

ABSTRACT
Osteoarthritis (OA) is characterized by apoptosis of chondrocytes and an imbalance of extracellular matrix (ECM) synthesis and catabolism. Emerging evidence has demonstrated that miRNAs are involved in OA pathologies, but the role of miR-296-5p in OA remains unclear. The present study proposes to reveal the functions and mechanisms of miR-296-5p in a cell model of OA. In this study, human chondrocytes were treated with 5 ml interleukin-1 beta (IL-1β) to induce apoptosis and cartilage degradation. Our results showed that miR-296-5p was downregulated in chondrocytes stimulated with IL-1β. Overexpressed miR-296-5p enhanced cell proliferation and inhibited apoptosis and matrix degrading enzyme expression in response to IL-1β stimulation, and knockdown of miR-296-5p showed the opposite effect. Further, we found that miR-296-5p directly targeted the 3′-untranslated region (3′-UTR) of TGF-β1 mRNA, and miR-296-5p inactivated the TGF-β1/CTGF/p38MAPK signaling pathway. Overexpression of TGF-β1 alleviated the inhibition of miR-296-5p on chondrocyte apoptosis and cartilage degradation. In conclusion, miR-296-5p inhibited the progression of OA through the CTGF/p38MAPK pathway by directly targeting TGF-β1.
KEYWORDS: Osteoarthritis, miR-296-5p, chondrocyte
Introduction
Osteoarthritis (OA) is one of the most prevalent degenerative joint diseases, which seriously affects the health of the elderly and is characterized by articular cartilage degradation [1,2]. Articular cartilage contains two components, articular cartilage chondrocytes and extracellular matrix (ECM) [3]. Normally, chondrocytes can regulate the synthesis and degradation of type II collagen, aggrecan, and glycoproteins, to maintain the dynamic balance of cartilage ECM and exert its biological functions [4,5]. However, during the onset of OA, chondrocytes activate several proinflammatory cytokines, such as interleukin-1β (IL-1β), which can stimulate the expression of collagenase (matrix metalloproteinases, MMPs) and aggrecanase (a disintegrin-like and metalloprotease with a thrombospondin type 1 motif, ADAMTS) [6]. These matrix degradation-related proteolytic enzymes contribute to the degradation of ECM and destruction of cartilage, and finally aggravate the pathologies of OA [5,7,8]. Therefore, IL-1β-induced chondrocytes are often used as a cell model of OA in vitro.
MicroRNAs (miRNAs), a family of endogenous noncoding RNA molecules containing about 17 to 25 nucleotides, are highly conserved and prevalent in eukaryotic cells [9]. miRNAs play important roles in post-transcriptional negative regulation of their target genes through base pairing [10]. Accumulating studies indicate that multiple miRNAs are aberrantly regulated in cartilage degradation, and several miRNAs have been proven to act in vital regulatory roles in OA progression [3,11]. For instance, miR-93-5p, miR-204-5p and miR-221-3p are downregulated in OA tissues, and they can ameliorate OA progression via suppression of chondrocyte apoptosis and cartilage degradation [3,12,13]. miR-296-5p, a member of the miR-296 family, is located on the chromosome 20q13.32 genomic locus and regarded as a tumor angiogenesis-related miRNA [14,15]. A recent study demonstrated that miR-296-5p is downregulated in OA patients [16], but its function remains unclear.
Transforming growth factor β1 (TGF-β1) plays a key role in the formation of articular cartilage during growth and development, and emerging studies indicate that its overexpression causes the degradation of proteoglycans, thereby leading to OA [17]. Connective tissue growth factor (CTGF/CCN2) is a 36 − 38 kDa cysteine-rich secreted protein that function as a downstream mediator of TGF-β1 in multiple tissues [18,19]. Upregulation of CTGF is frequently observed in arthritis, and an increasing number of articles have demonstrated that CTGF, mediated by TGF-β1, participates in the development of rheumatoid arthritis (RA), and inhibition of CTGF attenuates inflammatory responses and MMP levels in an IL-1β-stimulated knee OA model [20,21]. It has been reported that CTGF activates the p38MAPK signaling pathway in dilated cardiomyopathy, and CTGF induces matrix protein expression via the p38MAPK signaling pathway [22,23]. The P38MAPK pathway acts as a very important part of cell gene expression, proliferation, differentiation and adaptation to environmental stress, and inhibition of p38MAPK signaling ameliorates chondrocyte apoptosis and cartilage degradation in OA [24–26]. However, the molecular mechanism underlying the association between TGF-β1, CTGF and the p38MAPK pathway is still unclear.
This study aimed to explore the effects and underlying mechanism of miR-296-5p on an IL-1β-induced cell model of OA. In this study, we found that miR-296-5p was downregulated in IL-1β-evoked chondrocytes. Moreover, overexpression of miR-296-5p alleviated IL-1β-induced chondrocyte apoptosis and cartilage matrix degradation through targeting of TGF-β1 and inactivation of the connective CTGF/p38MAPK signaling pathway. These results indicated that miR-296-5p is a potential target for clinical prevention and therapeutic treatment of OA.
Material & methods
Cell culture and treatment
Normal human knee articular chondrocytes (NHAC-kn) were obtained from Lonza Bioscience (Walkersville, USA), and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, USA) containing 10% Fetal Bovine Serum (FBS, Thermo Fisher Scientific, USA) at 37°C with 5% CO2 in a humidified incubator. NHAC-kn cells were treated with IL-1β (PeproTech, NJ, USA) at a series of concentrations (1, 2, 5 and 10 μg/ml) for 12 h.
Cell transfection
MiR-295-5p mimics and mimic controls, miR-295-5p inhibitor and inhibitor control, small interfering RNA against TGF-β1 (TGF-β1 siRNA) and siRNA control, were synthesized and obtained from Gene Pharma (Shanghai, China). The TGF-β1 overexpression plasmid (pcDNA-TGF-β1) was constructed and obtained from Generay Biotech (Shanghai, China). IL-1β-stimulated NHAC-kn cells were transfected with Lipofectamine 3000 (Invitrogen, Carlsbad, USA) for 48 h.
Cell viability assay
The Cell Counting Kit-8 (CCK-8, Dojindo, Japan) was used to determine cell viability in this study. Briefly, NHAC-kn cells were seeded at a density of 2 × 103 cells per well in 96-well plates. After culturing for 48 h, 10 μl CCK-8 solution was added to each well and then incubated at 37°C for 2 h, and the optical density (OD) was measured using a 96-well microplate reader at 450 nm.
Cell apoptosis assay
The apoptotic rate of NHAC-kn cells was analyzed with an annexin V-fluorescein isothiocyanate (FITC) conjugate combined with the propidium iodide (PI) assay (Dojindo, Japan) and evaluated by fluorescence-activated cell sorting (BD Biosciences, San Diego, USA).
RNA extraction and quantitative reverse-transcription PCR (qRT-PCR)
The total RNA of treated or non-treated NHAC-kn cells was extracted with TRIzol Reagent (Invitrogen, Carlsbad, USA). The TaqMan MicroRNA Reverse Transcription Kit and TaqMan miRNA assay (Qiagen, China) were used to quantify miR-296-5p expression. U6 was used as the internal control. The PrimeScript RT reagent kit (Takara) was used to synthesize cDNA of the target mRNA, and SYBR Premix Ex Taq (Takara) was used for the analysis according to the manufacturer’s instructions, with β-actin as an internal control. The qRT-PCR procedure was as follows: 1 min at 95°C, 20 s at 95°C and 10 s at 56°C and 15 s at 72°C for 35 cycles then held at 4°C. Finally, we used the 2−ΔΔCT method to quantify the abundance of miRNAs or mRNA. The primers used were miR-296-5p: forward, 5′‐ GTATCCAGTGCAGGGTCCGA‐3′, reverse, 5′‐ CGACGAGGGCCCCCCCT‐3′; U6: forward, 5′-CGCTTCGGCAGCACATATAC-3′, reverse, 5′-AAATATGGAACGCTTCACGA-3′; MMP-13: forward, 5′-GACAGTGGAGGTGGCCTTAC-3′, reverse, 5′-ACAAGTGGGTAGATAAACAAGGT-3′; ADAMTS-5: forward, 5′-CTTTAGAGGGAGAAAATTCTGG-3′, reverse, 5′-AAAGATTTACCATTGGGTGG-3′; collagen II: forward, 5′-CTCAAGTCGCTGAACAACCA-3′, reverse, 5′-GTCTCCGCTCTTCCACTCTG-3′; aggrecan: forward, 5′-ACCCCAACACCTACAAGCACA-3′, reverse, 5′-AAAGCGACAAGAAGACACCA-3′; β-actin: forward, 5′-CCTCTATGCCAACACAGT-3′, reverse, 5′-AGCCACCAATCCACACAG-3′.
Western blot
NHAC-kn cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific) with a protease inhibitor cocktail (Millipore, MA, USA). The protein extraction was resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% resolving gel with a 5% stacking gel), and subsequently transferred onto a PVDF membrane (Millipore, MA, USA). After blocking in 5% nonfat milk for 1 h at room temperature, the membrane was incubated with primary antibody at 4°C overnight. After washing with PBS three times, the membranes were then treated with corresponding secondary antibodies conjugated to horseradish peroxidase for 2 h at room temperature and incubated with enhanced chemiluminescence solution (Bio-Rad, Hercules, USA). Finally, the densitometer technology Quantity One software (Bio-Rad, Hercules, USA) was used for signal visualization and quantification. The antibodies used in western blotting included anti-CDK2 (ab32147); anti-Cyclin D1 (ab16663); anti-Bcl-2 (ab32370); anti-Bax (ab32503); anti-cleaved-Caspase 9 (ab2324); anti-TGF-β1 (ab92486); anti-CTGF (ab231824); anti-p-p38 (ab133581); anti-p-ERK (ab 201015); anti-pJNK (ab207477); anti-GAPDH (ab181602), which were all purchased from Abcam (USA).
Enzyme‐linked immunosorbent assay (ELISA)
Samples were centrifuged, and the culture supernatant was collected for analysis. The concentrations of MMP-13 and ADAMTS-5 in NHAC-kn cells were determined by ELISA Kits (R&D Systems, Minneapolis, USA) according to the manufacturer’s instructions.
Luciferase activity assay
The wild‐type pGL3-TGF-β1-3′-UTR (WT-TGF-β1) and mutant type pGL3‐TGF-β1‐3′-UTR (MUT-TGF-β1) were constructed and purchased from Agilent Technologies (Santa Clara, USA). The plasmids were transfected into cells using Lipofectamine 3000 (Invitrogen, Carlsbad, USA) for 48 h. Luciferase activities were consecutively measured according to the Dual‐Luciferase Reporter Assay System manufacturer’s protocol (Promega, Madison, USA).
Statistical analysis
All experiments were repeated in triplicate. Statistical analysis was performed with SPSS 22.0 software (SPSS Inc., USA). The data are all indicated as the mean ± SD. The resulting value of *p < 0.05, **p < 0.01, #p < 0.05 or &p < 0.05 was considered statistically significant.
Results
miR-296-5p is downregulated in IL-1β-treated NHAC-kn cells
As shown in Figure 1(a,b), cell viability and miR-296-5p expression in NHAC-kn cells were both decreased in a dose-dependent manner after treatment with IL-1β in a concentration gradient (1, 2, 5 and 10 μg/ml) for 12 h (*p < 0.05, **p < 0.01).
Figure 1.
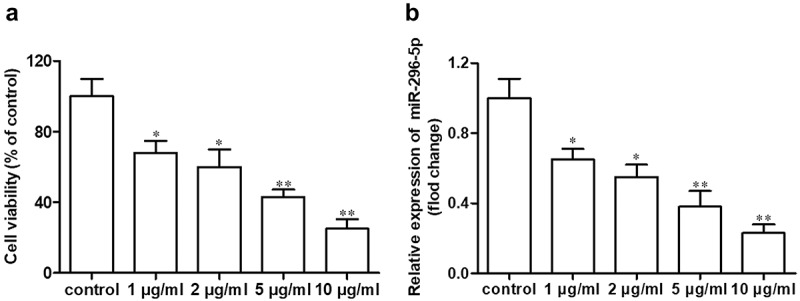
miR-296-5p is downregulated in IL-1β-treated NHAC-kn cells.
miR-296-5p inhibits apoptosis in IL-1β-treated NHAC-kn cells
Chondrocyte apoptosis and cartilage degradation are the main pathological features of OA [3,27]. Because miR-296-5p was downregulated in IL-1β-induced NHAC-kn cells, we speculated that miR-296-5p played a role in OA pathologies. The effects of miR-296-5p mimics and inhibitors on chondrocyte proliferation and apoptosis were determined using CCK-8, Annexin V FITC/PI and western blot assays. The results showed that miR-296-5p mimics and inhibitor were significantly elevated and reduced miR-296-5p levels, respectively (Figure 2(a), p < 0.05). After IL-1β stimulation, miR-296-5p mimics significantly improved cell viability and suppressed cell apoptosis rate, and miR-296-5p inhibitor remarkably reduced cell viability and increased the cell apoptosis rate (Figure 2(b,c), p < 0.05). With respect to the western blot data, miR-296-5p mimics facilitated the expression of cell cycle makers, including CDK2 and Cyclin D1, and attenuated the expression of apoptotic effector Bax/Bcl-2 and cleaved Caspase 9; moreover, miR-296-5p inhibitor showed the opposite effect (Figure 2(d–h), p < 0.05). These results indicated that miR-296-5p inhibited IL-1β-induced NHAC-kn apoptosis.
Figure 2.
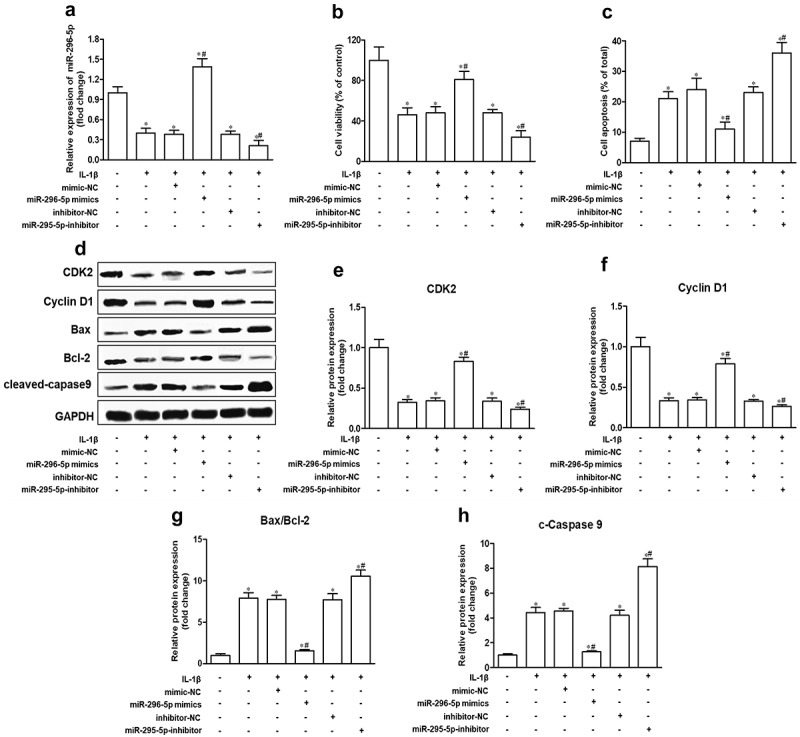
miR-296-5p inhibits apoptosis in IL-1β-treated NHAC-kn cells.
miR-296-5p alleviates cartilage degradation in IL-1β-treated NHAC-kn cells
To determine the effect of miR-296-5p on IL-1β-induced cartilage degradation, we analyzed the expression of major cartilage degradation-related genes, including MMP-13 and ADAMTS-5, collagen II and aggrecan, which are responsible for the balance of ECM catabolism and synthesis in NHAC-kn cells. The results showed that the mRNA and protein expression levels of MMP-13 and ADAMTS-5 were increased remarkably, whereas the mRNA and protein expression levels of collagen II and aggrecan were decreased remarkably by IL-1β stimulation. Further, miR-296-5p mimics reduced MMP-13 and ADAMTS-5 expression and enhanced collagen II and aggrecan expression, while miR-296-5p inhibitor improved MMP-13 and ADAMTS-5 expression and attenuated collagen II and aggrecan expression in NHAC-kn cells (Figure 3, p < 0.05). The results suggested that miR-296-5p inhibited cartilage matrix degradation in IL-1β-stimulated NHAC-kn cells.
Figure 3.
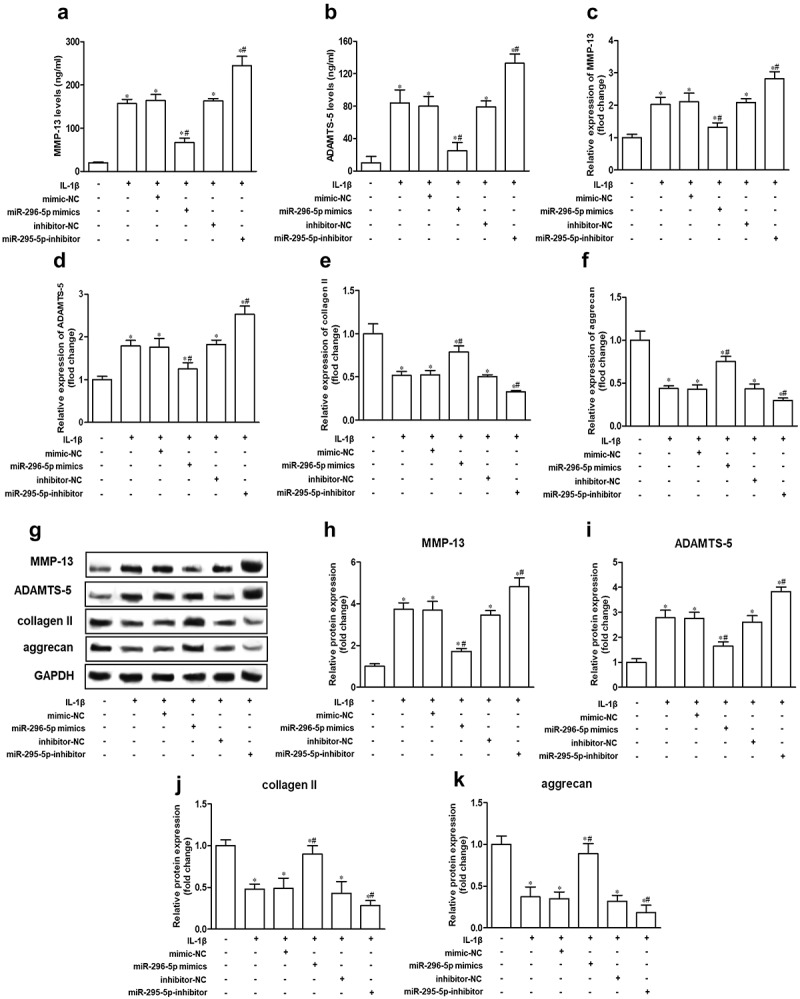
miR-296-5p alleviates cartilage matrix degradation in IL-1β-treated NHAC-kn cells.
miR-296-5p regulates the CTGF/p38MAPK axis via targeting of TGF-β1
Next, we attempted to understand the mechanisms of miR-296-5p in OA. Based on the bioinformatics software Starbase 2.0., we found that the 3′-untranslated region (3′-UTR) of TGF-β1 mRNA possesses a potential miR-296-5p binding site (Figure 4(a)). To determine the direct binding between miR-296-5p and TGF-β1, a luciferase reporter assay containing WT-TGF-β1 or MUT-TGF-β1 was constructed and transfected into NHAC-kn cells. The result showed that miR-296-5p mimics significantly attenuated the luciferase activity of WT-TGF-β1, rather than MUT-TGF-β1 (Figure 4(b), p < 0.05). Moreover, miR-296-5p mimics conspicuously attenuated the expression of TGF-β1 mRNA and protein in NHAC-kn cells (Figure 4(c,d), p < 0.05). In the present study, we found that small interfering RNA against TGF-β1 (TGF-β1 siRNA) ameliorated the promotion of miR-296-5p inhibitor on TGF-β1, CTGF, p-p38, p-ERK and p-JNK expression levels, and TGF-β1 overexpression vector (pcDNA-TGF-β1) counteracted the suppression of miR-296-5p mimics on the expression of these genes (Figure 4(e–j), p < 0.05). These data revealed that miR-296-5p was able to blunt the activation of the TGF-β1/CTGF/p38MAPK pathway in NHAC-kn cells.
Figure 4.
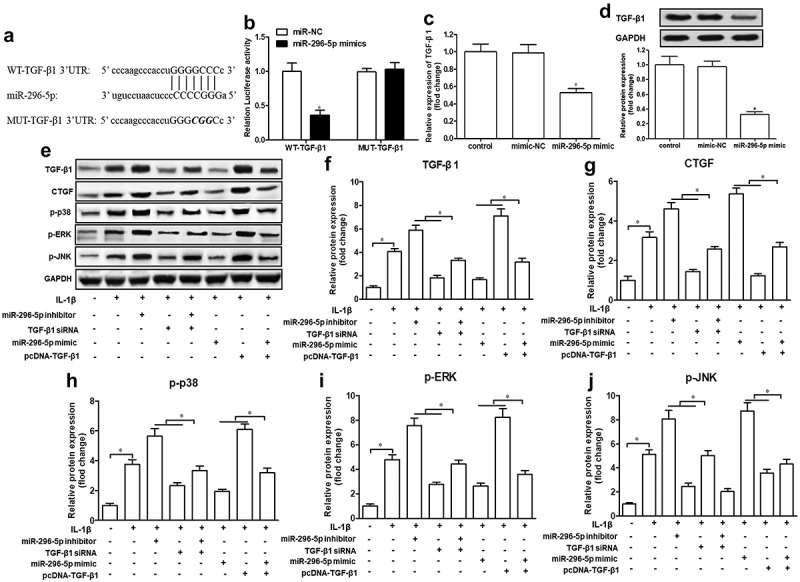
miR-296-5p regulates TGF-β1/CTGF/p38MAPK axis via targeting of TGF-β1.
TGF-β1 partly attenuates the suppressive effect of miR-296-5p on IL-1β-induced chondrocyte apoptosis and cartilage degradation
Lastly, we explored the role of TGF-β1 in the function of miR-296-5p on IL-1β-induced chondrocyte apoptosis and cartilage degradation. Our data demonstrated that TGF-β1 siRNA significantly abrogated the effect of miR-296-5p on cell viability, the apoptosis rate and MMP-13 and ADAMTS-5 levels, while pcDNA-TGF-β1 significantly impaired the function of miR-296-5p mimics on cell viability, the apoptosis rate and MMP-13 and ADAMTS-5 levels, in IL-1β-treated NHAC-kn cells (Figure 5(a–d), p < 0.05). Hence, our data suggested that miR-296-5p inhibited IL-1β-induced chondrocyte apoptosis and cartilage degradation via blocking activation of the TGF-β1/CTGF/p38MAPK pathway.
Figure 5.
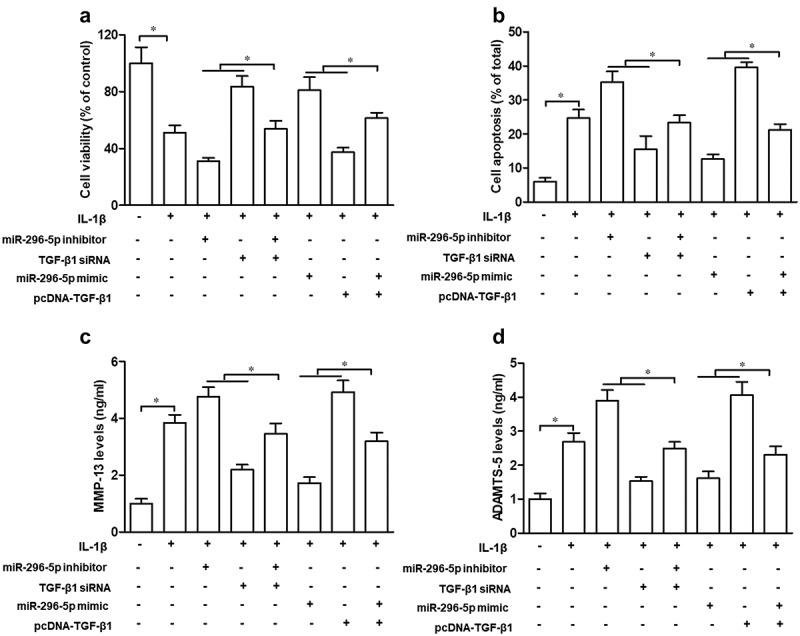
TGF-β1 partly attenuates the suppressive effect of miR-296-5p on IL-1β-induced chondrocyte apoptosis and cartilage degradation.
Discussion
The involvement of miRNAs in osteoarthritis (OA) pathogenesis has received much attention in recent years [3,11]. In this study, we aimed to characterize the potential function and mechanism of miR-296-5p in a cell model of OA. Our data demonstrated that miR-296-5p was significantly downregulated in IL-1β-induced chondrocytes, which is consistent with a study by Song et al., who found that miR-296-5p expression was decreased in OA tissues [16]. Previous studies have reported that miR-296-5p is aberrantly expressed in a variety of human diseases and exhibits tissue-specific differences [28–30]. Notably, by acting on different target genes, miR-296-5p plays a different role in many pathophysiological processes, such as cellular proliferation and apoptosis [31,32]. On the one hand, miR-296 suppresses cell proliferation and induces apoptosis in hepatocellular carcinoma and prostate cancer by targeting NRG1/ERBB2/ERBB3/RAS/MAPK/Fra-2 signaling and peptidyl-prolyl isomerase (Pin1), respectively [15,33]. On the other hand, miR-296-5p promotes cell proliferation in vascular smooth muscle cells (VSMCs) through inhibition of p53, p21WAF1 and p27; miR-296-5p attenuates apoptosis of gastric cancer by interacting with Caudal-related homeobox 1 (CDX1) [34,35]. In the present study, we found that miR-296-5p ameliorated IL-1β-induced chondrocyte apoptosis and cartilage degradation via targeting of TGF-β1.
A growing number of studies have shown that TGF-β1 plays a key role in the formation of articular cartilage during growth and development, and it can stimulate chondrocytes to synthesize and secrete proteoglycans and type II collagen [36]. Some evidence indicate that TGF-β1 downregulation can trigger the occurrence of OA, and it can play a protective role in articular cartilage [37]. However, other investigations have found that overexpression of TGF-β1 is closely related to the degradation of proteoglycans, thereby leading to OA occurrence [38]. Zhang et al. reported that the TGF-β1 receptor inhibitor sB-505124 can significantly reduce levels of cartilage degeneration-related markers, including MMP-9 and MMP-13, in anterior cruciate ligament (ACL)-induced OA mice [39]. Zhen et al. revealed that elevating TGF-β1 levels in osteoplastic can aggravate the development of OA, whereas down-regulating TGF-β1 expression in subchondral ameliorates cartilage degradation [40]. Chen et al. also indicated that inhibition of TGF-β1 signaling in the articular chondrocyte decreases cartilage degradation in OA mice [41]. In addition, TGF-β1 has been proven to induce apoptosis in several cancers cell by blocking cyclin-dependent kinase CDK2 and Cyclin D1 activity, and the suppression of TGF-β1 could significantly reverse the suppression of chondrocyte growth and the induction of apoptosis [36,42,43]. Herein, we found that miR-296-5p inhibited IL-1β-induced chondrocyte apoptosis and cartilage degradation by negatively regulating TGF-β1.
Emerging studies have reported that CTGF is an important downstream modulator and also acts as a downstream effector molecule of TGF-β1 in various tissues. Meanwhile, TGF-β1 plays some biological functions dependent on CTGF, including promoting cell proliferation and ECM synthesis [21,44,45]. Notably, CTGF plays an important role in TGFβ activation in response to cartilage injury [45]. It has been reported that CTGF contributes to myocardial fibrosis and induces matrix protein production by activating the p38MAPK signaling pathway [22,23]. What is more, CTGF has been proved that increases the intensity and persistence of synovitis, as well as the expression of catabolic factors in OA by the potentiation of p38 activation [46]. In this study, we found that miR-296-5p inhibited CTGF/p38MAPK signaling via suppression of TGF-β1. Therefore, miR-296-5p can protect chondrocytes from IL-1β-induced injury by downregulating TGF-β1/CTGF/p38MAPK expression.
In conclusion, the present study showed that miR-296-5p was downregulated in IL-1β-induced chondrocytes, and miR-296-5p inhibited IL-1β-induced chondrocyte apoptosis and matrix-degrading enzyme expression. An in-depth analysis suggested that TGF-β1 is a direct target of miR-296-5p, and miR-296-5p mediated OA development, possibly by blocking the TGF-β1/CTGF/p38MAPK pathway. Thus, our study suggested that miR-296-5p may serve as a therapeutic target for patients with OA.
Supplementary Material
Funding Statement
The study was funded by Yantai Science and Technology Plan (2017YD016).
Consent to publication
All authors have obtained consent to publish the data from the study.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
XJ S designed the experiments; ZL C, WG L, XY Q, XJ S, Q Y and G C performed the experiments; ZL C and WG L analyzed the data and wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Lu Z, Luo M, Huang Y.. lncRNA-CIR regulates cell apoptosis of chondrocytes in osteoarthritis. J Cell Biochem. 2018.;120:7229–7237 [DOI] [PubMed] [Google Scholar]
- [2].Zheng W, Zhang H, Jin Y, et al. Butein inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes and slows the progression of osteoarthritis in mice. Int Immunopharmacol. 2017;42:1–10. [DOI] [PubMed] [Google Scholar]
- [3].Zheng X, Zhao FC, Pang Y, et al. Downregulation of miR-221-3p contributes to IL-1β-induced cartilage degradation by directly targeting the SDF1/CXCR4 signaling pathway. J Mol Med. 2017;95(6):615–627. [DOI] [PubMed] [Google Scholar]
- [4].Zhang G, Wu Y, Xu D, et al. Long noncoding RNA UFC1 promotes proliferation of chondrocyte in osteoarthritis by acting as a sponge for miR-34a. DNA Cell Biol. 2016;35(11):691–695. [DOI] [PubMed] [Google Scholar]
- [5].Xia L, Zhang H-X, Xing M-L, et al. Knockdown of PRMT1 suppresses IL-1β-induced cartilage degradation and inflammatory responses in human chondrocytes through Gli1-mediated Hedgehog signaling pathway. Mol Cell Biochem. 2017;438:1–8. [DOI] [PubMed] [Google Scholar]
- [6].Zheng W, Feng Z, You S, et al. Fisetin inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes through activating SIRT1 and attenuates the progression of osteoarthritis in mice. Int Immunopharmacol. 2017;45:135–147. [DOI] [PubMed] [Google Scholar]
- [7].Haque Bhuyan MZ, Tamura Y, Sone E, et al. The intra-articular injection of RANKL-binding peptides inhibits cartilage degeneration in a murine model of osteoarthritis. J Pharmacol Sci. 2017;134(2):124–130. [DOI] [PubMed] [Google Scholar]
- [8].Qu Y, Zhou L, Wang C. Mangiferin inhibits IL-1β-induced inflammatory response by activating PPAR-γ in human osteoarthritis chondrocytes. Inflammation. 2017;40(1):52–57. [DOI] [PubMed] [Google Scholar]
- [9].Li JH, Liu S, Zhou H, et al. Starbase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale clip-seq data. Nucleic Acids Res. 2014;42(D1):D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li J, Li Q, Chen L, et al. Competitive endogenous RNA networks: integrated analysis of non-coding RNA and mRNA expression profiles in infantile hemangioma. Oncotarget. 2018;9:11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ko J-Y, Lee MS, Lian W-S, et al. MicroRNA-29a counteracts synovitis in knee osteoarthritis pathogenesis by targeting VEGF. Sci Rep. 2017;7(1):3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cao J, Han X, Qi X, et al. miR‑204‑5p inhibits the occurrence and development of osteoarthritis by targeting Runx2, International. J Mol Med. 2018;42:2560–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xue H, Tu Y, Ma T, et al. miR-93-5p attenuates IL-1β-induced chondrocyte apoptosis and cartilage degradation in osteoarthritis partially by targeting TCF4. Bone. 2019;123:129–136. [DOI] [PubMed] [Google Scholar]
- [14].Xu C, Li S, Chen T, et al. miR-296-5p suppresses cell viability by directly targeting PLK1 in non-small cell lung cancer. Oncol Rep. 2016;35(1):497. [DOI] [PubMed] [Google Scholar]
- [15].Shi DM, Li LX, Bian XY, et al. miR-296-5p suppresses EMT of hepatocellular carcinoma via attenuating NRG1/ERBB2/ERBB3 signaling. J Exp Clin Cancer Res. 2018;37(1):294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Song J, Kim D, Chang HL, et al. MicroRNA-488 regulates zinc transporter SLC39A8/ZIP8 during pathogenesis of osteoarthritis. J Biomed Sci. 2013;20(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yoon HJ, Kim SB, Somaiya D, et al. Type II collagen and glycosaminoglycan expression induction in primary human chondrocyte by TGF-β1. BMC Musculoskelet Disord. 2015;16(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tang X. The role of connective tissue growth factor (CTGF) in articular cartilage. Imperial College London; 2015. https://doi.org/10.25560/25292. [Google Scholar]
- [19].Arnott JA, Nuglozeh E, Rico MC, et al. Connective tissue growth factor (CTGF/CCN2) is a downstream mediator for TGF-beta1-induced extracellular matrix production in osteoblasts. J Cell Physiol. 2010;210(3):843–852. [DOI] [PubMed] [Google Scholar]
- [20].Wang Z, Qiu Y, Lu J, et al. Connective tissue growth factor promotes interleukin-1β-mediated synovial inflammation in knee osteoarthritis. Mol Med Rep. 2013;8(3):877–882. [DOI] [PubMed] [Google Scholar]
- [21].Zhang J, Li C, Zheng Y, et al. Inhibition of angiogenesis by arsenic trioxide via TSP-1–TGF-β1-CTGF–VEGF functional module in rheumatoid arthritis. Oncotarget. 2017;8(43):73529–73546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nagai N, Klimava A, Lee W-H, et al. CTGF is increased in basal deposits and regulates matrix production through the ERK (p42/p44 mapk) MAPK and the p38 MAPK signaling pathways. Invest Ophthalmol Vis Sci. 2009;50(4):1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hong L, Lai HL, Fang Y, et al. Silencing CTGF/CCN2 inactivates the MAPK signaling pathway to alleviate myocardial fibrosis and left ventricular hypertrophy in rats with dilated cardiomyopathy. J Cell Biochem. 2018;119(11):9519–9531. [DOI] [PubMed] [Google Scholar]
- [24].Le LTT, Swingler TE, Crowe N, et al. The microRNA-29 family in cartilage homeostasis and osteoarthritis. J Mol Med. 2016;94(5):583–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dubash AD, Kam CY, Aguado BA, et al. Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J Cell Biol. 2016;212(4):425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou Y, Liu SQ, Yu L, et al. Berberine prevents nitric oxide-induced rat chondrocyte apoptosis and cartilage degeneration in a rat osteoarthritis model via AMPK and p38 MAPK signaling. Apoptosis. 2015;20(9):1187–1199. [DOI] [PubMed] [Google Scholar]
- [27].Dong HC, Li PN, Chen CJ, et al. Sinomenine attenuates cartilage degeneration by regulating miR-223-3p/NLRP3 inflammasome signaling. Inflammation. 2019;42:1–11. [DOI] [PubMed] [Google Scholar]
- [28].Fang L, Ellims AH, Moore X, et al. Circulating microRNAs as biomarkers for diffuse myocardial fibrosis in patients with hypertrophic cardiomyopathy. J Transl Med. 2015;13(1):314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wei Y, Nazari-Jahantigh M, Chan L, et al. The microRNA-342-5p fosters inflammatory macrophage activation through an Akt1-and microRNA-155–dependent pathway during atherosclerosis. Circulation. 2013;127(15):1609–1619. [DOI] [PubMed] [Google Scholar]
- [30].Li S, Zhu J, Zhang W, et al. Signature microRNA expression profile of essential hypertension and its novel link to human cytomegalovirus infection. Circulation. 2011;124(2):175–184. [DOI] [PubMed] [Google Scholar]
- [31].Lee H, Shin CH, Kim HR, et al. MicroRNA-296-5p promotes invasiveness through downregulation of nerve growth factor receptor and caspase-8. Mol Cells. 2017;40(4):254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dinami R, Buemi V, Sestito R, et al. Epigenetic silencing of miR-296 and miR-512 ensures hTERT dependent apoptosis protection and telomere maintenance in basal-type breast cancer cells. Oncotarget. 2017;8(56):95674–95691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee KH, Lin FC, Hsu TI, et al. MicroRNA-296-5p (miR-296-5p) functions as a tumor suppressor in prostate cancer by directly targeting Pin1. Biochim Biophys Acta. 2014;1843(9):2055–2066. [DOI] [PubMed] [Google Scholar]
- [34].Li T, Lu YY, Zhao XD, et al. MicroRNA-296-5p increases proliferation in gastric cancer through repression of Caudal-related homeobox 1. Oncogene. 2014;33(6):783–793. [DOI] [PubMed] [Google Scholar]
- [35].A-Rum Y, Ran G, Zeenia K, et al. MicroRNA-296 is enriched in cancer cells and downregulates p21WAF1 mRNA expression via interaction with its 3ʹ untranslated region. Nucleic Acids Res. 2011;39(18):8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang R, Li G, Zeng C, et al. Mechanical stress contributes to osteoarthritis development through the activation of transforming growth factor beta 1 (TGF-β1). Bone Joint Res. 2018;7(11):587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shen J, Li S, Chen D. TGF-β signaling and the development of osteoarthritis. Bone Res. 2014;2(1):14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fang J, Xu L, Li Y, et al. Roles of TGF-beta 1 signaling in the development of osteoarthritis. Histol Histopathol. 2016;31(11):1161–1167. [DOI] [PubMed] [Google Scholar]
- [39].Zhang R-K, Li G-W, Zeng C, et al. Mechanical stress contributes to osteoarthritis development through the activation of transforming growth factor beta 1 (TGF-β1). Bone Joint Res. 2018;7(11):587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gehua Z, Chunyi W, Xiaofeng J, et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2016;19:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rebecca C, Michelle M, Martin F, et al. Attenuation of the progression of articular cartilage degeneration by inhibition of TGF-β1 signaling in a mouse model of osteoarthritis. Am J Pathol. 2015;185(11):2875–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kitisin K, Ganesan N, Tang Y, et al. Disruption of transforming growth factor-β signaling through β-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene. 2007;26(50):7103–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gong JG, Ammanamanchi S, Ko TC, et al. Transforming growth factor β1 increases the stability of p21/WAF1/CIP1 protein and inhibits CDK2 kinase activity in human colon carcinoma FET cells. Cancer Res. 2003;63(12):3340–3346. [PubMed] [Google Scholar]
- [44].Abreu JG, Ketpura NI, Reversade B, et al. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β. Nat Cell Biol. 2002;4(8):599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Arnott JA, Nuglozeh E, Rico MC, et al. Connective tissue growth factor (CTGF/CCN2) is a downstream mediator for TGF-β1-induced extracellular matrix production in osteoblasts. J Cell Physiol. 2007;210(3):843–852. [DOI] [PubMed] [Google Scholar]
- [46].Wang Z, Qiu Y, Lu J, et al. Connective tissue growth factor promotes interleukin‑1β‑mediated synovial inflammation in knee osteoarthritis. Mol Med Rep. 2013;8(3):877–882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.