

ABSTRACT
p97 has recently emerged as a therapeutic target for cancer due to its essential functions in protein homeostasis. CB-5083 is a first-in-class, potent and selective ATP-competitive p97 inhibitor that induces proteotoxic stress in cancer cells. Potential mechanisms regulating the sensitivity of cells to p97 inhibition remain poorly studied. Here, we demonstrate that Thrombospondin-1 (THBS1) is a CB-5083-upregulated gene that helps confer resistance of HCT116 cells to CB-5083. Our immunoblotting and immunofluorescence data showed that CB-5083 significantly increases the steady-state abundance of THBS1. Blockade of THBS1 induction sensitized cells to CB-5083-mediated growth inhibition. Suppression of THBS1 caused an increase of CB-5083-induced sub-G1 population and caspase 3/7 activity suggesting that its function is linked to the survival of cancer cells in response to p97 inhibition. Altogether our data provide new evidence that THBS1 is important for the susceptibility of cells to p97 inhibition.
KEYWORDS: Thrombospondin-1, p97, cb-5083, protein homeostasis
Introduction
Protein homeostasis has recently emerged as an oncogene-independent addiction mechanism that provides a basis for growth and invasion in cancer. Targeting this mechanism mitigates cancer cell growth even when diverse oncogenic mutations are involved. Protein homeostasis is a well-regulated cellular system that governs the balance between protein synthesis, degradation, folding and quality control. Cancer cells largely depend on this mechanism for growth and survival due to their high protein synthetic rate and rapid cell cycle. This protein ‘quality control’ mechanism provided by the ubiquitin-proteasome system (UPS) is thus one of the vulnerabilities in cancer, which can be targeted for a therapeutic purpose [1]. The relevance of targeting the UPS has been successfully demonstrated in the clinic for the treatment of leukemias and solid tumors [2]. However, relapses with frequent acquired resistance and lack of complete understanding of resistance biology support the need to develop inhibitors of other regulators of protein homeostasis as well as combination therapies [3–5].
The unfolded protein response (UPR) is a pathway that acts both to resolve unfolded protein stress and to trigger cell death when the buildup of such unfolded proteins becomes critical [6]. p97 (also known as Valosin-containing protein) has emerged as one of the most important key regulators of protein homeostasis [7]. p97 is an essential and conserved member of the AAA family of adenosine tri-phosphatases (ATPases). p97 mediates the retrieval of proteins primed for destruction by the UPS from organelles, chromatin, and protein complexes. p97 is also a key regulator of endoplasmic reticulum (ER)-associated degradation (ERAD), which is the main quality control mechanism for soluble, membrane-associated, glycosylated proteins as well as non-glycosylated proteins during their processing in ER [8,9]. Given the importance of UPR and ERAD pathways in the survival and growth of cancer cells and the key role of p97 in this mechanism, inhibition of p97 has become a well-accepted pharmacological approach to induce irresolvable proteotoxic stress as an anti-cancer therapy [10,11].
Drug discovery efforts have identified several classes of well-characterized ATP-competitive and allosteric inhibitors of p97 [12,13]. A number of other potential drug candidates have emerged from drug repositioning studies [14]. Despite their utility as experimental tools to better understand p97 biology, none have progressed to clinical application except for CB-5083, which represents a first-in-class ATP-competitive inhibitor of p97. CB-5083 has progressed through pre-clinical development and is currently being evaluated in two phase 1 clinical trials, in patients with relapsed and refractory multiple myeloma and in patients with advanced solid tumors [15]. These clinical trials have now terminated due to unexpected off-target effects, which require further optimization of the inhibitor [16].
The response mechanism of ER to stress is also the main source of resistance to proteotoxic therapies. Activation of the ER stress response sharply reduces protein synthesis in the ER, up-regulates protein degradation of damaged or misfolded proteins, and importantly induces expression of protective chaperones [17–19]. THBS proteins play a key role in the management of drug- or injury-induced ER stress. These secreted Ca-binding glycoproteins are involved in diverse biologic processes, and serve as interaction platforms in the extracellular matrix [18,20]. Indeed, the expression of THBS proteins is shown to mediate up-regulation of a unique profile of protective ER stress-response factors [21].
We have conducted a focused investigation on the resistance mechanism of CB-5083 in HCT116 cells. We report that THBS1 expression is upregulated in CB-5083-treated HCT116 cells, and is the source of drug resistance in these cells. We also report that the silencing of THBS1 sensitizes cells to CB-5083-mediated growth inhibition and cell death. Our findings may contribute to the basic understanding of resistance to CB-5083, and encourage further studies.
Materials and methods
TCGA data and statistical analysis
The TCGA dataset was downloaded from the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/). The RNA-Seq dataset represents a study of HCT116 cells treated for 8 hours with DMSO or 1uM CB-5083; data were produced on Illumina HiSeq 2500 sequencer [15]. Detailed information of the data is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE73588. We selected the level-3 data, which represent counts of gene expression such that the total number of reads for a given transcript is proportional to the expression level of the transcript. Data were normalized by TPM (transcripts parts per million) method and gene expression fold changes as Log2 ratios of CB-5083/DMSO treatments were calculated using independent t-test. Genes were ranked and a list of top differentially expressed genes (THBS1, DDIT3, ATF4, ATF3, HERPUD1, CYR61, CTGF and ANKRD1) was selected for further investigation. Independent t-test was performed for assessing the statistical significance of gene expression for further validation, reliably with p < 0.05.
Cells, reagents and antibodies
HCT116, HCT115, SW480, DLD1 and RKO cells were obtained from the American Type Culture Collection. CB-5083, Bortezomib and MLN4924 were purchased from Selleck. Anti-p97 (sc-57,492), anti-Thrombospondin-1 (sc-59,887), anti-p27 (sc-1641) and anti-Ubiquitin (sc-8017) were purchased from Santa Cruz Biotechnology. Anti-HERPUD1 (#26,730), anti-CHOP (#2895), anti-ATF4 (#11,815), anti-LC3B (#3868), anti-c-PARP (#9541), anti-p21 (#2947) and anti-CDT1 (#8064) were purchased from Cell signaling. Anti-beta-actin (ab8227) was purchased from Abcam. All primary antibodies were used at a dilution of 1:1000, whereas all secondary antibodies were used at a dilution of 1:3000.
RT-PCR, Immunoblotting and Immunofluorescence assays
Semi-quantitative RT-PCR was conducted as previously described [22]. Briefly, PCR was performed using 1 μl of 1:4 diluted cDNA for 28–36 cycles. The intensity was quantified and normalized to GAPDH. ddCT method was used to calculate fold changes in gene expression. The following forward and reverse primers were used for RT-PCR analysis:
THBS1 (ACTTCACCTTTGCCACCTC, AGACTCTGGAATGCGGTTG);
DDIT3 (GGTATGAGGACCTGCAAGAGGT, CTTGTGACCTCTGCTGGTTCTG);
ATF4 (CCTAGGTCTCTTAGATGACTATCTGGAGG, CCAGGTCATCCATTCGAAACAGAGCATCG);
ATF3 (GCTGGAGTCAGTCACCATCA, ACACTTGGCAGCAGCAA);
HERPUD1 (CGTTGTTATGTACCTGCATC, TCAGGAGGAGGACCATCATTT);
CYR61 (GAGTGGGTCTGTGACGAGGAT, GGTTGTATAGGATGCGAGGCT);
CTGF (CGACTGGAAGACACGT TTGG, AGGCTTGGAGATTTTGGGAG);
ANKRD1 (CGACTCCTGATTATGTATGGCGC, GCTTTGGTTCCATTCTGCCAGTG);
GAPDH (GTCTCCTCTGACTTCAACAGCG, ACCACCCTGTTGCTGTAGCCAA).
For immunoblotting, cells were lysed in RIPA lysis buffer and clarified by centrifugation (20 mM Tris-HCl (pH 7.5) 150 mM NaCl, 1 mM Na2EDTA 1 mM EGTA 1% NP-40 1% sodium deoxycholate 2.5 mM sodium pyrophosphate 1 mM β-glycerophosphate 1 mM Na3VO4 1 µg/ml leupeptin). Proteins were mixed with laemmli buffer, boiled, fractionated by SDS-PAGE and transferred to PVDF membrane using the iBlot 2 system (Life Technologies). The membrane was blocked in 5% nonfat dry milk for 1 h, incubated with primary antibodies at 4°C overnight and then with secondary antibodies for 1 h at room temperature. SuperSignal West Pico Plus (Thermo Fisher Scientific) was used for detection.
For immunofluorescence assay, HCT116 cells were seeded in chamber slides incubated in 0.5 ml RPMI medium supplemented with 2% FBS and 1 µM CB-5083. After 12 hours of incubation at 37°C, cells were washed and fixed with 4% paraformaldehyde in PBS. Cells were then probed with anti-THBS1 primary antibody, and then probed with Alexa Fluor-488 secondary antibody. Subsequently, cells were incubated with phalloidin rhodamine. Finally, mounting media with DAPI was used to attach No 1.5 cover slides. Fluorescence microscopy was performed with a Zeiss LSM 710 inverted confocal microscope [DAPI: λ(ex) = 420 nm, Alexa fluor: λ(ex) = 488 nm, phalloidin rhodamine: λ(ex) = 540]. Alexa fluor 488 signal was quantified to estimate the cellular distribution of THBS1, using an average of 20 cells per sample. Software packages ZEN lite and Fiji were used to collect the data and calculate the cellular level of THBS1.
Cell growth assay
Cell growth assays were performed as previously described [23]. Cell numbers were counted using FACSCOPE (Curiosis, Inc.) at 24 h intervals according to the manufacturer’s instructions. For cell cycle analysis, cells were fixed with 70% ethanol and resuspended in 1 ml of PBS containing RNase Propidium Iodide. The assay was conducted on a FACScan flow cytometer (Becton Dickinson), and the cell cycle profile was assessed using MultiCycle software (Phoenix Flow Systems). Cell viability assay was performed using Cell Titer-Glo (Promega) based on the previous report [24]. The Caspase activity was evaluated using Caspase-Glo 3/7 assay system (Promega) according to the manufacturer’s instruction.
Colony assay
HCT116, HCT115 and RKO cells were plated in 6-well plates with a seeding density of 3000 cells per well in RPMI 1640 medium supplemented with 2% FBS. Twenty-four hours later CB-5083 was added to the cells at concentrations 0.05 µM, 0.12 µM, 0.25 mM, 0.5 µM, 1.0 µM together with DMSO control. After 10 d of incubation, colonies were stained with crystal violet and colonies were counted.
Stress and toxicity 10-pathway reporter assay
Signal Finder stress and toxicity 10-Pathway Reporter Array (QIAGEN) included the following reporters for genotoxic stress with their corresponding transcription factors: ER stress (CBF/NF-Y/YY1), Antioxidant response (Nrf2/Nrf1), DNA damage (p53), NFkB (NFkB), Hypoxia (HIF1-a), Heavy metal stress (MTF1), Heat shock (HSF-1), Glucocorticoid (GR), MAPK/JNK (AP-1), Xenobiotic (AhR), together with a negative control and positive control. All reporters carried luciferase construct under the control of the corresponding transcription regulatory element together with constitutively expressing Renilla luciferase. Negative control carried a non-inducible firefly luciferase construct and a constitutively expressing Renilla luciferase construct, while positive control carried a constitutively expressing luciferase construct and a constitutively expressing Renilla luciferase construct. HCT116 cells, with wildtype and over-expressed THBS1, were transfected with the reporters for 24 hours, and then were with a dose range (0.1–30 µM) of CB-5083 for 24 hours. Luciferase signal was measured in a plate reader.
Overexpression and knockdown of THBS1
Human THBS1 cloned into pCMV3-Flag-THBS1 plasmid was purchased from Sino Biological (catalog number HG10508-NF). HCT116, HCT115 and RKO cells seeded in 6-well plate in RPMI medium supplemented in 10% FBS were transfected with the THBS1 construct using Lipofectamine method. Twenty-four hours after transfection, the medium was renewed and transformed cells were selected using 200 µg/ml hygromycin. After 72 hours, over-expression of THBS1 was confirmed using western blot analysis. Cells were kept in culture for 10–14 d and were subsequently used for further analyses.
For the knock-down of THBS1 expression, three different siRNAs were tested. HCT116 cells were transfected at 65% of confluence with 100 nM of siRNA control (siControl, GCAUGACCCUCGUCACAUATT) or THBS1 (siTHBS1-1, GCGGAGACAACAGCGUGUUUGACAUTT; siTHBS1-2, CCACAACGGAGUCCAGUAUdTdT; and siTHBS1-3, GGAUGCCUGUCCAAUCCUUdTdT) (Ambion, ThermoFisher Scientific) using Oligofectamine reagent following the manufacturer’s instructions (Life Technologies). After recovery of 24 hours, cells were first tested for the levels of THBS1 to assess the effect of siRNAs. Subsequently, cells were used for cell viability analysis with CB50-83 treatment, as described above.
Generation of CB5083-resistant cells
HCT116, HCT115 and RKO cells were plated in 10 cm plates in RPMI 1640 medium supplemented with 10% FBS. Upon 75% confluence, cells were treated with 1.5 µM of CB-5083 for 14 d. Medium and treatment were renewed every 3 d, and finally, the surviving colonies were pooled and tested for cell viability in comparison with CB-5083 cells. Cells with increased resistance for CB-5083 were tested for the status of THBS1 expression using western blot analysis.
RESULTS
P97 inhibition causes global mRNA expression changes
To explore how CB-5083 influences the mRNA expression profile, we sought to search for publicly disclosed sequencing data from NCBI’s Gene Expression Omnibus (GEO) databases. A previous study analyzed RNA-seq that reflects the whole transcriptome of HCT116 cells treated with or without CB-5083 [15]. Global mRNA expression changes upon CB-5083 treatment according to the previous data (GSE73588) are shown in (Figure 1A). We identified genes with a previously established link to ER stress such as DDIT3, ATF3, ATF4 and HERPUD1 [25,26] in a subset of highly induced mRNAs to CB-5083. In a further analysis of these data, we identified potentially interesting genes such as CYR61, THBS1, CTGF and ANKRD1, which were selected on the basis of their significant differential expression.
Figure 1.
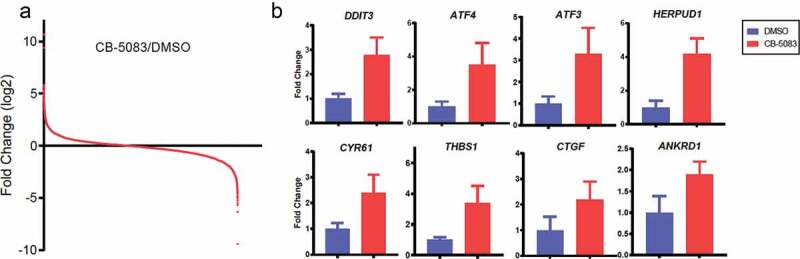
Identification and validation of genes upregulated by the p97 inhibitor CB-5083. (A) Summary of the GEO dataset evaluating whole transcriptome in HCT116 cells in response to CB-5083 (GSE73588). Independent t-test was performed to calculate Log2 ratios of CB-5083/DMSO treatments, with P < 0.0015. (B) HCT116 cells were treated with 1 μM CB-5083 for 8 h. Relative expressions of the indicated genes were analyzed by RT-PCR and quantified. p < 0.05 for differential gene expression. Data represent the mean (N = 3) and SD error.
To verify these, we examined the steady-state transcript levels of eight genes identified above. We measured 2.8 – 4.2-fold changes for DDIT3, ATF3, ATF4 and HERPUD1 in HCT116 cells treated with CB-5083 (Figure 1B). We also observed that CYR61, THBS1, CTGF and ANKRD1 are significantly up-regulated upon CB-5083 treatment (1.9 – 2.4-fold changes). Together, these data demonstrate that p97-targeted agent CB-5083 changes global mRNA expressions and a subset of previously known and unknown genes are successfully validated by RT-PCR.
Colon cancer cells acquire resistance to CB-5083
We initially tested CB-5083 on a number of colon cancer cells that represent various genetic backgrounds. In a dose–response assay, we observed IC50 values ranging from 0.8 µM to 3 µM (Supplementary Figure 1). The sensitive cell lines were HCT116, HCT115 and RKO with IC50 values of 1.18 µM, 0.82 µM and 0.88 µM, respectively. We focused on these three cell lines for the purpose of the further experiments. We subsequently generated CB-5083-resistant versions of these three cell lines and compared their colony-forming ability to that of their wild-type counterparts (Supplementary Figure 2). We observed a consistent 3–5-fold shift in IC50 between sensitive and resistant cell.
As a next step, we performed immune-blotting to detect THBS1 levels in the parental (sensitive) and resistant cells (Supplementary Figure 3). As expected, all three CB-5083-resistant cells showed increased THBS1 levels. Overall, we got convinced that THBS1 was involved in the CB-5083-resistant state, as we were able to proceed to the further experiments.
CB-5083 up-regulates the THBS1 level
For the sake of consistency with the TCGA data, we chose to work with HCT116 cells since they displayed high sensitivity to CB-5083. In the light of the recent studies [27,28], we conducted immunoblotting assays to show the response of ER stress signaling in response to CB-5083 treatment. HCT116 cells treated with increasing concentrations of CB-5083 (0.19–3 μM), Bortezomib (1 μM) or MLN4924 (3 μM) were analyzed by immunoblotting (Figure 2A). Accumulation of polyubiquitin, LC3B and cleaved PARP, known features of p97 inhibition, were remarkably increased by CB-5083 as shown in (Figure 2A). Also, biomarkers of p97 inhibition such as HERPUD1 (EC50: 0.35 μM), ATF4 (EC50: 0.68 μM) and CHOP (EC50: 0.74 μM) were strongly elevated by CB-5083 (Figure 2B). Likewise, we found that THBS1 was significantly induced by CB-5083 with 1.2 μM of EC50 (Figure 2C). p97 functions upstream of proteasome for recognition of ubiquitin-modified proteins thereby down-regulating p21, p27 and CDT1 [29–31]. While p27 and CDT1 were readily increased by Bortezomib and MLN4924, we were unable to observe their increases by CB-5083 (Figure 2D). Also, we found that p21 is only slightly increased by CB-5083 compared to Bortezomib and MLN4924 (Figure 2D) raising a question whether they are appropriate biomarkers of p97 inhibition in these cells.
Figure 2.

The steady-state level of THBS1 protein is increased by CB-5083. (A) HCT116 cells were treated with the indicated concentrations of CB-5083, Bortezomib and MLN4924 for 8 h. The steady-state levels of THBS1 and indicated proteins were analyzed by immunoblotting. Actin was used as a loading control. (B) Relative abundance of THBS1 protein was quantified (N = 3, SD error). (C) Relative abundance of known biomarkers of p97 inhibition (HERP1, CHOP and ATF4) was quantified (N = 3, SD error). (D) Mean fold changes (N = 3, SD error) for CDT1 and p21 with 3 μM CB-5083, 1 μM Bortezomib and 3 μM MLN4924. (E) An immunofluorescence assay for THBS1 (green), Actin (red) and DAPI (blue) in HCT116 cells treated with or without 1.5 μM CB-5083 for 8 h (N = 3, SD error).
In order to confirm these findings, we tested some of the key targets in HCT115 cells, which were treated in the same format (Supplementary Figure 4). In response to p97 inhibition by CB-5083, we observed comparable up-regulation of THBS1 together with accompanying increase in levels of cleaved PARP, HERPUD1 and CHOP. Altogether, we were able to draw parallels with the response from HCT116 cells.
Next, we tested whether THBS1 is regulated by CB-5083 at cellular level. Consistent with the immunoblotting result, the fluorescence intensity of THBS1 protein was greatly increased in response to CB-5083 (Figure 2E). Signal from DAPI and actin was comparable between DMSO control and CB-5083 treatment. Quantified cellular fluorescence analysis revealed at least 7-fold increase in THBS1 levels in response to the inhibitor. Collectively, we confirmed that THBS1 expression is significantly augmented with p97 inhibition in HCT116 cells.
THBS1 level affects response to CB-5083
To further investigate the role of THBS1 in acquired resistance for CB-5083, we pursued knock-down of THBS1. Due to its higher potency both on the gene expression and protein level of THBS1, we chose to work with siTHBS1-1, as compared to the other two siRNAs (Supplementary Figure 5). We next examined cell growth after cells were transfected with siRNA targeting endogenous THBS1 (Figure 3A.) Knockdown of THBS1 resulted in remarkable sensitization of cells to CB-5083, severely impacting the colony-forming ability of the cells under ER stress (Figure 3B). Consistently, cells with a decreased level of THBS1 were found to be more sensitive to CB-5083 (Figure 3C). Interestingly, the effect of THBS1 knock-down is only measurable when cells are treated with CB-5083, that is, under ER stress. While the growth of THBS1-deficient cells was not impacted under normal conditions, they failed to acquire resistance to CB-5083, thus suggesting that THBS1 induction conferred pro-survival response under genotoxic stress conditions. Within the first 72 hours of incubation, the effect of CB-5083 on suppression of cell viability was more evident in THBS1-depleted cells (Figure 3D). However, we anticipated that the effect of THBS1 depletion would be more pronounced in the long term.
Figure 3.

Knockdown of THBS1 sensitizes cells to CB-5083 and promotes apoptosis. (A) HCT116 cells transfected with siControl or siTHBS1-1 (shortly siTHBS1) were treated with or without 0.75 μM CB-5083 for 8 h. Total extracts were analyzed by immunoblotting. Actin was used as a loading control. (B) HCT116 cells transfected with siControl or siTHBS1 were treated with or without 0.375 μM CB-5083 for 72 h. Cells stained with crystal violet and are shown. (N = 3, SD error). *p < 0.05. (C) HCT116 cells transfected with siControl or siTHBS1 were treated with or without 0.375 μM CB-5083. Cell numbers were counted at 24 h intervals (N = 3, SD error). *p < 0.05. (D) HCT116 cells transfected with siControl or siTHBS1 were treated with indicated concentrations of CB-5083. Cell viability was analyzed by quantifying cellular ATP levels (N = 3, SD error). *p < 0.05. (E) HCT116 cells transfected with siControl or siTHBS1 were treated with indicated concentrations of CB-5083 for 72 h. Cell cycle was analyzed by flow cytometry and the percent Sub-G1 is shown (N = 3, SD error). *p < 0.05. (F) HCT116 cells transfected with siControl or siTHBS1 were treated with indicated concentrations of CB-5083 for 72 h. Caspase 3/7 activities were measured by immunofluorescence (N = 3, SD error). *p < 0.05.
To further elucidate the role of THBS1 dosage in conferring resistance to CB-5083, we over-expressed THBS1 in HCT116 cells. Cells with the added expression of THBS1 were selected over a period of 10 d to reflect its effect in the longer term (Supplementary Figure 6A and 6B). We found that THBS1-overexpressing HCT116 cells were 12.5-fold more resistant to CB-5083 as compared to the parental cells. This observation confirmed our hypothesis that THBS1 expression protected cells from CB8053-induced cell death.
We next questioned the mechanism of cell death induced by CB-5083. We investigated whether knockdown of THBS1 affected the apoptosis of cells in response to the inhibitor. Compared to the DMSO control, THBS-1-depleted cells treated with CB50-83 had increased sub-G1 population (2.6% vs 2.2%), together with measurably attenuated G1, S and G2/M phases (Figure 3E and Supplementary Figure 7). We reasoned that such accumulation in sub-G1 phase could be indicative of active apoptotic events, and therefore proceeded with measuring the Caspase 3/7 activity. Likewise, knockdown of THBS-1 resulted in elevated Caspase 3/7 activity indicating THBS-1 compromised the CB-5083’s pro-apoptotic effect (Figure 3F). Together, these data demonstrate that elevated level of THBS1 rescued cells from CB-5083-induced ER stress, in part by regulating apoptosis.
CB-5083 affects ER stress signaling
To interrogate the effect of CB-5083 on various genotoxic signaling pathways, as well as to shed light on the role of THBS1 on conferring resistance to CB-5083, we used 10-pathway luciferase-based reporter system. Wild-type and THBS1-over-expressing HCT116 cells were transfected with the reporter and the effect of CB-5083 was measured as reporter signal. We found that CB-5083 had a dose-dependent effect on ER reporter, which was more profound in wild-type cells. Reporters for antioxidant, heat shock and xenobiotic response were also affected, albeit at elevated doses. For the THBS1-over-expressing cells, only ER reporter showed a differential response (Supplementary Figure 8). In a further comparison, the EC50 of the inhibitor for the ER reporter was calculated as 8.5 µM and 67.6 µM for the wild-type and THBS1-over-expressing cells, respectively. This 8-fold difference in response further underlined the key role of THBS1 in counter-acting the effect of CB-5083 on ER stress (Supplementary Figure 9). Even though the EC50 values in this assay were elevated due to the un-physiological level of transfected THBS1 expression, the overall magnitude of fold change matched the fold change for cell viability we observed between CB-5083-sensitive and resistant cells. These observations further demonstrated the role of THBS1 in providing pro-survival and resistance mechanism to the genotoxic effect of the p97 inhibitor, CB-5083. These studies provide mechanistic insight into the cellular effect of CB-5083, as well as motivate further research.
Discussion
Resistance to proteasome inhibitors, in general, is a serious concern that renders such anti-cancer therapies ineffective in the clinic. Fast-adapting cancer cells abrogate long-term therapeutic efficacy of such novel therapies. It has previously been shown that antitumor activity of CB-5083 is strongly correlated to mRNA and protein expression levels of p97, whereby its enhanced levels may indicate acquired resistance. [15]. Our findings show that THBS1 provides a protective mechanism to p97 inhibition by increasing its protein levels, and thus suggest a novel mechanism of resistance to p97 inhibition therapy. THBS1 provides a protective ER stress response, plays an opposing function to ERAD, and provides an escape mechanism from apoptotic cell death.
Comparative genomic data analysis on a wide cell line panel has clearly demonstrated that CB-5083-induced ER stress produces the expected gene expression signature of ERAD pathway. The analyses have also revealed the factors that are involved in conferring resistance or sensitivity to p97 inhibition, suggesting that cells with high ERAD and UPR activity may be more vulnerable to p97 inhibition [15].
THBS1 has emerged as a factor that provides a defender role to p97 inhibition and thereby antagonizes clearance of misfolded proteins via ERAD. THBS1 has recently been shown to induce cell repair and renewal induced by genotoxic stress and injury in various cell types [21,32,33]. THBS1 is also shown to regulate inflammatory responses that contribute to colorectal carcinogenesis in mice [34]. Given, the ability of THBS1 to regulate cellular responses to a variety of stress conditions suggests that loss of THBS1 may also have a deeper effect on cancer cell metabolism, and may provide a path to regulate the sensitivity of cancers to therapies. One possible mechanism is that THBS1-dependent adaptive ER stress response enhances ER functionality, induces protective chaperones and up-regulates expression of key proteins in ERAD [21].
Intriguingly, CD47, which is a receptor for THBS1 and a counter-receptor for signal regulatory protein-α (SIRPα) on macrophages, is heavily involved in tumor invasion and metastasis in a number of solid cancers including colon carcinomas [35–37]. Inhibition of CD47 is likewise known to sensitize cancers to chemotherapy. CD47 blockade may control tumor growth by inhibiting THBS1 signaling or by preventing inhibitory SIRPα signaling in tumor-associated macrophages. Indeed, evidence suggests that pharmacological inhibition of CD47 impairs the self-renewal capacity of cancer-initiating cells and predisposes tumors to regression [38]. A number of recent studies have underlined the connection of THBS1 with CD47 in promoting drug resistance and metastatic behavior, together with the therapeutic utility of this interaction [39,40]. Other studies have investigated the expression patterns of THBS1 in metastatic cancers and have established THBS1 as a prognostic marker of metastatic colorectal cancer [41].
Importance of CD47 becomes even more apparent when considered in connection with glucose-regulated protein-78 (GRP78), a key regulator of fatty acid metabolism. CD47-GRP78 axis is shown to confer resistance to tamoxifen [42,43]. GRP78 inhibition, alone or in combination with tamoxifen, decreased CD47 expression and increased macrophage infiltration via interaction with THBS1 [44]. Therefore, there is an emerging link in which CD47 expression and function may be regulated by the UPR stress signaling and changes in lipid metabolism. Presumably, this mechanism works by exerting negative feedback on THBS1 and in turn reducing its protective effect on p97. Expectedly, our findings show that the depletion of THBS1 sensitizes HCT116 cells to CB-5083 inhibition.
One interesting aspect of THBS1 is its connection with calcium signaling. Indeed, Calcium signaling has been shown to play a role in acquired resistance to various drugs and genotoxic stress [45–47]. It has also been shown that CD47 interacts with both THBS1 and Calreticulin (CRT) to control calcium homeostasis in mitochondrial bioenergetics [48,49]. During times of cellular stress, THBS1 and CRTS are secreted from endoplasmic reticulum membrane in order to orchestrate cell death via apoptosis or necrosis. In cell culture, depletion of such key calcium-regulating factors is shown to have an effect on cell death in the long term under normal conditions. THBS1 plays important role in cell homeostasis, however, its effect on cell survival becomes apparent only under conditions of genotoxic stress. Consistent with this, we found that the effect of siTHBS1 was not apparent unless the cells were treated with CB-5083; under this treatment cells fail to acquire resistance to this p97 inhibitor. Similarly, the effect of THBS1 knockdown was not clear during the first 72 hours of incubation; this effect was pronounced at longer exposure to the drug. Also consistently with published reports, we found that CB8053 had a selective effect on ER reporter, which was over-rode with over-expressed THBS1.
Emerging reports already indicate the importance of THBS1 in enhancing the therapeutic sensitivity of existing proteasome inhibitors by smart therapy combinations to circumvent acquired chemo-resistance and extend the therapeutic benefit of these therapies for cancer patients [39,50,51]. Cancer cells develop resistance to chemotherapy in part via accumulation of mitochondria leading to an increase in reactive oxygen species that stimulate pro-survival pathways. CD47-THBS1 axis can be potentially targeted to sensitize cancer cells to chemotherapy by increasing mitochondrial turnover. Therefore, pharmacological inhibition of CD47 may potentiate the effect of proteasome inhibitors such as CB-5083 and sensitize cancer cells by regulating bioenergetics, thereby reducing the active glucose metabolism.
Our results show that THBS1 has a new role in conditioning and adaptation of ER to genotoxic stress, using which it provides a pro-survival mechanism to cancer cells exposed to CB-5083 treatment. Altogether, our study provides a new insight into the therapeutic potential of CB-5083 and emerging mechanisms for its resistance. We suggest further investigation to fully understand the mechanistic breadth of THBS1-related CB-5083 resistance and design prospective strategies to circumvent it.
Supplementary Material
Disclosure statement
The authors declare no competing interests.
Supplemental data
Supplemental data for this article can be accessed here.
References
- [1].Van Drie JH. Protein folding, protein homeostasis, and cancer [review]. Chin J Cancer. 2011. February;30(2):124–137. PubMed PMID: 21272445; PubMed Central PMCID: PMC4013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy [review]. Nat Rev Clin Oncol. 2017. July;14(7):417–433. PubMed PMID: 28117417; PubMed Central PMCID: PMC5828026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ruschak AM, Slassi M, Kay LE, et al. Novel proteasome inhibitors to overcome bortezomib resistance [research support, non-U.S. gov’t review]. J Natl Cancer Inst. 2011. July 6;103(13):1007–1017. PubMed PMID: 21606441. [DOI] [PubMed] [Google Scholar]
- [4].Milano A, Perri F, Caponigro F. The ubiquitin-proteasome system as a molecular target in solid tumors: an update on bortezomib. Onco Targets Ther. 2009. February 18;2:171–178. PubMed PMID: 20616904; PubMed Central PMCID: PMC2886336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wright JJ. Combination therapy of bortezomib with novel targeted agents: an emerging treatment strategy [research support, non-U.S. gov’t review]. Clin Cancer Res off J Am Assoc Cancer Res. 2010. August 15;16(16):4094–4104. CCR-09-2882. PubMed PMID: 20682705. [DOI] [PubMed] [Google Scholar]
- [6].Hetz C, Papa FR. The unfolded protein response and cell fate control [review]. Mol Cell. 2018. January 18;69(2):169–181. PubMed PMID: 29107536. [DOI] [PubMed] [Google Scholar]
- [7].Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system [research support, non-U.S. gov’t review]. Nat Cell Biol. 2012. February 2;14(2):117–123. PubMed PMID: 22298039. [DOI] [PubMed] [Google Scholar]
- [8].Rabinovich E, Kerem A, Frohlich KU, et al. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation [research support, non-U.S. gov’t research support, U.S. gov’t, non-P.H.S.]. Mol Cell Biol. 2002. January;22(2):626–634. PubMed PMID: 11756557; PubMed Central PMCID: PMC139744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol [research support, non-U.S. gov’t research support, U.S. gov’t, P.H.S.]. Nature. 2001. December 6;414(6864):652–656. PubMed PMID: 11740563. [DOI] [PubMed] [Google Scholar]
- [10].Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy [research support, N.I.H., extramural research support, non-U.S. gov’t review]. BMC Biol. 2014. November 11;12(1):94. PubMed PMID: 25385277; PubMed Central PMCID: PMC4226866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Oromendia AB, Amon A. Aneuploidy: implications for protein homeostasis and disease [review]. Dis Model Mech. 2014. January;7(1):15–20. PubMed PMID: 24396150; PubMed Central PMCID: PMC3882044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chou T-F, Li K, Frankowski KJ, et al. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase [research support, N.I.H., extramural research support, non-U.S. gov’t]. ChemMedChem. 2013. February;8(2):297–312. PubMed PMID: 23316025; PubMed Central PMCID: PMC3662613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chou T-F, Brown SJ, Minond D, et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways [research support, N.I.H., extramural research support, non-U.S. gov’t]. Proc Natl Acad Sci U S A. 2011. March 22;108(12):4834–4839. PubMed PMID: 21383145; PubMed Central PMCID: PMC3064330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Segura-Cabrera A, Tripathi R, Zhang X, et al. A structure- and chemical genomics-based approach for repositioning of drugs against VCP/p97 ATPase. Sci Rep. 2017. March 21;7(1):44912. PubMed PMID: 28322292; PubMed Central PMCID: PMC5359624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Anderson DJ, Le Moigne R, Djakovic S, et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis [research support, N.I.H., extramural research support, non-U.S. Gov’t]. Cancer Cell. 2015. November 9;28(5):653–665. PubMed PMID: 26555175; PubMed Central PMCID: PMC4941640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tang WK, Odzorig T, Jin W, et al. Structural basis of p97 inhibition by the site-selective anticancer compound CB-5083 [research support, N.I.H., intramural]. Mol Pharmacol. 2019. March;95(3):286–293. PubMed PMID: 30591537; PubMed Central PMCID: PMC6355941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response [research support, N.I.H., extramural research support, non-U.S. gov’t review]. Nature. 2008. July 24;454(7203):455–462. PubMed PMID: 18650916; PubMed Central PMCID: PMC2727659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kazerounian S, Yee KO, Lawler J. Thrombospondins: from structure to therapeutics. Cell Mol Life Sci. 2008. March;65(5):700–712. PubMed PMID: 18193162; PubMed Central PMCID: PMC2752021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Glembotski CC. Endoplasmic reticulum stress in the heart [research support, N.I.H., extramural review]. Circ Res. 2007. November 9;101(10):975–984. PubMed PMID: 17991891. [DOI] [PubMed] [Google Scholar]
- [20].Stenina OI, Topol EJ, Plow EF. Thrombospondins, their polymorphisms, and cardiovascular disease [research support, N.I.H., extramural research support, non-U.S. gov’t review]. Arterioscler Thromb Vasc Biol. 2007. September;27(9):1886–1894. PubMed PMID: 17569883. [DOI] [PubMed] [Google Scholar]
- [21].Lynch JM, Maillet M, Vanhoutte D, et al. A thrombospondin-dependent pathway for a protective ER stress response [research support, N.I.H., extramural research support, non-U.S. gov’t]. Cell. 2012. June 8;149(6):1257–1268. PubMed PMID: 22682248; PubMed Central PMCID: PMC3372931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Her NG, Jeong SI, Cho K, et al. PPARdelta promotes oncogenic redirection of TGF-beta1 signaling through the activation of the ABCA1-Cav1 pathway [research support, non-U.S. gov’t]. Cell Cycle. 2013. May 15;12(10):1521–1535. PubMed PMID: 23598720; PubMed Central PMCID: PMC3680532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee HC, Her NG, Kang D, et al. Radiation-inducible miR-770-5p sensitizes tumors to radiation through direct targeting of PDZ-binding kinase [research support, non-U.S. gov’t]. Cell Death Dis. 2017. March 23;8(3):e2693. PubMed PMID: 28333152; PubMed Central PMCID: PMC5386522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Her NG, Toth JI, Ma CT, et al. p97 composition changes caused by allosteric inhibition are suppressed by an on-target mechanism that increases the enzyme’s atpase activity. Cell Chem Biol. 2016. April 21;23(4):517–528. PubMed PMID: 27105284; PubMed Central PMCID: PMC4841942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Han J, Back SH, Hur J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death [research support, N.I.H., extramural research support, non-U.S. gov’t]. Nat Cell Biol. 2013. May;15(5):481–490. PubMed PMID: 23624402; PubMed Central PMCID: PMC3692270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Marchand A, Tomkiewicz C, Magne L, et al. Endoplasmic reticulum stress induction of insulin-like growth factor-binding protein-1 involves ATF4 [research support, non-U.S. gov’t]. J Biol Chem. 2006. July 14;281(28):19124–19133. PubMed PMID: 16687408. [DOI] [PubMed] [Google Scholar]
- [27].Cunha DA, Cito M, Carlsson PO, et al. Thrombospondin 1 protects pancreatic beta-cells from lipotoxicity via the PERK-NRF2 pathway [research support, non-U.S. gov’t]. Cell Death Differ. 2016. December;23(12):1995–2006. PubMed PMID: 27588705; PubMed Central PMCID: PMC5136495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cunha DA, Cito M, Grieco FA, et al. Pancreatic beta-cell protection from inflammatory stress by the endoplasmic reticulum proteins thrombospondin 1 and mesencephalic astrocyte-derived neurotrophic factor (MANF) [research support, non-U.S. gov’t]. J Biol Chem. 2017. September 8;292(36):14977–14988. PubMed PMID: 28698383; PubMed Central PMCID: PMC5592674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou HL, Geng C, Luo G, et al. The p97-UBXD8 complex destabilizes mRNA by promoting release of ubiquitinated HuR from mRNP [research support, N.I.H., extramural research support, non-U.S. gov’t research support, U.S. gov’t, non-P.H.S.]. Genes Dev. 2013. May 1;27(9):1046–1058. PubMed PMID: 23618873; PubMed Central PMCID: PMC3656322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bastola P, Neums L, Schoenen FJ, et al. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with salubrinal [research support, N.I.H., extramural research support, U.S. gov’t, non-P.H.S. research support, non-U.S. gov’t]. Mol Oncol. 2016. December;10(10):1559–1574. PubMed PMID: 27729194; PubMed Central PMCID: PMC5423134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Raman M, Havens CG, Walter JC, et al. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction [research support, N.I.H., extramural research support, non-U.S. gov’t]. Mol Cell. 2011. October 7;44(1):72–84. PubMed PMID: 21981919; PubMed Central PMCID: PMC3190166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kaur S, Soto-Pantoja DR, Stein EV, et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors [research support, N.I.H., extramural research support, N.I.H., intramural research support, non-U.S. gov’t]. Sci Rep. 2013;3:1673. PubMed PMID: 23591719; PubMed Central PMCID: PMC3628113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee JH, Bhang DH, Beede A, et al. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis [research support, N.I.H., extramural research support, non-U.S. gov’t]. Cell. 2014. January 30;156(3):440–455. PubMed PMID: 24485453; PubMed Central PMCID: PMC3951122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Soto-Pantoja DR, Sipes JM, Martin-Manso G, et al. Dietary fat overcomes the protective activity of thrombospondin-1 signaling in the Apc(Min/+) model of colon cancer. Oncogenesis. 2016. May 30;5(5):e230. PubMed PMID: 27239962; PubMed Central PMCID: PMC4945754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhao H, Wang J, Kong X, et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci Rep. 2016. July 14;6:29719. PubMed PMID: 27411490; PubMed Central PMCID: PMC4944213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang Y, Sime W, Juhas M, et al. Crosstalk between colon cancer cells and macrophages via inflammatory mediators and CD47 promotes tumour cell migration [research support, non-U.S. gov’t]. Eur J Cancer. 2013. October;49(15):3320–3334. PubMed PMID: 23810249. [DOI] [PubMed] [Google Scholar]
- [37].Tseng D, Volkmer JP, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response [research support, N.I.H., extramural research support, non-U.S. gov’t]. Proc Natl Acad Sci U S A. 2013. July 2;110(27):11103–11108. PubMed PMID: 23690610; PubMed Central PMCID: PMC3703977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee TK, Cheung VC, Lu P, et al. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma [research support, non-U.S. gov’t]. Hepatology. 2014. July;60(1):179–191. PubMed PMID: 24523067. [DOI] [PubMed] [Google Scholar]
- [39].Daubon T, Leon C, Clarke K, et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development [research support, non-U.S. gov’t]. Nat Commun. 2019. March 8;10(1):1146. DOI: 10.1038/s41467-019-08480-y. PubMed PMID: 30850588; PubMed Central PMCID: PMC6408502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kamijo H, Miyagaki T, Takahashi-Shishido N, et al. Thrombospondin-1 promotes tumor progression in cutaneous T-cell lymphoma via CD47. Leukemia. 2019. November 11;34(3):845–856. DOI: 10.1038/s41375-019-0622-6. PubMed PMID: 31712778. [DOI] [PubMed] [Google Scholar]
- [41].Marisi G, Scarpi E, Passardi A, et al. IL-8 and thrombospondin-1 as prognostic markers in patients with metastatic colorectal cancer receiving bevacizumab. Cancer Manag Res. 2018;10:5659–5666. PubMed PMID: 30532588; PubMed Central PMCID: PMC6241685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Clarke R, Cook KL, Hu R, et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate [research support, N.I.H., extramural research support, non-U.S. gov’t research support, U.S. gov’t, non-P.H.S. review]. Cancer Res. 2012. March 15;72(6):1321–1331. PubMed PMID: 22422988; PubMed Central PMCID: PMC3313080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cook KL, Shajahan AN, Warri A, et al. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness [research support, N.I.H., extramural research support, U.S. gov’t, non-P.H.S.]. Cancer Res. 2012. July 1;72(13):3337–3349. PubMed PMID: 22752300; PubMed Central PMCID: PMC3576872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cook KL, Soto-Pantoja DR, Clarke PA, et al. Endoplasmic reticulum stress protein GRP78 modulates lipid metabolism to control drug sensitivity and antitumor immunity in breast cancer [research support, U.S. gov’t, non-P.H.S. research support, N.I.H., extramural research support, N.I.H., intramural]. Cancer Res. 2016. October 1;76(19):5657–5670. PubMed PMID: 27698188; PubMed Central PMCID: PMC5117832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Busselberg D, Florea AM. Targeting intracellular calcium signaling ([Ca(2+)]i) to overcome acquired multidrug resistance of cancer cells: a mini-overview [review]. cancers. 2017May9;9(5). DOI:10.3390/cancers9050048. PubMed PMID: 28486397; PubMed Central PMCID: PMC5447958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bong AHL, Monteith GR. Calcium signaling and the therapeutic targeting of cancer cells [research support, non-U.S. gov’t review]. Biochim Biophys Acta Mol Cell Res. 2018. November;1865(11 Pt B):1786–1794. PubMed PMID: 29842892. [DOI] [PubMed] [Google Scholar]
- [47].Bazzazi H, Isenberg JS, Popel AS. Inhibition of VEGFR2 activation and its downstream signaling to ERK1/2 and calcium by thrombospondin-1 (TSP1): in silico investigation. Front Physiol. 2017;8:48. PubMed PMID: 28220078; PubMed Central PMCID: PMC5292565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Soto-Pantoja DR, Kaur S, Roberts DD. CD47 signaling pathways controlling cellular differentiation and responses to stress [research support, N.I.H., intramural review]. PubMed PMID: 25708195; PubMed Central PMCID: PMC4822708 Crit Rev Biochem Mol Biol. 2015;50(3):212–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Feng M, Marjon KD, Zhu F, et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer [research support, N.I.H., extramural research support, non-U.S. gov’t]. Nat Commun. 2018. August 10;9(1):3194. DOI: 10.1038/s41467-018-05211-7. PubMed PMID: 30097573; PubMed Central PMCID: PMC6086865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kaur S, Schwartz AL, Jordan DG, et al. Identification of schlafen-11 as a target of CD47 signaling that regulates sensitivity to ionizing radiation and topoisomerase inhibitors. Front Oncol. 2019;9:994. PubMed PMID: 31632920; PubMed Central PMCID: PMC6781860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Denefle T, Pramil E, Gomez-Morales L, et al. Homotrimerization approach in the design of thrombospondin-1 mimetic peptides with improved potency in triggering regulated cell death of cancer cells. J Med Chem. 2019. September 12;62(17):7656–7668. PubMed PMID: 31403795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.