

ABSTRACT
Macroautophagy (hereafter autophagy) plays an important role in maintaining cellular homeostasis under stress conditions. We previously demonstrated that conditional autophagy deficiency in adult mice causes selective tissue damage, is lethal upon fasting, and shortens lifespan to less than three months primarily due to neurodegeneration, but not all the mechanisms are known. We conditionally deleted Trp53/p53 and/or the essential autophagy gene Atg7 throughout adult mice to test whether TRP53 is responsible for any of these phenotypes. atg7Δ/Δ trp53Δ/Δ mice have extended lifespan due to delayed tissue damage and neurodegeneration, and are resistant to death upon fasting compared to atg7Δ/Δ mice. Atg7 also suppresses apoptosis induced by the TRP53 activator Nutlin-3 in liver and brain. We then deleted Atg7 in the presence or absence of the master regulator of antioxidant defense NFE2L2/NRF2 (nuclear factor, erythroid derived 2, like 2) to test if increased oxidative stress causes TRP53 activation in atg7Δ/Δ mice. nfe2l2−/-atg7Δ/Δ mice die rapidly due to intestinal damage, which is not rescued by trp53 co-deletion. Therefore, these data demonstrate the tissue specificities and functional dependencies between autophagy, TRP53 and NFE2L2 stress response mechanisms.
KEYWORDS: Apoptosis, atg7, autophagy, brain, DNA damage, intestine, liver, nrf2, oxidative stress, p53
Autophagy is critical for cellular homeostasis under stress conditions by maintaining organelle function, degrading toxic cellular waste products, and sustaining cell metabolism through recycling. Conditional whole-body autophagy deficiency in adult mice causes liver, brain and muscle damage, prevents depletion of white adipose tissue (WAT), and shortens life span to less than three months due to susceptibility to infection and neurodegeneration. Although adult mice tolerate autophagy deficiency in the fed state in the short term, fasting is lethal due to hypoglycemia. These findings indicate that autophagy is required to maintain systemic mammalian metabolism and survival by mitigating metabolic stress during nutrient deprivation. Moreover, in contrast to other tissues, brain, liver, muscle and WAT display prominent tissue-specific autophagy dependencies. The mechanisms by which autophagy protects these tissues and sustains survival are still poorly understood.
TRP53 is a transcription factor and tumor suppressor that responds to stress including oxidative stress, hypoxia, DNA damage and oncogene activation, and there is mounting evidence on functional interaction between TRP53 and autophagy pathways. TRP53 and its target genes can turn on autophagy, and autophagy loss causes TRP53 induction in lung, breast, pancreatic tumors and neurons correlating with increased apoptosis. Another stress-responsive gene product is the master regulator of antioxidant defense NFE2L2, which is induced by oxidative stress and autophagy deficiency. Therefore, we sought to test whether TRP53 and NFE2L2 modulate the degenerative phenotypes induced by conditional autophagy deficiency in adult mice. We recently reported that autophagy suppresses TRP53 activation and TRP53-induced damage in liver and brain, whereas NFE2L2 limits intestinal damage due to autophagy deficiency by a TRP53-independent mechanism, demonstrating the tissue-specific functional interaction between these stress-regulated pathways [1] (Figure 1A).
Figure 1.
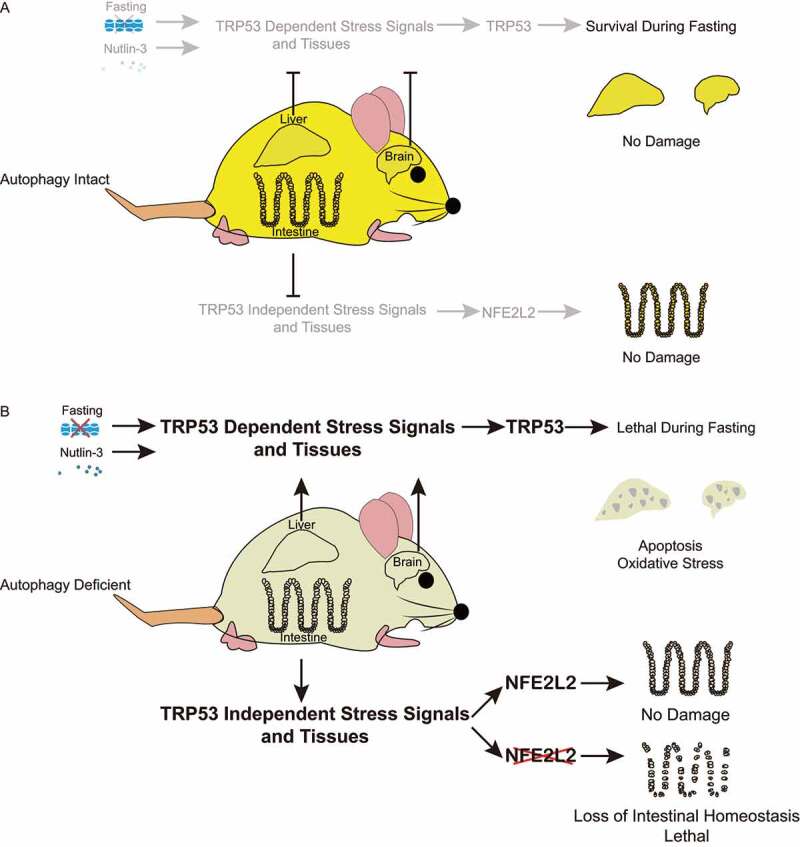
Autophagy protects tissues by interaction with TRP53 and NFE2L2 stress-response mechanisms in a tissue-specific manner. (A) Autophagy protects liver and brain, and sustains survival during fasting by inhibiting TRP53 (top), and maintains intestinal homeostasis (bottom). (B) When autophagy is deficient, TRP53 activation causes lethality upon fasting, and liver and brain damage that is further increased by Nutlin-3, whereas NFE2L2 alleviates intestinal damage in a TRP53-independent manner. In the absence of NFE2L2, autophagy deficiency is synthetically lethal due to severe intestinal damage.
We demonstrated the interaction between autophagy and TRP53 by utilizing genetically engineered mouse models (GEMMs) to conditionally delete essential autophagy gene Atg7 and/or Trp53 systemically using tamoxifen (TAM). Adult mice with co-deletion of essential autophagy gene Atg7 and Trp53 (atg7Δ/Δ trp53Δ/Δ) have extended lifespan and live up to six months, sustained survival during fasting, and show decreased tissue damage, apoptosis, and DNA damage in liver and brain in comparison to mice with deletion of Atg7 only (atg7Δ/Δ). Remarkably, atg7Δ/Δ trp53Δ/Δ mice have fewer spontaneous tumors than trp53Δ/Δ mice consistent with the pro-tumorigenic role of autophagy. Autophagy also inhibits TRP53 activation by the MDM2 inhibitor and TRP53 activator, Nutlin-3, which protects liver and brain from TRP53 hyperactivation and apoptosis. Therefore, we concluded that autophagy is essential to protect liver and brain from apoptosis and sustain adult mice during fasting by limiting TRP53 activation (Figure 1B).
Because we found increased oxidative stress in atg7Δ/Δ mice, we hypothesized that increased oxidative stress is responsible for tissue damage caused by TRP53 activation. We addressed this by generating GEMMs and used TAM to delete Atg7 in the presence and absence of Nfe2l2. Surprisingly, nfe2l2−/ – atg7Δ/Δ mice have a lifespan of less than seven days, and the only tissue with significant damage is the intestine. Intestines from nfe2l2−/- atg7Δ/Δ mice display increased oxidative stress and apoptosis, with abnormal Paneth cells and loss of goblet cells. Moreover, the intestinal stem cells are significantly decreased, and these cells are responsible for the renewal of other cell types, and their loss likely caused death. By deleting Trp53 together with Atg7 in the absence of Nfe2l2, nfe2l2−/- trp53Δ/Δ atg7Δ/Δ mice do not survive longer compared to nfe2l2−/- atg7Δ/Δ mice, suggesting that the intestinal damage in the nfe2l2−/- atg7Δ/Δ mice is not dependent on TRP53 (Figure 1B).
In summary, our recent work illustrated the tissue-specific interaction between autophagy, TRP53 and NFE2L2 stress response pathways by using the conditional whole body GEMMs. We provided evidence that Atg7 is a tissue-specific negative regulator of TRP53 and contributes to a negative feedback loop to limit TRP53 activation specifically in liver and brain, even when TRP53 activation is forced by Nutlin-3. Atg7 also restricts TRP53 activation to protect mice from lethality during fasting. These data indicated that autophagy responds to both endogenous and exogenous stresses by inhibiting TRP53. In the intestine, loss of Atg7 in the absence of NFE2L2 causes death within a week, independent of TRP53 before liver and brain damage can be observed, indicating NFE2L2 is specifically required to protect intestinal stem cells from oxidative stress. Therefore, our findings demonstrated the functional interaction and tissue specificity of different stress-regulated pathways.
Funding Statement
This work was supported by the National Institutes of Health grants: [R01 CA130893], [R01 CA188096] and [R01 CA163591] (to E.W.)
Disclosure statement
The authors declare no potential conflicts of interest to report.
Reference
- [1].Yang Y, Karsli-Uzunbas G, Poillet-Perez L, et al. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes Dev. 2020;34(9–10):688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]