

ABSTRACT
Defective macroautophagy/autophagy and mitochondrial dysfunction are known to stimulate senescence. The mitochondrial regulator PPARGC1A (peroxisome proliferator activated receptor gamma, coactivator 1 alpha) regulates mitochondrial biogenesis, reducing senescence of vascular smooth muscle cells (VSMCs); however, it is unknown whether autophagy mediates PPARGC1A-protective effects on senescence. Using ppargc1a−/- VSMCs, we identified the autophagy receptor SQSTM1/p62 (sequestosome 1) as a major regulator of autophagy and senescence of VSMCs. Abnormal autophagosomes were observed in VSMCs in aortas of ppargc1a−/- mice. ppargc1a−/- VSMCs in culture presented reductions in LC3-II levels; in autophagosome number; and in the expression of SQSTM1 (protein and mRNA), LAMP2 (lysosomal-associated membrane protein 2), CTSD (cathepsin D), and TFRC (transferrin receptor). Reduced SQSTM1 protein expression was also observed in aortas of ppargc1a−/- mice and was upregulated by PPARGC1A overexpression, suggesting that SQSTM1 is a direct target of PPARGC1A. Inhibition of autophagy by 3-MA (3 methyladenine), spautin-1 or Atg5 (autophagy related 5) siRNA stimulated senescence. Rapamycin rescued the effect of Atg5 siRNA in Ppargc1a+/+, but not in ppargc1a−/- VSMCs, suggesting that other targets of MTOR (mechanistic target of rapamycin kinase), in addition to autophagy, also contribute to senescence. Sqstm1 siRNA increased senescence basally and in response to AGT II (angiotensin II) and zinc overload, two known inducers of senescence. Furthermore, Sqstm1 gene deficiency mimicked the phenotype of Ppargc1a depletion by presenting reduced autophagy and increased senescence in vitro and in vivo. Thus, PPARGC1A upregulates autophagy reducing senescence by a SQSTM1-dependent mechanism. We propose SQSTM1 as a novel target in therapeutic interventions reducing senescence.
Abbreviations
3-MA: 3 methyladenine; ACTA2/SM-actin: actin, alpha 2, smooth muscle, aorta; ACTB/β-actin: actin beta; AGT II: angiotensin II; ATG5: autophagy related 5; BECN1: beclin 1; CAT: catalase; CDKN1A: cyclin-dependent kinase inhibitor 1A (P21); Chl: chloroquine; CTSD: cathepsin D; CYCS: cytochrome C, somatic; DHE: dihydroethidium; DPBS: Dulbecco’s phosphate-buffered saline; EL: elastic lamina; EM: extracellular matrix; FDG: fluorescein-di-β-D-galactopyranoside; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; γH2AFX: phosphorylated H2A histone family, member X, H2DCFDA: 2ʹ,7ʹ-dichlorodihydrofluorescein diacetate; LAMP2: lysosomal-associated membrane protein 2; MASMs: mouse vascular smooth muscle cells; MEF: mouse embryonic fibroblast; NBR1: NBR1, autophagy cargo receptor; NFKB/NF-κB: nuclear factor of kappa light polypeptide gene enhancer in B cells; MTOR: mechanistic target of rapamycin kinase; NFE2L2: nuclear factor, erythroid derived 2, like 2; NOX1: NADPH oxidase 1; OPTN: optineurin; PFA: paraformaldehyde; PFU: plaque-forming units; PPARGC1A/PGC-1α: peroxisome proliferator activated receptor, gamma, coactivator 1 alpha; Ptdln3K: phosphatidylinositol 3-kinase; RASMs: rat vascular smooth muscle cells; ROS: reactive oxygen species; SA-GLB1/β-gal: senescence-associated galactosidase, beta 1; SASP: senescence-associated secretory phenotype; SIRT1: sirtuin 1; Spautin 1: specific and potent autophagy inhibitor 1; SQSTM1/p62: sequestosome 1; SOD: superoxide dismutase; TEM: transmission electron microscopy; TFEB: transcription factor EB; TFRC: transferrin receptor; TRP53/p53: transformation related protein 53; TUBG1: tubulin gamma 1; VSMCs: vascular smooth muscle cells; WT: wild type
KEYWORDS: Aging, autophagy, oxidative stress, senescence, SQSTM1, vascular biology
Introduction
Mitochondrial function and autophagy, two critical processes involved in protection against oxidative stress, decline with age [1,2]. Increased mitochondrial dysfunction during aging, in part by decreased biogenesis, promotes oxidative stress that causes damage to DNA, proteins and lipids, and is a major inducer of vascular senescence. Expression of mitochondrial electron transport chain components decreases with age [3], as does PPARGC1A [4], a central organizer of metabolic function, oxidative states, and mitochondrial biogenesis and function [5]. The PPARGC1 family of transcriptional co-activators (PPARGC1A, PPARGC1B and PPRC1) induces a robust upregulation of transcriptional activity and mitochondrial function [5]. PPARGC1A, the best-studied member of this family, controls mitochondrial biogenesis by co-activating major transcription factors regulating mitochondrial genes, including NFE2L2 (nuclear factor, erythroid derived 2, like 2) [5]. We reported a novel function for PPARGC1A as a negative regulator of vascular senescence [6], a process that was associated with increased oxidative stress, telomere dysfunction and atherosclerosis [7]. However, little is known about the contribution of PPARGC1A to autophagy and the role of defective autophagy in senescence induced by Ppargc1a gene deficiency.
Autophagy is a process that mediates the degradation of cytoplasmic components, such as dysfunctional mitochondria (mitophagy) [8] and protein aggregates, through lysosome-mediated degradation, thus promoting cellular homeostasis [9]. Autophagy has also been associated with reduced senescence. In primary human fibroblasts, depletion of Atg12, Atg7 or Lamp2 causes senescence, which is associated with increased levels of reactive oxygen species (ROS) and reduced expression of autophagy genes, including SQSTM1 [10]. Interestingly, both replicative senescence and senescence induced by defective autophagy correlated with reduced SQSTM1 protein levels [10]. Similarly, in fibroblasts, senescence induced by oxidative stress was mediated by defective autophagy caused by lysosomal dysfunction [11]. In the cardiovascular system, defective autophagy by Atg7 depletion led to the accumulation of SQSTM1 and senescence of VSMCs, and to diet-induced atherosclerosis [12]. SQSTM1 is an autophagy receptor involved in the targeting of cargo into autophagosomes. After the fusion of autophagosomes with lysosomes, the autophagosome content including SQSTM1 is degraded [13]. Thus, SQSTM1 protein levels have been used as an indicator of autophagic flux.
TFEB (transcription factor EB) is a master regulator of lysosomal biogenesis [14] and autophagy [15]. PPARGC1A and TFEB co-regulate each other, as seen in mouse embryonic fibroblasts [16], suggesting that PPARGC1A may regulate autophagy through a TFEB-dependent mechanism; however, evidence of the direct role of PPARGC1A in promoting autophagy and the possible mechanism involved are lacking.
sqstm1−/- mice present increased mitochondrial oxidative stress, decreased lifespan [17], mature-onset obesity, and insulin resistance [18]. Sqstm1 depletion was also shown to increase atherosclerosis in apoe−/- mice fed high fat diet [19]. It is unknown, however, whether PPARGC1A and SQSTM1 functionally interact to regulate autophagy and cellular senescence. Here, we demonstrated that PPARGC1A regulates autophagy and SQSTM1 expression and that SQSTM1 downregulation mediates PPARGC1A deficiency-induced senescence.
Results
Using an apoe−/- ppargc1a−/- animal model, we previously demonstrated that PPARGC1A is a negative regulator of vascular senescence [6]. Increased vascular senescence in this animal model is associated with accelerated deposition of plaque in the aorta, DNA damage and telomere shortening [7]. However, it is unknown whether defective autophagy contributes to these effects. To assess signs of dysfunctional autophagy and its contribution to senescence, we focused on VSMCs in the aorta of mice. We previously demonstrated that senescent cells are observed in all layers of the aorta of ppargc1a−/- mice, including VSMCs in the media and fibroblasts in the adventitia [6]. Histological examination of Ppargc1a+/+ (Figures 1A and B) and Ppargc1a± (Figures 1C and D) aorta samples revealed that the media of Ppargc1a± samples contain less organized VSMCs with more deposition of extracellular matrix (EM) compared with wild type (WT). The media of the aorta contains several layers of elastic laminae (EL; dark blue lines labeled with arrowheads in Figures 1A and 1C). VSMCs between two layers of elastic laminae were traced in Figures 1A and D to highlight the overall organization of VSMCs. Additionally, structures resembling apoptotic bodies were also seen in the Ppargc1a± aorta (Figure 1C, arrow). To better assess structural changes, we examined transverse sections of aortas by transmission electron microscopy (TEM) (Figure 1E–G). The most striking differences in overall organization were observed in smooth muscle cells in the media, which can be seen between two layers of EL. We observed less cell-cell contacts and disorganized mitochondria in Ppargc1a-deficient (Figure 1F and G) compared with WT (Figure 1E) mice. These changes were associated with increased EM deposition in Ppargc1a-deficient mice (30.8 ± 3.4%, n = 10) compared with WT (15.9 ± 7.7%, n = 4) (Figure 1H), which is consistent with the senescence-associated secretory phenotype (SASP) of senescent cells. Mitochondria were abundant in both genotypes, suggesting that Ppargc1b could be upregulated to induce mitochondrial biogenesis, as a compensatory mechanism. To test this possibility, we isolated mouse vascular smooth muscle cells (MASMs) from both genotypes and found that Ppargc1b mRNA was not upregulated (Fig. S1A and B), suggesting that degradation of mitochondria by autophagy could be inhibited. Consistent with previous reports, ppargc1a−/- MASMs showed reduced expression of the antioxidant enzyme SOD2 (superoxide dismutase 2, mitochondrial) increased expression of the senescent marker CDKN1A/p21 (cyclin dependent kinase inhibitor 1A [P21]) (Fig. S1C), reduced cell proliferation (Fig. S1D) and increased expression of the DNA damage marker phosphorylated γH2AFX/H2AX (H2A histone family, member X) in nuclear foci (Fig. S1E).
Figure 1.
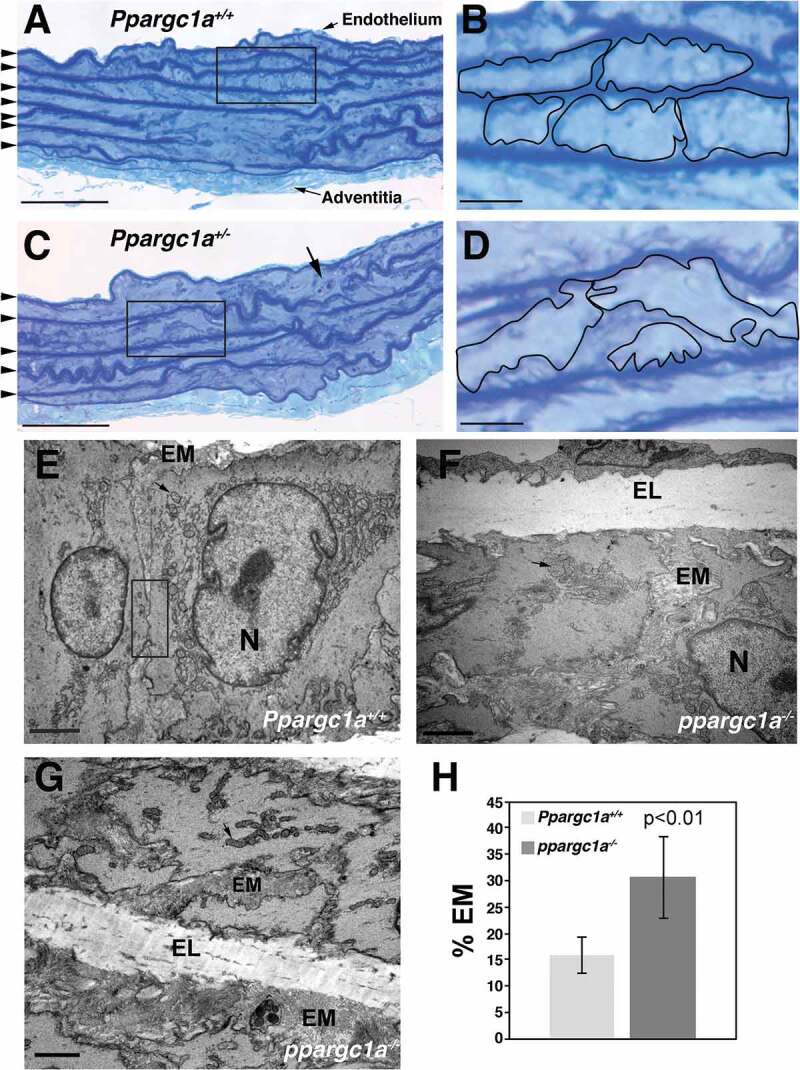
Structural differences in the media of ppargc1a−/- mouse aorta. Aortas of 6-month-old Ppargc1a+/+ (A and B) and Ppargc1a± (C and D) male mice were fixed, stained with 0.5% aqueous Toluidine Blue O and photographed at low (A and C) and high (B and D) magnification. Dark blue lines of elastic lamina are indicated with arrowheads in A and C. VSMCs in B and D were traced to indicate that more extracellular material is present in Ppargc1a± samples. TEM images of VSMCs in the media of Ppargc1a+/+ (E) and ppargc1a−/- (F and G) aortas were acquired using a Hitachi H-7500 electron microscope. Quantification of EM for both genotypes is shown in H. EM, extracellular matrix; N, nucleus; EL, elastic lamina. Bar: 50 μm in A and C, 10 μm in B and D, 2 μm in E-G.
Autophagy is downregulated by Ppargc1a deficiency
To test whether autophagy is regulated by PPARGC1A, we used MASMs isolated from aortas of WT and ppargc1a−/- mice and measured ROS levels, senescence, and autophagy markers in response to its overexpression using adenovirus containing Ppargc1a (Ad.PGC1A). Compared with WT, ROS levels were more than three-fold higher in ppargc1a−/- cells (n = 13, p < 0.01) and were significantly downregulated by overexpression of PPARGC1A (Figure 2A). Similarly, the increase in activity of the senescence associated GLB1/β-galactosidase (SA-GLB1) observed in ppargc1a−/- cells was significantly reduced (n = 13, p < 0.01) by PPARGC1A overexpression (Figure 2B), which was also associated with upregulation of cell proliferation (Figure 2C) and SOD2 expression (Figures 2D and E), compared with control ppargc1a−/- cells. Significant differences were also observed in SOD2 expression in WT cells in response to PPARGC1A overexpression, but not for ROS levels (Figure 2A), SA-GLB1 (Figure 2B) or cell proliferation (Figure 2C).
Figure 2.
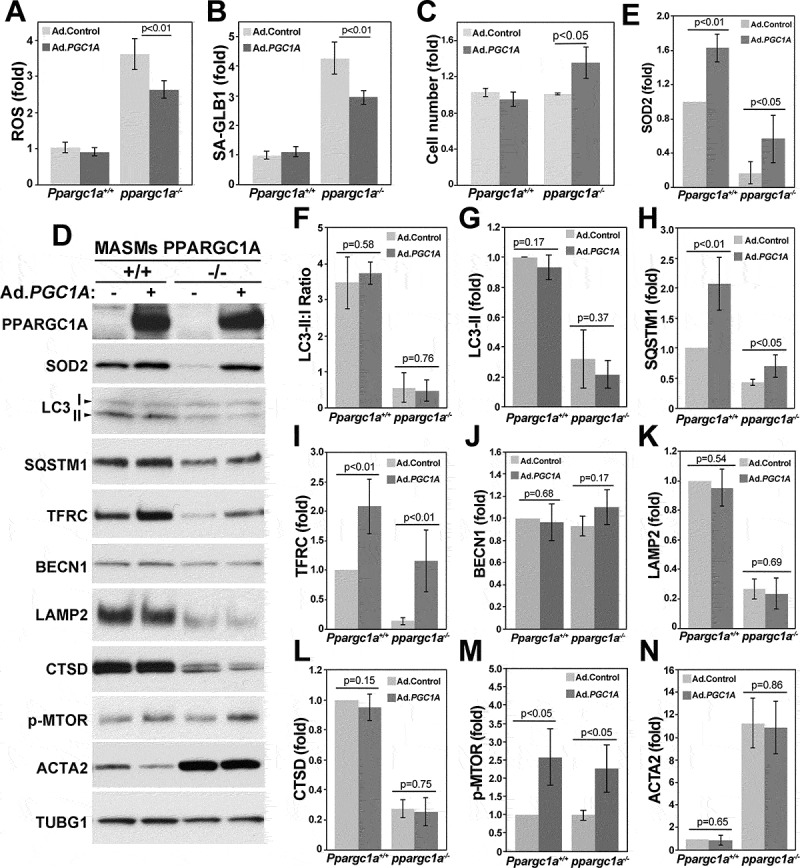
Ppargc1a deficiency increases senescence and reduces autophagy in VSMCs. Ppargc1a+/+ or ppargc1a−/- VSMCs were infected with 5 × 107 plaque-forming units (PFU)/ml of adenovirus control (Ad.Control) or adenovirus containing PPARGC1A (Ad.PGC1A). 3 d after infection, cells were processed for measurements of ROS levels using H2DCFDA (A), SA-GLB1 activity using FDG (B), cell proliferation (C) or protein expression by western blot (D). ROS levels, SA-GLB1 and protein expression shown in D, adjusted by TUBG1 (F-N), were calculated as fold change, compared with WT cells infected with Ad.Control in 3 to 4 independent experiments performed in cells between passage 4 and 7.
To determine whether autophagy is impaired in ppargc1a−/- VSMCs, we measured the conversion of LC3-I to II and expression of SQSTM1. Reduced LC3 conversion and accumulation of SQSTM1 are hallmarks of defective autophagy [20]. LC3-II:I ratio was strongly downregulated in ppargc1a−/- cells, compared with WT (n = 3, p < 0.01), and the ratio in both cell types was not altered by PPARGC1A overexpression (Figures 2D and F). Reduced LC3-II:I ratio was associated with reduced levels of LC3-II, which was not affected by PPARGC1A overexpression (Figure 2G). Surprisingly, reduced LC3-I to II conversion and LC3-II levels were not associated with SQSTM1 accumulation. Instead SQSTM1 behaved as a PPARGC1A-regulated gene (Figures 2D and H), similar to SOD2 (Figures 2D and E) and TFRC [21,22] (Figures 2D and I). Expression of both SQSTM1 and TFRC was reduced in ppargc1a−/- cells and was increased by PPARGC1A overexpression in WT and in Ppargc1a-deficient cells (n = 4, p < 0.01). Expression of the autophagy-related gene BECN1/Beclin1 was not affected by Ppargc1a deficiency or its overexpression (Figure 2D and J). Similar to LC3, expression of the lysosomal protein LAMP2 and the precursor forms (52 kDa and 48 kDa) of the lysosomal protease CTSD (Figure 2D, K and L) was strongly reduced in ppargc1a−/- cells (n = 4, p < 0.01, respectively). Expression of these markers was not upregulated by PPARGC1A overexpression, suggesting that lysosomal dysfunction could be a consequence of senescence and not directly regulated by PPARGC1A. It is also possible that expression of PPARGC1A above physiologic levels may activate a signaling pathway preventing the upregulation of lysosomal proteins and LC3 conversion. In fact, we observed a significant increase in phosphorylated/active MTOR in response to PPARGC1A overexpression in WT and Ppargc1a-deficient cells (n = 3, p < 0.05) (Figures 2D and M). Activation of MTOR, an autophagy inhibitor, may explain in part why LC3 conversion is not upregulated in PPARGC1A-overexpressing cells.
In addition to changes in the expression of autophagy and lysosomal genes, the level of ACTA2/SM-actin (actin, alpha 2, smooth muscle, aorta), mainly found at the cell periphery and in the cortical actin cytoskeleton, was strongly upregulated (n = 3, p < 0.01) by Ppargc1a deficiency, and was not significantly reduced by PPARGC1A overexpression (Figure 2D and N). Upregulation of ACTA2 is also observed during senescence in other cell types, including fibroblasts [23], suggesting that ACTA2 can be used as an additional marker of cellular senescence in VSMCs.
Altogether these data suggest that PPARGC1A-induced senescence is associated with impaired autophagy and lysosomal dysfunction. The fact that increased ROS levels and senescence induced by Ppargc1a deficiency were partially rescued by PPARGC1A overexpression suggests that upregulation of SQSTM1 and/or antioxidant genes like SOD2 may contribute to these effects.
To assess whether defective autophagy can be observed in vivo, we processed aortas of WT and ppargc1a−/- mice by TEM. This analysis revealed that autophagosome structures, characterized by a double membrane organelle containing cargo, were observed in WT VSMCs (Figure 3A, arrow shows an autophagosome containing a mitochondrion) although in low number (0.3 ± 0.1, n = 20 fields). ppargc1a−/- samples, on the other hand, showed abnormal autophagosome-like structures surrounded by a double membrane and containing electron-dense material and concentric membrane layers (Figure 3B). Interestingly, autophagosome-like structures were also observed in the EM (Figure 3C). On average, ppargc1a−/- aortas showed a tendency toward higher number of autophagosome-like structures/field (1.05 ± 0.44, n = 17 fields) with no significant differences, compared to WT (p = 0.085) (Figure 3D). These data suggest that the formation or maturation of autophagosomes and/or degradation of autophagic content could be altered by Ppargc1a deficiency. Overall, TEM analysis demonstrated the presence of defective autophagosome-like structures in vivo in aortas of ppargc1a−/- animals.
Figure 3.
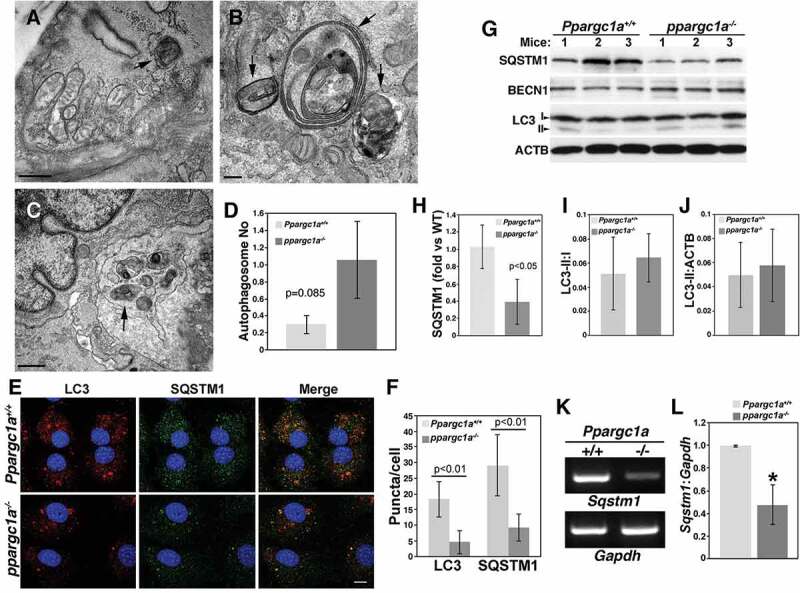
Abnormal autophagosomes and reduced SQSTM1 expression in the aorta of ppargc1a−/- mice. Aortas of Ppargc1a+/+ (A) and ppargc1a−/- mice (B and C) were analyzed by TEM. Arrows indicate autophagosomes. Bar: 0.5 μm in A and C, and 0.2 μm in B. Graph presenting the overall average of autophagosomes in both genotypes is shown in D. Ppargc1a+/+ and ppargc1a−/- VSMCs were fixed and incubated with mouse LC3 and rabbit SQSTM1 antibodies (E). Images in E were acquired using a confocal microscope for the quantification of LC3 and SQSTM1 puncta. Bar: 10 μm. (F). 34 cells and 30 cells were analyzed for LC3 in WT and knockout, respectively, while 23 and 24 cells were analyzed for SQSTM1 puncta in WT and knockout, respectively. Aortas of 6-month-old Ppargc1a+/+ and ppargc1a−/- male mice were lysed and processed for western blot analysis (G). SQSTM1, LC3-I and LC3-II levels were adjusted by ACTB and expressed as fold change, compared with WT (H-J). mRNA was isolated from Ppargc1a+/+ and ppargc1a−/- VSMCs and used to measure Sqstm1 and Gapdh mRNA levels (K and L). 3 independent mRNA preparations per genotype. * denotes p < 0.01 in L.
Next, we measured the number of autophagosomes in WT and ppargc1a−/- VSMCs in vitro by immunofluorescence. Consistent with the notion that PPARGC1A regulates autophagy, LC3- and SQSTM1-positive compartments (puncta) were significantly diminished in the absence of PPARGC1A (Figure 3E and F). The contradictory observation in the number of autophagosome structures in vivo and in vitro may be a consequence of differences in autophagic flux of cells in the aorta, which are in a quiescent state, compared with VSMCs in culture, which are in a proliferating state.
To test whether reduced SQSTM1 expression and SQSTM1-positive puncta in vitro correlated with reduced SQSTM1 expression in vivo, we measured its protein levels in aortas. Similar to VSMCs in culture, SQSTM1 level was significantly reduced in aortas of ppargc1a−/-, compared with WT mice (n = 3 per genotype, p < 0.05) (Figures 3G and H). In contrast, LC3-I to II conversion, as well as LC3-II levels were not affected (Figures 3G, I and J). To test whether SQSTM1 is regulated at the transcriptional level by PPARGC1A, we measured mRNA levels in VSMCs from both genotypes and found a significant reduction of approximately 50% in knockout cells (Figures 3K and L).
Altogether these data showed that Ppargc1a deficiency reduced SQSTM1 protein and mRNA levels and the number of autophagosomes and LC3-II levels in VSMCs in vitro and promoted the formation of dysfunctional autophagosome-like structures in VSMCs in aortas in vivo.
SQSTM1 is reduced by age and by Ppargc1a deficiency in mouse brain
To determine whether PPARGC1A regulates SQSTM1 expression and autophagy in other tissues, we isolated brain samples from young (6 months) and old (18 months) WT and ppargc1a−/- mice (Figure 4). The expression of SQSTM1 and LC3-I was significantly downregulated by age and by Ppargc1a depletion (n = 4, p < 0.01), which was not further reduced by age in knockout mice (Figure 4A-C). A longer exposure of the LC3 western blot revealed that LC3-II is expressed at lower levels in these samples. This observation is in line with data from other investigators showing that LC3-I expression is more prominent than LC3-II in brain homogenates [24–26]. Although TFRC expression was reduced by age, which did not reach significance (p = 0.13), it was strongly downregulated in young and old (n = 4, p < 0.01) ppargc1a−/- mice (Figures 4A and D). Similar effects, compared with SQSTM1 and LC3-I, were observed for SOD2 expression (Figure 4A and E).
Figure 4.
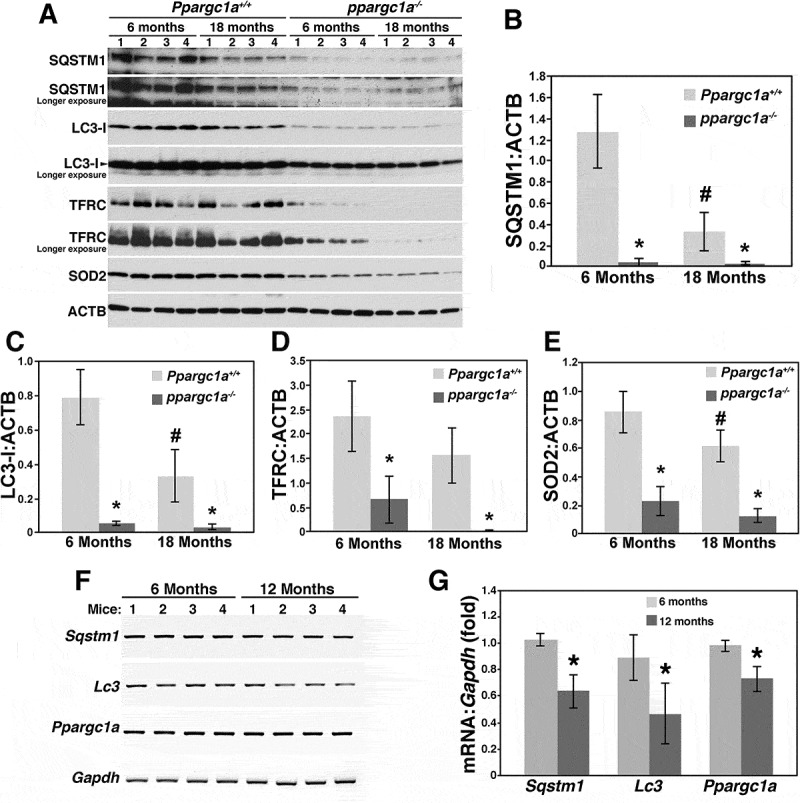
Reduced LC3 and SQSTM1 expression in the brain of ppargc1a−/- deficient mice. Brain samples from 6- and 18-month-old Ppargc1a+/+ and ppargc1a−/- male mice were homogenized and total extracts separated in 4–20% precast Criterion gels for the analysis of SQSTM1, LC3, TFRC and SOD2 expression (A). Protein expression of each gene was adjusted by ACTB (B-E). * and ‡ denote significant differences between WT and ppargc1a−/-, and between 6- and 18-month-old WT, respectively. mRNA from brain samples of 6- and 1-month-old male C57Bl/6 mice were tested for the expression of Sqstm1, Lc3 and Ppargc1a (F). mRNA levels were adjusted by Gpdh and expressed as fold change compared with animals of 6 months of age (G). * denote p < 0.05.
Expression of Sqstm1 mRNA is reduced during aging (17 months, compared with 3 months) in liver samples of C57Bl/6 mice [17]. Thus, we tested whether reduced protein levels of SQSTM1, LC3 and PPARGC1A correlated with reduced mRNA content. Consistent with the report by Kwon et al. [17], a reduction of about 40%, 50% and 30%, respectively, was seen for these three markers (Figure 4F and G).
Ppargc1a deficiency impairs lysosomal function and autophagic flux
As mentioned before, CTSD is a lysosomal protease that is synthetized as a 52-kDa precursor that is cleaved into a mature 48-kDa form in the lysosome. This form is further processed to produce the active enzyme formed by a heavy chain of 34 kDa and a light chain of 14 kDa. To further assess lysosomal function, we measured the expression of these two mature forms, as well as lysosomal morphology. Similar to the precursor forms, the 34-kDa form was also significantly reduced, while the 14-kDa form was undetected in ppargc1a−/- VSMCs (Figure 5A and B). As expected, reduced lysosomal function led to the accumulation of the autophagy receptor NBR1 (Figures 5A and C). Expression of these markers was not affected by PPARGC1A overexpression.
Figure 5.
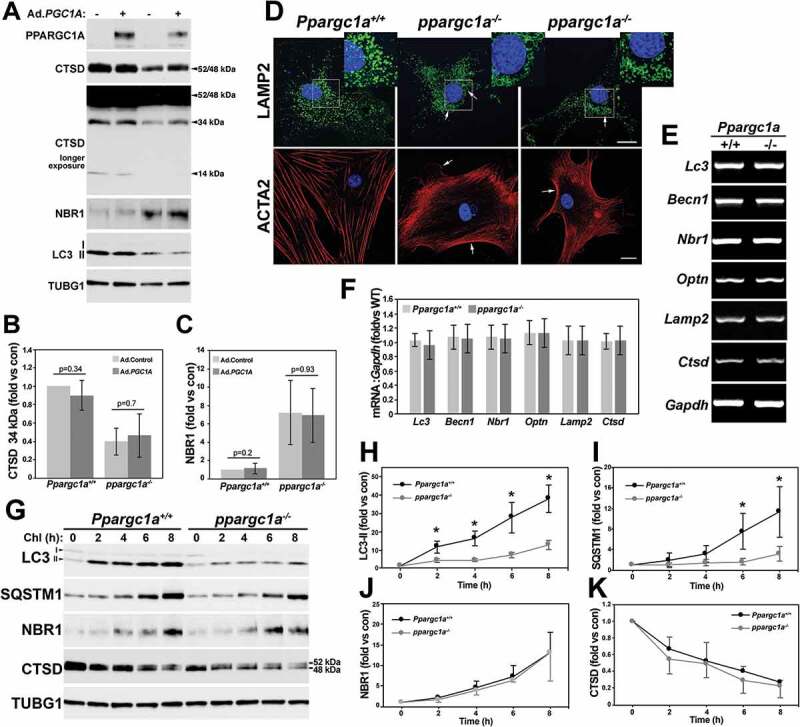
PPARGC1A regulates autophagic flux. Ppargc1a+/+ and ppargc1a−/- VSMCs were infected with 5 × 107 PFU of Ad.Control or Ad.PGC1A. 3 d after infection cells were lysed and samples tested for the indicated antibodies (A). The expression of the 34-kDa form of CTSD and NBR1 was adjusted by TUBG1 and expressed as fold change, compared with WT cells infected with Ad.Control (B and C). Ppargc1a+/+ and ppargc1a−/- VSMCs were fixed in 4% PFA and incubated with antibodies against LAMP2 or ACTA2, and images were acquired using a confocal microscope. Bar: 10 μm (D). mRNA from both genotypes was tested for the expression of the autophagy and lysosomal markers shown in E (n = 3 samples per genotype). mRNA levels were adjusted by Gapdh and expression expressed as fold change, compared with WT cells (F). Cells were treated with 50 μM Chl in DMEM containing 0.5% FBS for 2 to 8 h. Cells were then lysed and separated in 4–20% Precast Criterion gels (G). LC3-II, SQSTM1, NBR1, and CTSD protein levels were adjusted by TUBG1 (H-K). Expression of each marker in untreated cells was assigned as 1. Fold changes were then calculated for each genotype to assess the rate of protein accumulation or reduction. * denotes p < 0.05.
In terms of lysosomal function, reduced expression of CTSD and LAMP2 was associated with enlarged lysosomes (Figure 5D), a characteristic of lysosomal dysfunction [27]. Swollen lysosomes can be observed at the perinuclear area in ppargc1a−/- VSMCs (see Figure 5D inserts). Next, we determined whether changes in the expression of autophagy and lysosomal genes were also regulated at the mRNA level. In contrast to SQSTM1, mRNA expression of Lc3, Becn1, Nbr1, the autophagy receptor Optn (optineurin), Lamp2 and Ctsd was not affected by Ppargc1a deficiency (Figure 5E and F).
Next, we measured autophagic flux by treating cells with 50 μM chloroquine (Chl), an inhibitor of lysosomal degradation, for 2 to 8 h. As expected, the rate of accumulation of LC3-II and SQSTM1 over time was significantly higher in WT compared with ppargc1a−/- cells (Figure 5G-I), suggesting that decreased expression and not increased lysosomal degradation mediates SQSTM1 downregulation. Although NBR1 protein levels were elevated in knockout cells at baseline, the rate of NBR1 accumulation was similar between genotypes (Figure 5G and J). Interestingly, inhibition of lysosomal degradation reduced CTSD expression at a similar rate in both cell types, suggesting that reduced expression of the lysosomal proteins LAMP2 and CTSD could be a consequence of lysosomal dysfunction and not directly regulated by PPARGC1A.
Reduced autophagy induces senescence
To determine whether defective autophagy contributes to senescence development in VSMCs, we tested whether inhibition of autophagy can induce senescence and whether rapamycin can reduce the effects of AGT II, a well-known inducer of oxidative stress and senescence in VSMCs [28]. To inhibit autophagy, we used 3-MA and spautin-1 (specific and potent autophagy inhibitor-1), which inhibit autophagy through different molecular mechanisms. 3-MA inhibits class III Ptdln3K (phosphatidylinositol 3-kinase) [29] reducing the formation of Ptdln3P on the phagophore membrane, a process needed for the elongation of this membrane and the initiation of autophagy. Spautin-1 inhibits USP10 and USP13, two ubiquitin peptidases that deubiquitinate BECN1 in the class III Ptdln3K-PIK3C3/VPS34 complex leading to degradation of BECN1 and inhibition of the PIK3C3 complex in HeLa cells and mouse embryonic fibroblasts [30]. We used rapamycin, an MTOR inhibitor, to stimulate autophagy. MTOR inhibits autophagy by targeting the ULK1 complex [31], which is needed for the formation of the autophagosome.
We have used rat aortic smooth muscle cells (RASMs) to study the mechanism by which AGT II and zinc overload induce senescence [28,32]. We showed that AGT II induces the acetylation/inactivation of PPARGC1A leading to reduced CAT (catalase) [33] and SIRT1 (Sirtuin 1) expression and increased senescence. Thus, we first tested whether PPARGC1A also regulates SQSTM1 expression in RASM cells (Fig. S2). Similar to MASMs, PPARGC1A increased the expression of known targets including CAT, SOD2, TFRC and CYCS (cytochrome c, somatic), while downregulating CDKN1A expression, as we previously reported [6] (Fig. S2A). PPARGC1A also upregulated the expression of SQSTM1 and phosphorylated MTOR, but not LC3-II (Figs. S2A-D). It is likely that increased MTOR activity prevented the upregulation of LC3-II. Further, similar to MASMs Lc3 mRNA was unaffected by PPARGC1A (Figs. S2E and F). As we previously reported [28,34], AGT II increased SA-GLB1 activity compared with control cells, which was significantly downregulated by rapamycin in RASMs (Figures 6A and B). 3-MA increased SA-GLB1 activity (Figures 6A and B) and CDKN1A expression (Figures 6C and D) in basal conditions, effects that were not further increased by AGT II (Figures 6A and B). Both AGT II- and 3-MA-induced senescence was associated with elevated MTOR phosphorylation (Figures 6E and F). Similarly, spautin-1 upregulated SA-GLB1 activity basally and showed no additive effects in AGT II-induced senescence (Figures 6G and H). Next, we tested whether spautin-1 could also induce the degradation of BECN1 in RASMs. Surprisingly, a robust downregulation in the expression of SQSTM1, NBR1 and LC3-II, but not BECN1, was observed in spautin-1-treated cells (Figure 6I and J). AGT II showed a modest, but significant increase in SQSTM1 and NBR1 expression in control cells, but not in cells treated with spautin-1 (Figure 6I and J). Similar to SA-GLB1, CDKN1A expression was significantly upregulated by AGT II and spautin-1 and no additive effects were seen in AGT II- and spautin-1-treated cells (Figure 6I and K). Spautin-1 also upregulated SA-GLB1 activity in WT MASMs and showed a modest but significant effect in ppargc1a−/- cells (Figure 6L), suggesting that impaired autophagy is a major driver of senescence in the absence of Ppargc1a.
Figure 6.
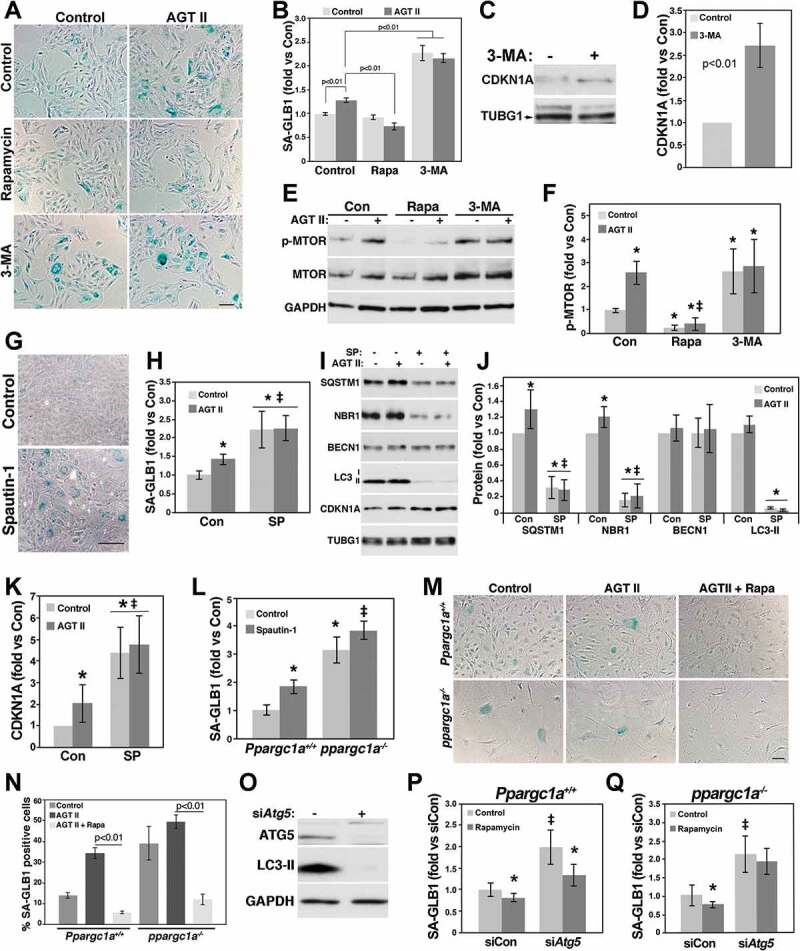
Impaired autophagy induces senescence of VSMCs. RASMs treated with and without 10 mM 3-MA or 100 nM rapamycin (Rapa) were treated with and without 100 nM AGT II for 3 d. Cells were then fixed and tested for SA-GLB1 activity measurements (A and B) or were lysed for the analysis of CDKN1A expression (C and D) and MTOR phosphorylation (E and F) by western blot. RASMs were incubated with 10 μM spautin-1 (SP) for 3 d for measurements of SA-GLB1 activity (G and H) or were treated with and without 100 nM AGT II and 10 μM spautin-1 for 3 d for protein expression analysis (I). Protein levels shown in I were adjusted by TUBG1 and expressed as fold change compared with control untreated cells (J and K). * and ‡ denote significant differences compared with control and AGT II, respectively. Ppargc1a+/+ and ppargc1a−/- VSMCs were treated with 10 μM spautin-1 (L) or with and without 100 nM AGT II and 100 nM rapamycin (M and N) for 3 d for SA-GLB1 activity measurements. WT MASMs were transfected with 200 nM siControl or siAtg5 and protein expression of ATG5 was determined by western blot (O). Ppargc1a+/+ and ppargc1a−/- VSMCs transfected with siControl or siAtg5 were treated with 100 nM rapamycin for 3 d (P and Q). SA-GLB1 was then expressed a fold change compared with untreated siControl cells. * and ‡ denote significant differences between cells treated with and without rapamycin and between siControl and siAtg5-transfected cells, respectively. Bar: 20 μm in A and 100 μm in G and M.
Next, we tested whether rapamycin could also reduce senescence induced by AGT II and Ppargc1a deficiency in MASMs (Figure 6M and N). Similar to RASMs, AGT II increased SA-GLB1 activity in WT MASMs, which was significantly downregulated by rapamycin. In contrast, SA-GLB1 activity was elevated basally in ppargc1a−/- cells but was not significantly upregulated by AGT II. Rapamycin reduced AGT II-induced senescence, as well as basal senescence (7.7 ± 3.4%, n = 3, p < 0.01). As expected, MTOR phosphorylation was reduced by rapamycin in both WT and ppargc1a−/- cells (Fig. S3A). Further, LC3-II and SQSTM1 levels were upregulated by rapamycin and reduced by 3-MA (Figs. S3B-D). The unexpected effects of these drugs in SQSTM1 protein levels suggest that SQSTM1 is not a good indicator of autophagic flux in VSMCs.
Since MTOR can regulate other processes, in addition to autophagy, that could also be involved in senescence, we tested the effect of rapamycin in cells depleted of ATG5, a core component of the autophagy machinery. Similar to spautin-1, Atg5 siRNA showed a robust downregulation in LC3-II levels (Figure 6O), and upregulation in SA-GLB1 activity (Figure 6P), an effect that was significantly reduced by rapamycin. Interestingly, rapamycin was unable to reduce senescence in ppargc1a−/- cells depleted of ATG5 (Figure 6Q).
Altogether these data showed that autophagy impairment promotes senescence of VSMCs and that upregulation of autophagy by rapamycin reduces senescence induced by AGT II and by Ppargc1a deficiency. Further, these data also suggest that MTOR regulates senescence by autophagy-dependent and independent mechanisms.
Sqstm1 deficiency induces senescence
Ppargc1a deficiency is associated with dysfunctional autophagy and increased senescence that can be rescued by rapamycin, as well as with reduced SQSTM1 expression. To determine the contribution of SQSTM1 to Ppargc1a deficiency effects, we downregulated Sqstm1 by siRNA in RASM cells and determined senescence in response to AGT II and zinc overload, another inducer of senescence in VSMCs [28,32]. SiSqstm1 reduced SQSTM1 protein expression by 80% compared with siControl-treated cells (n = 4, p < 0.01) (Figure 7A and B), while increasing CDKN1A levels more than 2-fold (Figures 7A and C). Basal SA-GLB1 activity was upregulated by Sqstm1 siRNA compared with siControl-treated cells (n = 12, p < 0.01) (Figure 7D). Senescence induced by AGT II and zinc was also elevated in siSqstm1-treated cells (Figure 7D). These additive effects suggest that SQSTM1 acts through a different mechanism compared with AGT II and zinc to regulate senescence of VSMCs. Next, we determined whether senescence induced by Ppargc1a deficiency could be rescued by SQSTM1. Overexpression of SQSTM1 using adenovirus (Ad.SQSTM1) promoted a small but significant reduction in ROS levels (Figure 7E) and SA-GLB1 activity (Figure 7F). Ad.SQSTM1 increased SQSTM1 expression more than 8-fold and LC3-II levels more than 2-fold, compared with cells treated with Ad.Control (Figure 7G-I). In contrast, ppargc1a−/- cells infected with Ad.SQSTM1 in the same conditions as WT cells showed a less robust, but significant upregulation of SQSTM1 expression (Figure 7G and H) and LC3-II levels (Figures 7G and I), compared with control cells. These data suggest that overexpressed SQSTM1 is degraded in ppargc1a−/- cells by an unknown mechanism. This effect can explain the small reduction in ROS levels and senescence observed in ppargc1a−/- cells overexpressing SQSTM1 (Figure 7E and F).
Figure 7.
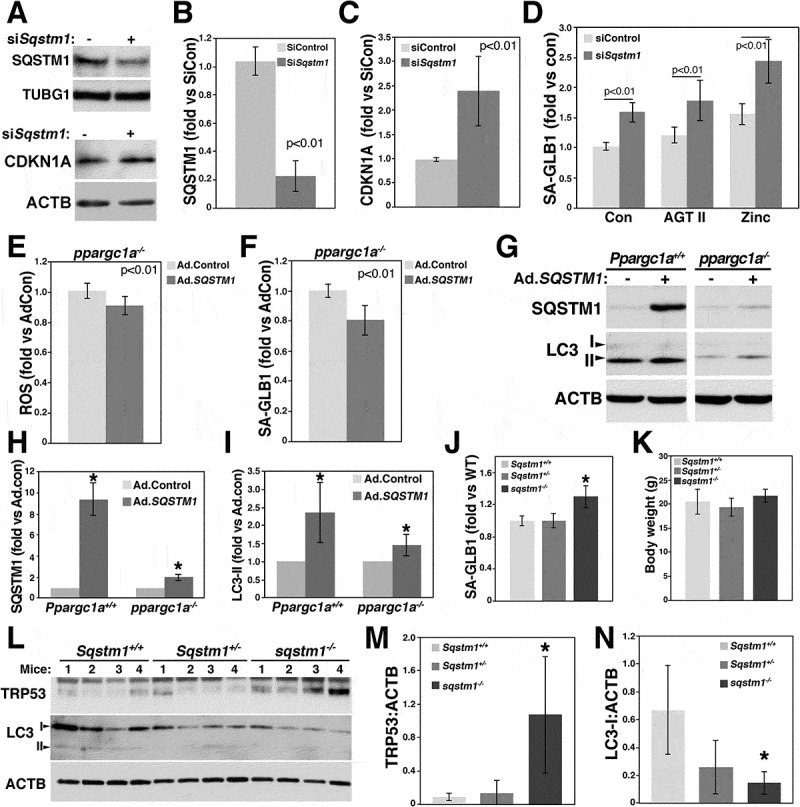
Reduced SQSTM1 expression increases ROS levels and senescence of VSMCs. RASMs were transfected with siRNA control or siSqstm1 for measurements of SQSTM1 and CDKN1A expression (A-C), and SA-GLB1 activity, using FDG, in response to 100 nM AGT II or 50 μM zinc for 3 d (D). ppargc1a−/- VSMCs were infected with 1 × 107 PFU of adenovirus control (Ad.Control) or adenovirus containing human SQSTM1 gene (Ad.SQSTM1) for ROS measurements (E) and SA-GLB1 activity using FDG (F). Ppargc1a+/+ and ppargc1a−/- VSMCs infected with Ad.Control or Ad.SQSTM1 were lysed for analysis of SQSTM1 and LC3-II expression. (G-I). Expression was expressed as fold change compared with siControl-transfected cells. Aortas of 2-month-old mice Sqstm1+/+, Sqstm1± and sqstm1−/- in the C57Bl/6 background (8 males each) were cleaned, fixed and tested for SA-GLB1 activity using FDG (J). Fluorescence was adjusted by the weight of aortas. * denotes p < 0.01. Body weight for each genotype is expressed in grams (g) (K). Aortas of each genotype (2-month-old, males) were cleaned of periadventitial fat, lysed and 80 μg of total extracts were separated in a 4–20% precast Criterion gels (L). Quantification of TRP53 and LC3-II adjusted by ACTB is shown in M and N, respectively. * denotes p < 0.05.
To determine the physiological significance of SQSTM1 in vivo, we measured senescence in aortas of 2-month-old sqstm1−/- male mice. SA-GLB1 activity was upregulated (n = 8, p < 0.01) in sqstm1−/-, compared with WT C57Bl/6 and Sqstm1 heterozygote mice (Figure 7J). The senescence phenotype of sqstm1−/- mice was not associated with the obesity phenotype of this animal model (Figure 7K) since body weight of WT (20.5 ± 2.6 g, n = 8), heterozygote (19.32 ± 1.9 g, n = 8) and sqstm1−/- (21.7 ± 1.4 g, n = 8) mice was not significantly different at this age. sqstm1−/- mice display mature-onset obesity with increased body weight at about 5 months of age, compared with WT C57Bl/6 mice [18]. Consistent with elevated SA-GLB1 activity, TRP53/p53 (transformation related protein 53) protein expression was also upregulated in aortas of sqstm1−/- (n = 4, p < 0.05), but not heterozygote (n = 4, p = 0.56), compared with WT mice (Figures 7L and M). These changes were associated with a significant reduction in LC3-I expression in sqstm1−/-, but not Sqstm1±, compared with WT mice (Figures 7L and N). Similar to brain homogenates, LC3-II was barely detected in aorta samples. Quantification of LC3-II was not possible since no bands were detected in some of the samples.
To determine whether Sqstm1- and Ppargc1a-deficient VSMCs share the same or similar phenotypes, we isolated VSMCs from aortas of WT and sqstm1−/- mice. Similar to Ppargc1a deficiency, Sqstm1 depletion resulted in reduced cell proliferation (Figure 8A), increased SA-GLB1 activity (Figures 8B and C) and ROS levels (Figures 8D and E), and defective autophagy (Figure 8F-J). sqstm1−/- cells showed NBR1 accumulation, and reduced LC3-II levels and no changes in BECN1 expression (Figures 8F and G). Different from Ppargc1a deficiency, cells lacking Sqstm1 expressed more MTOR and its phosphorylated form (Figures 8F and G). Autophagic flux was also impaired in these cells, as measured by the increased and reduced accumulation of NBR1 and LC3-II, respectively, in response to Chl treatment (Figure 8H-J).
Figure 8.
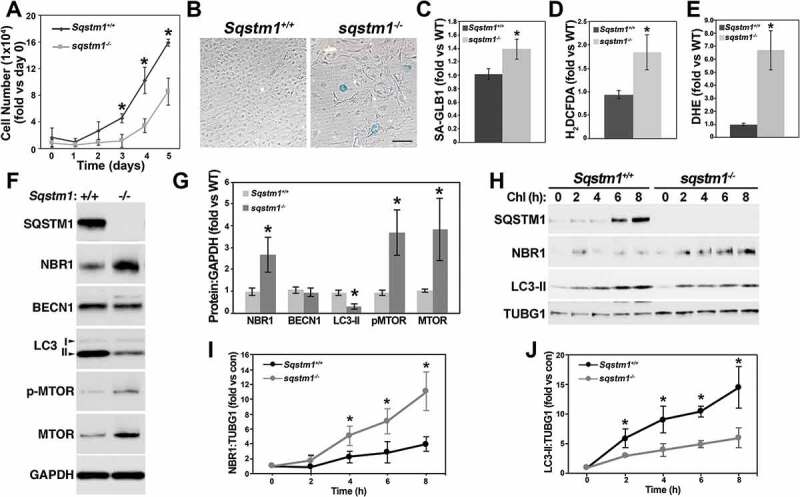
Sqstm1 deficiency increases ROS levels and senescence and reduces autophagic flux. WT and sqstm1−/- VSMCs were cultured in DMEM with 10% FBS for cell proliferation experiments (A) (triplicates in 3 independent experiments), analysis of SA-GLB1 activity (B and C), and hydrogen peroxide (D) and superoxide (E) levels, or for analysis of protein expression (F). Expression of markers in F was adjusted by GAPDH and expressed as fold change, compared with WT cells (G). WT and sqstm1−/- VSMCs were treated with 50 μM Chl for 2 to 8 h for the analysis of autophagic flux (H). The expression of NBR1 (I) and LC3-II (J) levels was adjusted by TUBG1. The value of untreated cells was calculated as 1. Fold change compared with untreated cells was then calculated for each genotype. * denoted p < 0.05. Bar: 100 μm in B.
Altogether these data demonstrate that downregulation of SQSTM1 mediates Ppargc1a deficiency-induced senescence and that PPARGC1A and SQSTM1 have opposite effects in MTOR function.
Discussion
PPARGC1A is a well-known regulator of mitochondrial biogenesis and antioxidant capacity among other functions. However, little is known about the role of PPARGC1A in autophagy in the cardiovascular system. Here, we report that Ppargc1a deficiency causes defective autophagy, which was associated with reduced LC3-I to II conversion, LC3-II levels, SQSTM1 expression (protein and mRNA levels), autophagosome number, autophagic flux, and with accumulation of NBR1 in VSMCs in culture. Reduced SQSTM1 expression and the presence of abnormal autophagosome-like structures were also observed in VSMCs in aortas of ppargc1a−/- mice in vivo. As we previously reported [6], Ppargc1a deficiency reduced cell proliferation and increased ROS levels and senescence. Our data suggest that impaired autophagy promoted the senescence phenotype of Ppargc1a depleted cells since inhibition of autophagy by 3-MA, spautin-1 or Atg5 siRNA caused senescence. The upregulation in SA-GLB1 seen in ppargc1a−/- cells was further increased by spautin-1 and Atg5 depletion. This effect is consistent with the observation that autophagic flux is reduced, but not completely inhibited, in ppargc1a−/- cells. Thus, blockade of that residual autophagy by spautin-1 and Atg5 siRNA upregulated senescence even further.
Interestingly, senescence induced by Atg5 depletion was partially rescued by rapamycin in WT, but not in ppargc1a−/- VSMCs cells. These data suggest that MTOR also regulates senescence by a mechanism different from its effect on autophagy. For instance, MTOR activates NFKB/NF-κB (nuclear factor of kappa light polypeptide gene enhancer in B cells) [35], a major driver of the SASP [36]. We have demonstrated that activation of NFKB mediates the upregulation of NOX1 (NADPH oxidase 1) leading to increased ROS levels that promoted senescence of VSMCs [32]. Thus, inhibition of MTOR by rapamycin could also inhibit senescence by reducing oxidative stress. It is unknown, however, whether NADPH oxidases are upregulated by Ppargc1a deficiency. The fact that rapamycin was unable to rescue senescence induced by Atg5 siRNA in ppargc1a−/- cells could be related with the higher levels of ROS in the knockout cells. Multiple sources of ROS can contribute to this elevated oxidative stress since PPARGC1A regulates the expression of several antioxidant enzymes, including SOD1, SOD2 and CAT, while regulating mitochondrial function. We demonstrated that downregulation of CAT increased ROS levels and senescence of VSMCs [28].
We hypothesized that SQSTM1 downregulation mediated the effect of Ppargc1a deficiency. In support of this hypothesis, we found that sqstm1−/- cells mimicked the phenotype of Ppargc1a deficiency by presenting reduced proliferation, increased ROS levels, reduced LC3-II expression and autophagic flux and increased levels of NBR1 and senescence. In contrast to ppargc1a−/-, sqstm1−/- cells expressed higher levels of MTOR and its phosphorylated form, compared with WT cells. Further, in vivo, aortas of sqstm1−/- mice showed more senescence and reduced levels of LC3-I.
Thus, altogether our data summarized in Figure 9 suggest that PPARGC1A upregulates autophagy by controlling the expression of SQSTM1 and that upregulation of mitochondrial biogenesis and autophagy reduces senescence of VSMCs. These findings are in part in line with previous observations in skeletal muscle. Brandt et al. recently showed that low and moderate intensity exercise training increased LC3-II:I ratio in WT, but not in ppargc1a−/- mice [37]. However, only low-intensity exercise training showed reduced SQSTM1 levels in ppargc1a−/- muscle. These observations suggest that PPARGC1A is required for the upregulation of autophagy in response to exercise in skeletal muscle; however, no differences in LC3-II:I ratio or in SQSTM1 levels were observed between untrained control WT and ppargc1a−/- mice. In a skeletal muscle-specific Ppargc1a transgenic mouse, Halling et al. showed upregulated LC3-II:I ratio at rest and after exercise, compared with WT mice [38]. Interestingly, SQSTM1 level was strongly downregulated in the transgenic animal, suggesting that the upregulation of autophagy caused SQSTM1 degradation. Further, Vainshtein et al. showed that exercise training upregulated Sqstm1 mRNA levels in skeletal muscle of WT, but not in ppargc1a−/- mice [39]. No difference at the mRNA level was observed in skeletal muscle of WT and ppargc1a−/- mice in untrained control conditions [39]. In contrast to these studies, SQSTM1 protein and mRNA levels, as well as LC3 conversion, were significantly downregulated by Ppargc1a deficiency in VSMCs in our study. Although no difference in LC3-II:I ratio was observed in aortas of both genotypes in vivo, cargo containing autophagosome-like structures of various sizes were identified in VSMCs, as well as in the extracellular space of the media of the aorta. Accumulation of these structures suggests that the fusion with lysosomes and/or degradation in autolysosomes, but not the formation of autophagosomes, is inhibited in the absence of Ppargc1a in vivo. With respect to extracellular autophagosomes, it is possible that vesicle compartments containing autophagosome-like structures can fuse with the plasma membrane releasing autophagosomes into the extracellular media. In fact, autophagosomes can fuse with the plasma membrane under starvation conditions, a process that is regulated by the SNARE protein VAMP7 (vesicle-associated membrane protein 7) and is associated with the release of ATP [40]. Autophagosomes can also fuse with multivesicular bodies, organelles containing internal vesicles and exosomes, that fuse with the plasma membrane, releasing these vesicles into the extracellular media [41]. Furthermore, extracellular vesicles, including exosomes, can be secreted by senescent cells as part of the SASP [42]. Thus, increased accumulation of EM and the presence of extracellular vesicles in aortas of ppargc1a−/- mice suggest that the SASP may contribute to tissue dysfunction in vivo. Whether PPARGC1A regulates the secretion of extracellular vesicles remains to be elucidated.
Figure 9.
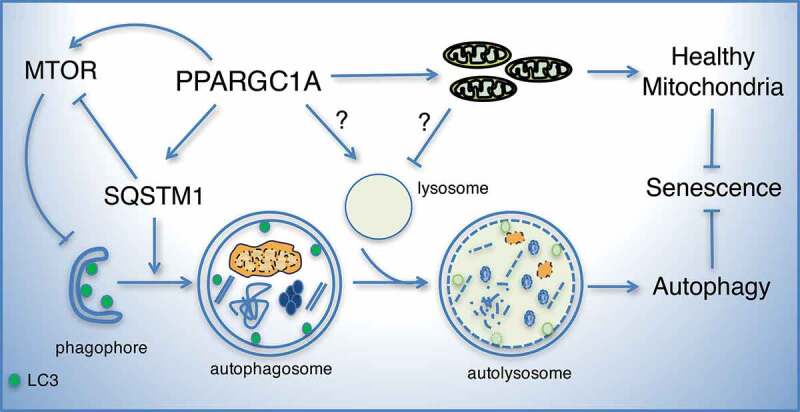
Model of the role of PPARGC1A and SQSTM1 in autophagy and senescence. Our data suggest that PPARGC1A regulates SQSTM1 expression leading to the upregulation of LC3-II and the stimulation of autophagy. Downregulation of PPARGC1A or SQSTM1 reduces LC3-II levels, autophagic flux, and cell proliferation and increases ROS levels and senescence. Elevated PPARGC1A expression or SQSTM1 deficiency increases MTOR phosphorylation creating a regulatory loop that may restrain excessive autophagy. It is unknown whether lysosomal dysfunction seen in PPARGC1A deficient cells also contributes to impaired autophagy. Altogether, the known functions of PPARGC1A, as a regulator of mitochondrial function and antioxidant capacity, together with its novel role, as a regulator of autophagy, contribute to reduced oxidative stress and senescence of VSMCs.
In addition to the aorta tissue, SQSTM1 and LC3 expression was also downregulated in brain samples of Ppargc1a-deficient mice and in old compared with young C57BL/6 WT mice. TFRC and SOD2, two genes regulated by PPARGC1A in VSMCs, were also significantly reduced by the lack of Ppargc1a in the brain. Reduced protein expression of these genes during aging correlated with reduced mRNA levels. Brain samples from 1-year-old mice showed lower levels of Sqstm1, Lc3 and Ppargc1a mRNA, compared with brains of 6-month-old animals. Interestingly, PPARGC1A is downregulated in postmortem brain samples of patients with Alzheimer disease and in neurons of the Tg2576 Alzheimer disease animal model [43]. Overexpression of PPARGC1A in these neurons reduced the formation of β-amyloid [43]. Our data suggest that secretion of autophagosomes and/or exosomes containing pathological aggregates, by reduced PPARGC1A expression, may contribute to the progression of Alzheimer disease. Thus, it is tempting to speculate that PPARGC1A could be protective to the brain, not only by maintaining mitochondrial function and the antioxidant capacity, but also by upregulating autophagy.
SQSTM1 protein expression was downregulated by Ppargc1a deficiency and upregulated by PPARGC1A overexpression, suggesting that SQSTM1 is a direct target of PPARGC1A; however, the molecular mechanism of this effect is unknown. Sqstm1 transcription is regulated by NFE2L2 [44], a transcription factor co-activated by PPARGC1A [45], and by TFEB [14], a central regulator of lysosomal biogenesis [15]. The fact that ppargc1a−/- VSMCs show reduced expression of the lysosomal proteins LAMP2 and CTSD and enlarged lysosomes suggests that TFEB may regulate both autophagy and lysosome biogenesis in VSMCs and that the function of TFEB may be altered by the absence of Ppargc1a. Interestingly, in SH-SY5Y neuroblastoma cells, overexpression of TFEB increased PPARGC1A expression leading to upregulation of mitochondrial biogenesis [46]. Similarly in skeletal muscle, exercise upregulated TFEB in a PPARGC1A-dependent manner [47], suggesting that TFEB and PPARGC1A can regulate each other. In this scenario, it is possible that upregulation of TFEB by PPARGC1A may upregulate autophagy by increasing SQSTM1 expression leading to LC3 conversion and may increase autophagic flux by increasing lysosome biogenesis, as suggested in our model (Figure 9). However, no changes in mRNA levels for lysosomal genes (Lamp2 and Ctsd), or autophagy genes (Lc3, Becn1 and Nbr1) were observed in ppargc1a−/- cells. Only Sqstm1 mRNA was significantly downregulated in these cells, suggesting that TFEB is likely not involved in this pathway.
ppargc1a−/- cells also showed reduce expression of CTSD and LAMP2 and enlarged lysosomes, suggesting that lysosomal function may be impaired. Interestingly, treatment of both WT and ppargc1a−/- cells with Chl, a drug that raises the pH of lysosomes [48], reduced CTSD protein levels. It is unknown whether changes in lysosome acidification may cause lysosomal dysfunction and/or reduced expression of CTSD and LAMP2 in cells lacking Ppargc1a. In fact, activation of the DNA damage response stimulates senescence [49] and mediates the phosphorylation and inactivation of the vacuolar ATPase [50] leading to reduced lysosomal acidification. Increased DNA damage and telomere shortening were observed in ppargc1a−/- VSMCs [7], suggesting that impaired function of the vacuolar ATPase could cause defective lysosomal function in these cells.
Lysosome dysfunction could be also caused by defective mitochondrial function, as suggested by Demers-Lamarche et al. Inhibition of mitochondrial function in this study resulted in impaired lysosome function and enlarged lysosomes [27]. It is also possible that reduced expression of lysosomal enzymes, like CTSD, can be caused by secretion of the enzyme. In fact, CTSD is secreted at low levels in physiologic conditions [51] and at high levels in cancer cells [52].
Autophagy is a protective process critically involved in VSMC function [53] that has been shown to reduce senescence of VSMCs [54,55] and to limit atherosclerosis [56]. We showed that inhibition of autophagy by 3-MA or spautin-1 induces senescence, while upregulation of autophagy by rapamycin reduces senescence induced by AGT II, a known inducer of cellular senescence in VSMCs [28,57]. However, overexpression of PPARGC1A significantly upregulated MTOR phosphorylation preventing LC3 conversion. In contrast, Sqstm1 deficiency promoted a robust upregulation of MTOR. Thus, PPARGC1A and SQSTM1 seem to regulate a feedback loop mechanism, as shown in Figure 9, to maintain MTOR function and autophagy at physiological levels. This mechanism could be of importance to reduce autophagy and prevent autophagic cell death.
SQSTM1 is an autophagy receptor involved in the targeting of ubiquitinated proteins and organelles, like mitochondria, to autophagosomes for degradation. sqstm1−/- mice have increased mitochondrial oxidative stress, decreased lifespan [17], mature-onset obesity and insulin resistance [18] and features of neurodegeneration [58]. Also, Sqstm1 mutations are associated with Paget bone disease [59] and were also found in frontotemporal lobar degeneration and amyotrophic lateral sclerosis [60]. Here, we demonstrated that lack of Sqstm1 is also associated with vascular senescence and that SQSTM1 downregulation mediates senescence caused by Ppargc1a deficiency. SQSTM1 downregulation by siRNA increased ROS levels, CDKN1A expression and SA-GLB1 activity basally and in response to AGT II and zinc overload, while SQSTM1 overexpression reduced ROS levels and SA-GLB1 activity in ppargc1a−/- VSMCs. Also, overexpression of SQSTM1 upregulated LC3-II levels in WT and ppargc1a−/- cells, suggesting that LC3 conversion is downstream of SQSTM1. Interestingly, lower levels of overexpressed SQSTM1 protein were observed in ppargc1a−/- cells. Endogenous and overexpressed SQSTM1 accumulated at a similar level in ppargc1a−/- cells treated with Chl (data not shown), suggesting that SQSTM1 is degraded by a lysosome-independent pathway.
Sqstm1 deficiency induces mature-onset obesity and insulin resistance at about 5 months of age [18]. Our observation that 2-month-old sqstm1−/- male mice showed increased senescence in the aorta tissue suggests that senescence is not a consequence of the obesity phenotype of this animal model. Recently, increased atherosclerosis was also observed in 2-month-old apoe−/-sqstm1−/-, compared with apoe−/- mice fed high fat diet [19]. It is unknown, however, whether accelerated aging by the lack of Sqstm1 promotes atherosclerosis. It is also unknown whether the molecular mechanism involved in Sqstm1 deficiency-induced senescence depends mainly on the inhibition of autophagy. Since SQSTM1 overexpression reduces ROS levels in ppargc1a−/- VSMCs and sqstm1−/- cells showed increased ROS levels, it is likely that other mechanisms are also involved.
Telomere dysfunction causes mitochondrial dysfunction by a TRP53-dependent downregulation of PPARGC1A and PPARGC1B leading to mitochondrial dysfunction and increased ROS production [61]. This evidence supports a unified view of aging in which DNA damage to telomeres induces metabolic and mitochondrial compromise leading to accelerated aging [62,63]. PPARGC1A is central to this regulatory network [64] by regulating mitochondrial function, the expression of antioxidant enzymes, and as shown here, autophagy.
Altogether, our data demonstrate that senescence induced by Ppargc1a deficiency is mediated by impaired autophagy caused by SQSTM1 downregulation. The phenotype of sqstm1−/- and ppargc1a−/- mice is similar in terms of accelerated vascular aging and atherosclerosis. Our data also suggest that measurements of SQSTM1 protein expression, as an indicator of successful autophagy, should be evaluated carefully since in our study reduced SQSTM1 expression was associated with defective autophagy, but not with upregulation of autophagic flux.
Materials and methods
Antibodies and reagents
Rabbit antibodies against LC3 (2765), BECN1 (3738), phosphorylated MTOR (Ser2448, 2971) and MTOR (2983) were from Cell Signaling Technology. Goat anti-CTSD (C-20, sc-6486), and rabbit anti-SQSTM1 (H290, sc-25575) and anti-CDKN1A (sc397) were from Santa Cruz Biotechnology. Rabbit antibodies against LAMP2 (L0668), CAT (219010) and ATG5 (A0856), and mouse antibodies against TUBG1 (T5326), ACTB (A5441), and ACTA2 (A5228) were from Sigma-Aldrich. Mouse TRP53 (BML-SA293-0050) and rabbit SOD2 (ADI-SOD-110) antibodies were from Enzo Life Science. Mouse TFRC antibody was from Invitrogen (136800). Rabbit CTSD (ab75852) and mouse SQSTM1 (ab56416) and NBR1 (ab55474) antibodies were from Abcam. Mouse (MA5-15738) and rabbit (PA1-988) GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibodies were from Thermo Fisher Scientific. PPARGC1A antibody was from Dr. D. Kelly (Perelman School of Medicine, University of Pennsylvania), as described previously [33].
AGT II (A6402), 3-MA (M9281), Chl (C6628), rapamycin (553210), spautin-1 (SML0440-5MG) and protease inhibitor cocktail (P340-5ML) were from Sigma-Aldrich. Fetal bovine serum (FBS, 97,068–075) was from Seradigm. 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, C6827) and dihydroethidium (DHE, D11347) were from Invitrogen. Fluorescein-di-β-D-galactopyranoside (FDG, MO250) was from Marker Gene Technologies. Collagenase type 2 (LS004204) and elastase (LS006363) were from Worthington Biochemical Corporation. Adenovirus containing PPARGC1A was obtained from Dr. Wayne Alexander at Emory University [33]. Adenovirus containing HA-tagged human SQSTM1 (4 x 1010 plaque-forming units [PFU]/ml) was from Vector Biolabs.
Mice
Animal experiments using Ppargc1a+/+ and ppargc1a−/- (obtained from Dr. Wayne Alexander at Emory University [6]) and sqstm1−/- mice in the C57BL/6 background (obtained from Dr. Toru Yanagawa at the University of Tsukuba, Japan [65]) were performed in compliance with the Institutional Animal Care and Use Committee of Emory University and Florida State University, respectively. Animals were euthanized using CO2 and aortas were isolated for measurements of protein expression or for isolation of VSMCs.
Isolation of vsmcs, cell culture and adenovirus transduction
RASMs were isolated by enzymatic digestion as previously described [66]. Briefly, aortas from 5 male Sprague-Dawley rats (2-month-old) were cleaned from periadventitial fat, washed in Hanks balanced salt buffer (Corning, 21–021-CV) and digested with 175 units/ml collagenase for 30 min at 37°C to remove the adventitial layer. The endothelium was removed using a sterile cotton swab and aortas were incubated in DMEM (Corning, 10–014-CV) overnight, and then digested with 175 units/ml collagenase and 0.5 mg/ml elastase for 1 h at 37°C. VSMCs released after digestion were centrifuged and cell pellets resuspended in DMEM media. Isolation of MASMs was performed using a similar protocol, with the exception that after collagenase digestion and overnight incubation, aortas were cut into small pieces, which were allowed to attach to culture plates for at least 3 d. Both RASMs and MASMs were cultured in DMEM containing 1 g/L glucose, 100 U/ml penicillin, 100 mg/ml streptomycin, 2 mM glutamine (Corning, 30–009-CI) and 10% FBS at 37ºC in a 5% CO2 incubator. Contamination of cell cultures with fibroblasts was assessed by immunofluorescence using fibroblast antigen antibodies, as shown in Fig. S4. Cells were used for experiments between passages 4 and 12, as we previously reported [6,28,34]. For AGT II and zinc treatment, cells were starved in DMEM containing 0.5% FBS for 24 h and then incubated in the same media with 100 nM AGT II or 50 μM zinc (Sigma-Aldrich, Z0251) for 3 d, as previously described [67]. Cells were treated with 50 μM Chl, 10 mM 3-MA, 100 nM rapamycin or 10 μM spautin-1 in DMEM 0.5% FBS.
For adenovirus infection, cells were incubated with adenovirus containing PPARGC1A (Ad.PGC1A), SQSTM1 (Ad.SQSTM1) or empty adenovirus for 1 h in plain DMEM medium. Cells were allowed to recover in complete medium for 48 h prior to experimental treatment.
Transmission electron microscopy (TEM)
Immediately after CO2 euthanasia, mice were perfused from the left ventricle and aortas prepared for TEM analysis, as previously reported [68]. Aorta samples were fixed overnight in 2.5% glutaraldehyde (Electron Microscopy Sciences, 111–30-8) in 0.1 M sodium cacodylate (Electron Microscopy Sciences, 12300) buffer, followed by postfixation in 1% osmium tetroxide (Electron Microscopy Sciences, 19112) in 0.1 M sodium cacodylate buffer, dehydration in a graded ethanol series followed by propylene oxide (Electron Microscopy Sciences, 20410), and embedding in Epon-812 resin (Sigma-Aldrich, 45345). Semi-thin transverse sections were stained with 0.5% aqueous Toluidine Blue O (Sigma-Aldrich, 89640) and brightfield images were taken with a Zeiss Axioskop equipped with an RT Slider Spot Camera. Ultrathin, transverse sections were poststained with uranyl acetate and lead citrate. Images were taken on a Hitachi H-7500 electron microscope operated at 75 kV in the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University. The images were imported into Image-Pro Plus v. 6.2 software (MediaCybernetics, 41N51000-LUSB) for morphometric area measurements of EM.
Morphometric measurements
Area measurements of aortic images were performed using Image-Pro Plus 6.2 software. First, using the Measurements/Create Polygon feature of the software, total area between two elastin lamellae (total interlamellar area) was traced and was measured. Within these interlamellar zones, fibrillar/amorphous areas of EM were also measured. These area measurements were expressed in μm2. The EM areas were divided by the total medial layer area and expressed as % EM in the medial layers.
Senescence associated GLB1/β-galactosidase (SA-GLB1) staining
SA-GLB1 activity was determined as previously described [34]. Cells were washed with PBS (Corning, 21–40-CV) and then fixed with 0.2% glutaraldehyde in PBS for 10 min at room temperature. After fixation, cells were washed with PBS and were incubated overnight at 37°C in 40 mM phosphate buffer (4.8 mM Na2HPO4, 35.2 NaH2PO4, pH 6.0) supplemented with 1 mg/ml X-Gal (Amresco, X1205), 2 mM MgCl2, 150 mM NaCl, 5 mM K4Fe(CN)6 (Sigma-Aldrich, P3289) and 5 mM K3Fe(CN)6 (Sigma-Aldrich, P8131). Images were taken using an Axio Observer.A1 microscope (Carl Zeiss) using a 10× objective. SA-GLB1-positive cells were expressed as a percentage of total cell number. In addition, FDG (Ex/Em: 485/515 nm), a fluorescent analog of X-gal, was used to measure SA-GLB1 using a Synergy H1 multi-mode plate reader (BioTek Instruments).
Immunofluorescence
Immunofluorescence was performed in cells fixed with 4% paraformaldehyde (PFA) (Sigma-Aldrich, P6148), as previously described [69]. Coverslips were washed with PBS and permeabilized in buffer containing 2% BSA (Sigma-Aldrich, A7906), 1% fish skin gelatin (Sigma-Aldrich, G7041), 0.02% saponin (Sigma-Aldrich, S4521) and 15% horse serum (Corning, 35–030-CV) in PBS. Secondary antibodies included Alexa Fluor® 568 and Alexa Fluor® 488 (Molecular Probes). 4ʹ,6ʹ-diamidino-2phenylindole (DAPI) (Biotium, 40,011) was used to stain the nuclei. Images were acquired using the 488- and 543-nm lines of the argon ion as well as green HeNe lasers with 515/30-nm band pass and 585-nm-long pass filters, respectively, in a confocal imaging system (Zeiss LSM 510 META) in the Department of Medicine at Emory University. Quantification of LC3 and SQSTM1 puncta was performed using ImageJ software (NIH). Briefly, each cell was traced, and the image was converted into an 8-bit grayscale. Then, using the edit/clear outside feature of the software, background was removed and the image was inverted to quantify puncta as black dots. A threshold was then applied, the size of the particles to be quantified was set and the number of puncta was determined using the analyzed particle feature of the software.
Reactive oxygen species measurements
ROS levels were measured using H2DCFDA and DHE, which were described previously [34]. Cells were washed once with DPBS (Dulbecco’s phosphate-buffered saline, Corning, 21–030-CV) and incubated with 10 μM H2DCFDA or DHE in DPBS for 30 min at 37°C. Fluorescence intensity for DHE (Ex/Em: 500/530 nm) and for H2DCFDA (Ex/Em: 492/520 nm) was determined using a Synergy H1 multi-mode plate reader (BioTek Instruments).
Western blotting
VSMCs were lysed in buffer A (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EGTA, 0.1 mM MgCl2) supplemented with 1% Triton X-100 (Sigma-Aldrich, X100-500ML), 2 mM sodium orthovanadate (Enzo Life Sciences, 400–032-G025), 10 mM sodium pyrophosphate (Sigma-Aldrich, 221368), 10 mM sodium fluoride (J. T. Baker, 3688–01), and protease cocktail inhibitor for 30 min on ice. Aortas were homogenized with 15 strokes of a Teflon/glass homogenizer in buffer A. Cell extracts were sonicated and equal amounts of protein from total extracts were separated using 10% SDS-PAGE gels or 4–20% Criterion precast gels (Bio-Rad, 5671094). Gels were transferred to PVDF (Thermo Scientific, PI88518) membranes and blocked for 20 min with 1% non-fat dry milk (Bio-Rad, 170–6404) in TBS buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4) supplemented with 0.05% Triton X-100 (TBS-T). Membranes were incubated with primary antibodies in PBS containing 3% BSA, washed 3 times with TBS-T 10 min each and incubated with HRP-conjugated secondary antibodies (Invitrogen, G-21234 [goat anti rabbit], R-21459 [rabbit anti goat], G-21040 [goat anti mouse]). After 3 washes with TBS-T, membranes were developed by enhanced chemiluminescence. Quantification of bands in western blots, including loading controls, within the linear range was performed using ImageJ software.
Reverse transcription PCR (RT-PCR) and RNA interference
Total RNA was extracted using the RNeasy kit (Qiagen, 74104) according to the manufacturer’s instructions. The SuperScriptTM One Step RT-PCR system (Invitrogen, 109280) was used to measure the mRNA levels of Ppargc1a, Ppargc1b, Sqstm1, Nbr1, Becn1, Optn, Ctsd, Lamp2, Lc3 and Gapdh. Oligonucleotides, listed in Table S1, were obtained from Sigma-Aldrich.
Downregulation of rat Sqstm1 in RASM cells was achieved using the following siRNA duplex from Invitrogen: CGGUGAAGGCCUAUCUACUGGGCAA and UUGCCCAGUAGAUAGGCCUUCACCG. Cells were transfected using the basic Nucleofector® kit for smooth muscle cells from Amaxa Biosystems. Downregulation of mouse Atg5 in MASMs was done using siRNAControl#1 and siAtg5 from Thermo Fisher Scientific (GGUUAUGAGACAAGAAGAUtt and AUCUUCUUGUCUCAUAACCtt).
Cell proliferation
Ppargc1a+/+ and ppargc1a−/- MASMs (5,000 cells/per well) were grown in complete media at 37°C for 2 d. Cells were starved for 48 h in DMEM supplemented with 0.5% FBS and then incubated in complete media with 10% FBS and counted every day for 4 d.
Statistical analysis
Data were analyzed by student’s T-test and one-way analysis of variance (ANOVA). Values were expressed as mean ± standard deviation (SD). p ≤ 0.05 was considered as statistically significant. For western blots, protein expression was adjusted by the level of loading controls (TUBG1, GAPDH or ACTB) in at least 3 independent experiments.
Supplementary Material
Acknowledgments
We thank Dr. Wayne Alexander at the Department of Medicine Emory University for his support. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Funding Statement
This work was supported in part by the American Federation for Aging Research, the American Heart Association (14GRNT20180028), the USDA (2018-67017-27518) and the Robert P. Apkarian Integrated Electron Microscopy Core (RPAIEMC), which is subsidized by the Emory College of Arts and Sciences and the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR000454 and from the National Institute of Health award numbers UO1 HL80711 and HL60728 of Dr. Wayne Alexander.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material:
Supplemental data for this article can be accessed here.
References
- [1].Hubbard VM, Valdor R, Macian F, et al. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology. 2012;13(1):21–35. [DOI] [PubMed] [Google Scholar]
- [2].Cuervo AM, Dice JF.. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275(40):31505–31513. [DOI] [PubMed] [Google Scholar]
- [3].Zahn JM, Sonu R, Vogel H, et al. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006;2(7):e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vina J, Gomez-Cabrera MC, Borras C, et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev. 2009;61(14):1369–1374. [DOI] [PubMed] [Google Scholar]
- [5].Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27(7):728–735. [DOI] [PubMed] [Google Scholar]
- [6].Xiong S, Salazar G, Patrushev N, et al. Peroxisome proliferator-activated receptor gamma coactivator-1alpha is a central negative regulator of vascular senescence. Arterioscler Thromb Vasc Biol. 2013;33(5):988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xiong S, Patrushev N, Forouzandeh F, et al. PGC-1alpha modulates telomere function and DNA damage in protecting against aging-related chronic diseases. Cell Rep. 2015;12(9):1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3–5. [DOI] [PubMed] [Google Scholar]
- [9].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kang HT, Lee KB, Kim SY, et al. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One. 2011;6(8):e23367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tai H, Wang Z, Gong H, et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy. 2017;13(1):99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Grootaert MO, Da Costa Martins PA, Bitsch N, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. 2015;11(11):2014–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. [DOI] [PubMed] [Google Scholar]
- [14].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- [16].Scott I, Webster BR, Chan CK, et al. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem. 2014;289(5):2864–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kwon J, Han E, Bui C-B, et al. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012;13(2):150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rodriguez A, Durán A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3(3):211–222. [DOI] [PubMed] [Google Scholar]
- [19].Sergin I, Bhattacharya S, Emanuel R, et al. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci Signal. 2016;9(409):ra2–ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pugsley HR. Assessing autophagic flux by measuring LC3, p62, and LAMP1 Co-localization using multispectral imaging flow cytometry. J Vis Exp. 2017. 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].O’Hagan KA, Cocchiglia S, Zhdanov AV, et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci U S A. 2009;106(7):2188–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rensvold JW, Ong S-E, Jeevananthan A, et al. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis. Cell Rep. 2013;3(1):237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mellone M, Hanley CJ, Thirdborough S, et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging (Albany NY). 2016;9(1):114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu CL, Chen S, Dietrich D, et al. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28(4):674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Viscomi MT, D’Amelio M, Cavallucci V, et al. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8(2):222–235. [DOI] [PubMed] [Google Scholar]
- [26].Nikoletopoulou V, Sidiropoulou K, Kallergi E, et al. Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 2017;26(1):230–242 e5. [DOI] [PubMed] [Google Scholar]
- [27].Demers-Lamarche J, Guillebaud G, Tlili M, et al. Loss of mitochondrial function impairs lysosomes. J Biol Chem. 2016;291(19):10263–10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Patrushev N, Seidel-Rogol B, Salazar G. Angiotensin II requires zinc and downregulation of the zinc transporters ZnT3 and ZnT10 to induce senescence of vascular smooth muscle cells. PLoS One. 2012;7(3):e33211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu YT, Tan H-L, Shui G, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nazio F, Strappazzon F, Antonioli M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15(4):406–416. [DOI] [PubMed] [Google Scholar]
- [32].Salazar G, Huang J, Feresin RG, et al. Zinc regulates Nox1 expression through a NF-kappaB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic Biol Med. 2017;108:225–235. [DOI] [PubMed] [Google Scholar]
- [33].Xiong S, Salazar G, San Martin A, et al. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem. 2010;285(4):2474–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Feresin RG, Huang J, Klarich DS, et al. Blackberry, raspberry and black raspberry polyphenol extracts attenuate angiotensin II-induced senescence in vascular smooth muscle cells. Food Funct. 2016;7(10):4175–4187. [DOI] [PubMed] [Google Scholar]
- [35].Dan HC, Cooper MJ, Cogswell PC, et al. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22(11):1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chien Y, Scuoppo C, Wang X, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brandt N, Dethlefsen MM, Bangsbo J, et al. PGC-1alpha and exercise intensity dependent adaptations in mouse skeletal muscle. PLoS One. 2017;12(10):e0185993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Halling JF, Ringholm S, Nielsen MM, et al. PGC-1alpha promotes exercise-induced autophagy in mouse skeletal muscle. Physiol Rep. 2016;4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vainshtein A, Tryon LD, Pauly M, et al. Role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308(9):C710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fader CM, Aguilera MO, Colombo MI. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy. 2012;8(12):1741–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fader CM, Sánchez DG, Mestre MB, et al. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793(12):1901–1916. [DOI] [PubMed] [Google Scholar]
- [42].Kadota T, Fujita Y, Yoshioka Y, et al. Emerging role of extracellular vesicles as a senescence-associated secretory phenotype: Insights into the pathophysiology of lung diseases. Mol Aspects Med. 2018;60:92–103. [DOI] [PubMed] [Google Scholar]
- [43].Qin W, Haroutunian V, Katsel P, et al. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66(3):352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jain A, Lamark T, Sjøttem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol. 2005;25(4):1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ivankovic D, Chau K-Y, Schapira AHV, et al. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136(2):388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Erlich AT, Brownlee DM, Beyfuss K, et al. Exercise induces TFEB expression and activity in skeletal muscle in a PGC-1alpha-dependent manner. Am J Physiol Cell Physiol. 2018;314(1):C62–C72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Seglen PO, Gordon PB. Effects of lysosomotropic monoamines, diamines, amino alcohols, and other amino compounds on protein degradation and protein synthesis in isolated rat hepatocytes. Mol Pharmacol. 1980;18(3):468–475. [PubMed] [Google Scholar]
- [49].Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. Embo J. 2011;30(8):1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kang HT, Park JT, Choi K, et al. Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol. 2017;13(6):616–623. [DOI] [PubMed] [Google Scholar]
- [51].Zuhlsdorf M, Imort M, Hasilik A, et al. Molecular forms of beta-hexosaminidase and cathepsin D in serum and urine of healthy subjects and patients with elevated activity of lysosomal enzymes. Biochem J. 1983;213(3):733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dubey V, Luqman S. Cathepsin D as a promising target for the discovery of novel anticancer agents. Curr Cancer Drug Targets. 2017;17(5):404–422. [DOI] [PubMed] [Google Scholar]
- [53].Tai S, Hu X-Q, Peng D-Q, et al. The roles of autophagy in vascular smooth muscle cells. Int J Cardiol. 2016;211:1–6. [DOI] [PubMed] [Google Scholar]
- [54].Luo Z, Xu W, Ma S, et al. Moderate autophagy inhibits vascular smooth muscle cell senescence to stabilize progressed atherosclerotic plaque via the mTORC1/ULK1/ATG13 signal pathway. Oxid Med Cell Longev. 2017;2017:3018190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sung JY, Lee KY, Kim JR, et al. Interaction between mTOR pathway inhibition and autophagy induction attenuates adriamycin-induced vascular smooth muscle cell senescence through decreased expressions of p53/p21/p16. Exp Gerontol. 2017;109:51–58. [DOI] [PubMed] [Google Scholar]
- [56].Grootaert MOJ, Moulis M, Roth L, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res. 2018;114(4):622–634. [DOI] [PubMed] [Google Scholar]
- [57].Kunieda T, Minamino T, Nishi J-I, et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114(9):953–960. [DOI] [PubMed] [Google Scholar]
- [58].Ramesh Babu J, Lamar Seibenhener M, Peng J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106(1):107–120. [DOI] [PubMed] [Google Scholar]
- [59].Laurin N, Brown JP, Morissette J, et al. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70(6):1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rubino E, Rainero I, Chiò A, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79(15):1556–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kelly DP. Cell biology: Ageing theories unified. Nature. 2011;470(7334):342–343. [DOI] [PubMed] [Google Scholar]
- [63].Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. 2012;13(6):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Salazar G. NADPH oxidases and mitochondria in vascular senescence. Int J Mol Sci. 2018;19(5):1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. [DOI] [PubMed] [Google Scholar]
- [66].Griendling KK, Taubman MB, Akers M, et al. Characterization of phosphatidylinositol-specific phospholipase C from cultured vascular smooth muscle cells. J Biol Chem. 1991;266(23):15498–15504. [PubMed] [Google Scholar]
- [67].Feresin RG, Pourafshar S, Huang J, et al. Extraction and purification of polyphenols from freeze-dried berry powder for the treatment of vascular smooth muscle cells in vitro. JoVE. 2017;125:e55605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sutliff RL, Hilenski LL, Amanso AM, et al. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler Thromb Vasc Biol. 2013;33(9):2154–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhao Y, Feresin RG, Falcon-Perez JM, et al. Differential targeting of SLC30A10/ZnT10 heterodimers to endolysosomal compartments modulates EGF-induced MEK/ERK1/2 activity. Traffic. 2016;17(3):267–288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.