complex inhibits NDC80 end-on MT-binding capacity to prevent stabilization of KT-MT attachments. However, DYNEIN can relieve this inhibitory activity by shedding RZZ of KTs in a SPINDLY-dependent manner. In order to avoid premature stabilization of erroneous attachments, POLO/PLK1 promotes the uncoupling of RZZ and SPINDLY-DYNEIN and delays RZZ removal from KTs. Therefore, POLO kinase is an important modulator of the RZZ-SPINDLY-DYNEIN axis and of its function in KT-MT attachment formation. By regulating the turnover of KT-MT interactions, POLO/PLK1 fine-tunes an essential feature of mitosis and ensures the preservation of genomic integrity.
Funding Statement
This work was supported by the Fundação para a Ciência e a Tecnologia [SFRH/BD/87871/2012]; Fundação para a Ciência e a Tecnologia [Norte-01-0145-FEDER- 000029]; Fundação para a Ciência e a Tecnologia [IF/01755/2014]; Fundação para a Ciência e a Tecnologia [UID/BIM/04293/2019].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Musacchio A, Desai A.. A molecular view of kinetochore assembly and function. Biology (Basel). 2017;6(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schukken KM, Foijer F. CIN and aneuploidy: different concepts, different consequences. Bioessays. 2018;40(1):1700147. [DOI] [PubMed] [Google Scholar]
- [3].Cimini D, Howell B, Maddox P, et al. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol. 2001;153(3):517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci USA. 2011;108(44):17974–17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bakhoum SF, Genovese G, Compton DA. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol. 2009;19(22):1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bakhoum SF, Thompson SL, Manning AL, et al. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol. 2009;11(1):27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tanaka TU. Kinetochore-microtubule Interactions: steps towards bi-orientation. Embo J. 2010;29(24):4070–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lampson MA, Grishchuk EL. Mechanisms to avoid and correct erroneous kinetochore-microtubule attachments. Biology (Basel). 2017;6(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Monda JK, Cheeseman IM. The kinetochore-microtubule interface at a glance. J Cell Sci. 2018;131(16). DOI: 10.1242/jcs.214577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Alushin GM, Ramey VH, Pasqualato S, et al. The Ndc80 kinetochore complex forms oligomeric arrays along microtubules. Nature. 2010;467(7317):805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ciferri C, Pasqualato S, Screpanti E, et al. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell. 2008;133(3):427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cheeseman IM, Chappie JS, Wilson-Kubalek EM, et al. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006;127(5):983–997. [DOI] [PubMed] [Google Scholar]
- [13].Barisic M, Paulo Aguiar P, Geley S, et al. Kinetochore motors drive congression of peripheral polar chromosomes by overcoming random arm-ejection forces. Nat Cell Biol. 2014;16(12):1249–1256. [DOI] [PubMed] [Google Scholar]
- [14].Itoh G, Ikeda M, Iemura K, et al. Lateral attachment of kinetochores to microtubules is enriched in prometaphase rosette and facilitates chromosome alignment and bi-orientation establishment. Sci Rep. 2018;8(1):3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Magidson V, O’Connell CB, Lončarek J, et al. The spatial arrangement of chromosomes during prometaphase facilitates spindle assembly. Cell. 2011;146(4):555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Maiato H, Gomes AM, Sousa F, et al. Mechanisms of chromosome congression during mitosis. Biology (Basel). 2017;6(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kalantzaki M, Kitamura E, Zhang T, et al. Kinetochore-microtubule error correction is driven by differentially regulated interaction modes. Nat Cell Biol. 2015;17(4):421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shrestha RL, Conti D, Tamura N, et al. Aurora-B kinase pathway controls the lateral to end-on conversion of kinetochore-microtubule attachments in human cells. Nat Commun. 2017;8(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shrestha RL, Draviam VM. Lateral to end-on conversion of chromosome-microtubule attachment requires kinesins CENP-E and MCAK. Curr Biol. 2013;23(16):1514–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang Y, Lin L, Liu X, et al. BubR1 phosphorylates CENP-E as a switch enabling the transition from lateral association to end-on capture of spindle microtubules. Cell Res. 2019;29(7):562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sacristan C, Ahmad MD, Keller J, et al. Dynamic kinetochore size regulation promotes microtubule capture and chromosome biorientation in mitosis. Nat Cell Biol. 2018;20(7):800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pereira C, Reis RM, Gama JB, et al. Self-assembly of the RZZ complex into filaments drives kinetochore expansion in the absence of microtubule attachment. Curr Biol. 2018;28(21):3408–3421.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rodriguez-Rodriguez JA, Lewis C, McKinley KL, et al. Distinct roles of RZZ and Bub1-KNL1 in mitotic checkpoint signaling and kinetochore expansion. Curr Biol. 2018;28(21):3422–3429.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Magidson V, Paul R, Yang N, et al. Adaptive changes in the kinetochore architecture facilitate proper spindle assembly. Nat Cell Biol. 2015;17(9):1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zaytsev AV, Grishchuk EL. Basic mechanism for biorientation of mitotic chromosomes is provided by the kinetochore geometry and indiscriminate turnover of kinetochore microtubules. Mol Biol Cell. 2015;26(22):3985–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Moura M, Conde C. Phosphatases in mitosis: roles and regulation. Biomolecules. 2019;9(2):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sherwin D, Wang Y. The opposing functions of protein kinases and phosphatases in chromosome bipolar attachment. Int J Mol Sci. 2019;20(24):6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Krenn V, Musacchio A. The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front Oncol. 2015;5:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Agarwal S, Smith KP, Zhou Y, et al. Cdt1 stabilizes kinetochore-microtubule attachments via an Aurora B kinase-dependent mechanism. J Cell Biol. 2018;217(10):3446–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].DeLuca JG, Gall WE, Ciferri C, et al. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell. 2006;127(5):969–982. [DOI] [PubMed] [Google Scholar]
- [31].DeLuca KF, Lens SM, DeLuca JG. Temporal changes in Hec1 phosphorylation control kinetochore-microtubule attachment stability during mitosis. J Cell Sci. 2011;124(Pt 4):622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Welburn JP, Vleugel M, Liu D, et al. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010;38(3):383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zaytsev AV, Mick JE, Maslennikov E, et al. Multisite phosphorylation of the NDC80 complex gradually tunes its microtubule-binding affinity. Mol Biol Cell. 2015;26(10):1829–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Joglekar AP. A cell biological perspective on past, present and future investigations of the spindle assembly checkpoint. Biology (Basel). 2016;5(4):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol. 2015;25(20):R1002–18. [DOI] [PubMed] [Google Scholar]
- [36].Lampson MA, Cheeseman IM. Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Bio. 2011;21(3):133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu D, Vader G, Vromans MJ, et al. Sensing chromosome bi-orientation by spatial separation of Aurora B kinase from kinetochore substrates. Science. 2009;323(5919):1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Broad AJ, DeLuca KF, DeLuca JG. Aurora B kinase is recruited to multiple discrete kinetochore and centromere regions in human cells. J Cell Biol. 2020;219(3):pii: e201905144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hadders MA, Hindriksen S, Truong MA, et al. Untangling the contribution of haspin and Bub1 to Aurora B function during mitosis. J Cell Biol. 2020;219(3):pii: e201907087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yoo TY, Choi JM, Conway W, et al. Measuring NDC80 binding reveals the molecular basis of tension-dependent kinetochore-microtubule attachments. Elife. 2018;7:pii: e36392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Foley EA, Maldonado M, Kapoor TM. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol. 2011;13(10):1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kruse T, Zhang G, Larsen MS, et al. Direct binding between BubR1 and B56-PP2A phosphatase complexes regulate mitotic progression. J Cell Sci. 2013;126(Pt 5):1086–1092. [DOI] [PubMed] [Google Scholar]
- [43].Suijkerbuijk SJ, Vleugel M, Teixeira A, et al. Integration of kinase and phosphatase activities by BUBR1 ensures formation of stable kinetochore-microtubule attachments. Dev Cell. 2012;23(4):745–755. [DOI] [PubMed] [Google Scholar]
- [44].Xu P, Raetz EA, Kitagawa M, et al. BUBR1 recruits PP2A via the B56 family of targeting subunits to promote chromosome congression. Biol Open. 2013;2(5):479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cheerambathur DK, Gassmann R, Cook B, et al. Crosstalk between microtubule attachment complexes ensures accurate chromosome segregation. Science. 2013;342(6163):1239–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Amin MA, McKenney RJ, Varma D. Antagonism between the dynein and Ndc80 complexes at kinetochores controls the stability of kinetochore–microtubule attachments during mitosis. J Biol Chem. 2018;293(16):5755–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Barbosa J, Martins T, Bange T, et al. Polo regulates spindly to prevent premature stabilization of kinetochore-microtubule attachments. Embo J. 2020;39(2):e100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Barisic M, Sohm B, Mikolcevic P, et al. Spindly/CCDC99 is required for efficient chromosome congression and mitotic checkpoint regulation. Mol Biol Cell. 2010;21(12):1968–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gassmann R, Essex A, Hu JS, et al. A new mechanism controlling kinetochore-microtubule interactions revealed by comparison of two dynein-targeting components: SPDL-1 and the Rod/Zwilch/Zw10 Complex. Genes Dev. 2008;22(17):2385–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Karess R. Rod-Zw10-Zwilch: A key player in the spindle checkpoint. Trends Cell Biol. 2005;15(7):386–392. [DOI] [PubMed] [Google Scholar]
- [51].Civril F, Wehenkel A, Giorgi FM, et al. Structural analysis of the RZZ complex Reveals common ancestry with multisubunit vesicle tethering machinery. Structure. 2010;18(5):616–626. [DOI] [PubMed] [Google Scholar]
- [52].Mosalaganti S, Keller J, Altenfeld A, et al. Structure of the RZZ complex and molecular basis of its interaction with spindly. J Cell Biol. 2017;216(4):961–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Famulski JK, Vos L, Sun X, et al. Stable hZW10 kinetochore residency, mediated by hZwint-1 interaction, is essential for the mitotic checkpoint. J Cell Biol. 2008;180(3):507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kasuboski JM, Bader JR, Vaughan PS, et al. Zwint-1 Is a Novel Aurora B substrate required for the assembly of a dynein-binding platform on kinetochores. Mol Biol Cell. 2011;22(18):3318–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Starr DA, Saffery R, Li Z, et al. HZwint-1, a novel human kinetochore component that interacts with HZW10. J Cell Sci. 2000;113(Pt 11):1939–1950. [DOI] [PubMed] [Google Scholar]
- [56].Varma D, Wan X, Cheerambathur D, et al. Spindle assembly checkpoint proteins are positioned close to core microtubule attachment sites at kinetochores. J Cell Biol. 2013;202(5):735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang H, Hu X, Ding X, et al. Human Zwint-1 specifies localization of Zeste White 10 to kinetochores and is essential for mitotic checkpoint signaling. J Biol Chem. 2004;279(52):54590–54598. [DOI] [PubMed] [Google Scholar]
- [58].Zhang G, Lischetti T, Hayward DG, et al. Distinct domains in Bub1 Localize RZZ and BubR1 to kinetochores to regulate the checkpoint. Nat Commun. 2015;6:7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pauleau AL, Bergner A, Janko Kajtez J, et al. The checkpoint Protein Zw10 Connects CAL1-dependent CENP-A centromeric loading and mitosis duration in drosophila cells. PLoS Genet. 2019;15(9):e1008380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Famulski JK, Chan GK. Aurora B kinase-dependent recruitment of hZW10 and hROD to tensionless kinetochores. Curr Biol. 2007;17(24):2143–2149. [DOI] [PubMed] [Google Scholar]
- [61].Matson DR, Stukenberg PT. CENP-I and Aurora B act as a molecular switch That Ties RZZ/Mad1 recruitment to kinetochore attachment status. J Cell Biol. 2014;205(4):541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chan YW, Fava LL, Uldschmid A, et al. Mitotic control of kinetochore-associated dynein and spindle orientation by human Spindly. J Cell Biol. 2009;185(5):859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gassmann R, Holland AJ, Varma D, et al. Removal of spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev. 2010;24(9):957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kardon JR, Vale RD. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol. 2009;10(12):854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Whyte J, Bader JR, Tauhata SBF, et al. Phosphorylation regulates targeting of cytoplasmic dynein to kinetochores during mitosis. J Cell Biol. 2008;183(5):819–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Starr DA, Williams BC, Hays TS, et al. ZW10 helps recruit dynactin and dynein to the kinetochore. J Cell Biol. 1998;142(3):763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Griffis ER, Stuurman N, Vale RD. Spindly, a novel protein essential for silencing the spindle assembly checkpoint, recruits dynein to the kinetochore. J Cell Biol. 2007;177(6):1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gama JB, Pereira C, Simões PA, et al. Molecular mechanism of dynein recruitment to kinetochores by the Rod-Zw10-Zwilch complex and spindly. J Cell Biol. 2017;216(4):943–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Celestino R, Henen MA, Gama JB, et al. A transient helix in the disordered region of dynein light intermediate chain links the motor to structurally diverse adaptors for cargo transport. PLoS Biol. 2019;17(1):e3000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hoogenraad CC, Akhmanova A. Bicaudal D family of motor adaptors: linking dynein motility to cargo binding. Trends Cell Biol. 2016;26(5):327–340. [DOI] [PubMed] [Google Scholar]
- [71].Holland AJ, Reis RM, Niessen S, et al. Preventing farnesylation of the dynein adaptor spindly contributes to the mitotic defects caused by farnesyltransferase inhibitors. Mol Biol Cell. 2015;26(10):1845–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Moudgil DK, Westcott N, Famulski JK, et al. A novel role of farnesylation in targeting a mitotic checkpoint protein, human spindly, to kinetochores. J Cell Biol. 2015;208(7):881–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Maciejowski J, Drechsler H, Grundner-Culemann K, et al. Mps1 regulates kinetochore-microtubule attachment stability via the ska complex to ensure error-free chromosome segregation. Dev Cell. 2017;41(2):143–156.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Auckland P, McAinsh AD. CENP-F controls force generation and the dynein-dependent stripping of CENP-E at kinetochores. Biorxiv. 2019. DOI: 10.1101/627380 [DOI] [Google Scholar]
- [75].Dudka D, Noatynska A, Smith CA, et al. Complete microtubule-kinetochore occupancy favours the segregation of merotelic attachments. Nat Commun. 2018;9(1):2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Beck J, Maerki S, Posch M, et al. Ubiquitylation-dependent Localization of PLK1 in mitosis. Nat Cell Biol. 2013;15(4):430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Conde C, Osswald M, Barbosa J, et al. Drosophila polo regulates the spindle assembly checkpoint through Mps1-dependent BubR1 phosphorylation. Embo J. 2013;32(12):1761–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Dumitru AM, Rusin SF, Clark AE, et al. Cyclin A/Cdk1 modulates Plk1 Activity in prometaphase to regulate kinetochore-microtubule attachment stability. eLife. 2017;6. DOI: 10.7554/eLife.29303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liu D, Davydenko O, Lampson MA. Polo-like kinase-1 regulates kinetochore-microtubule dynamics and spindle checkpoint silencing. J Cell Biol. 2012;198(4):491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Cordeiro MH, Smith RJ, Saurin AT. Kinetochore phosphatases suppress autonomous kinase activity to control the spindle assembly checkpoint. Biorxiv. 2019. DOI: 10.1101/856773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kabeche L, Compton DA. Cyclin A regulates kinetochore microtubules to promote faithful chromosome segregation. Nature. 2013;502(7469):110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hood EA, Kettenbach AN, Gerber SA, et al. Plk1 regulates the kinesin-13 protein Kif2b to promote faithful chromosome segregation. Mol Biol Cell. 2012;23(12):2264–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Moutinho-Santos T, Conde C, Sunkel CE. POLO ensures chromosome bi-orientation by preventing and correcting erroneous chromosome-spindle attachments. J Cell Sci. 2011;125(Pt 3):576–583. [DOI] [PubMed] [Google Scholar]
- [84].Paschal CR, Maciejowski J, Jallepalli PV. A stringent requirement for Plk1 T210 Phosphorylation during K-fiber assembly and chromosome congression. Chromosoma. 2012;121(6):565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Salimian KJ, Ballister ER, Smoak EM, et al. Feedback control in sensing chromosome biorientation by the Aurora B Kinase. Curr Biol. 2011;21(13):1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yu R, Wu H, Ismail H, et al. Methylation of PLK1 by SET7/9 ensures accurate kinetochore-microtubule dynamics. J Mol Cell Biol. 2019;pii: mjz107. DOI: 10.1093/jmcb/mjz107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Carmena M, Lombardia MO, Ogawa H, et al. Polo kinase regulates the localization and activity of the chromosomal passenger complex in meiosis and mitosis in Drosophila melanogaster. Open Biol. 2014;4(11):140162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cane S, Ye AA, Luks-Morgan SJ, et al. Elevated polar ejection forces stabilize kinetochore-microtubule attachments. J Cell Biol. 2013;200(2):203–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Drpic D, Pereira AJ, Barisic M, et al. Polar ejection forces promote the conversion from lateral to end-on kinetochore-microtubule attachments on mono-oriented chromosomes. Cell Rep. 2015;13(3):460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Sumara I, Giménez-Abián JF, Gerlich D, et al. Roles of Polo-Like Kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14(19):1712–1722. [DOI] [PubMed] [Google Scholar]
- [91].Hara M, Fukagawa T. Kinetochore assembly and disassembly during mitotic entry and exit. Curr Opin Cell Biol. 2018;52:73–81. [DOI] [PubMed] [Google Scholar]
- [92].Pesenti ME, Prumbaum D, Auckland P, et al. Reconstitution of a 26-subunit human kinetochore reveals cooperative microtubule binding by CENP-OPQUR and NDC80. Mol Cell. 2018;71(6):923–939.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Park CH, Park JE, Kim TS, et al. Mammalian Polo-like Kinase 1 (Plk1) promotes proper chromosome segregation by phosphorylating and delocalizing the PBIP1·CENP-Q complex from kinetochores. J Biol Chem. 2015;290(13):8569–8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kang YH, Park CH, Kim TS, et al. Mammalian polo-like kinase 1-dependent regulation of the PBIP1-CENP-Q complex at kinetochores. J Biol Chem. 2011;286(22):19744–19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kang YH, Park JE, Yu LR, et al. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol Cell. 2006;24(3):409–422. [DOI] [PubMed] [Google Scholar]
- [96].Matsumura S, Toyoshima F, Nishida E. Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J Biol Chem. 2007;282(20):15217–15227. [DOI] [PubMed] [Google Scholar]
- [97].Lera RF, Potts GK, Suzuki A, et al. Decoding Polo-like kinase 1 signaling along the kinetochore-centromere axis. Nat Chem Biol. 2016;12(6):411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]