

Abstract
Receptor tyrosine kinases (RTKs) and Yes‐associated protein (YAP) are critical driving factors in tumors. Currently, the regulation of RTKs in the Hippo‐YAP pathway has been recognized as an important issue. However, the relationship between AXL, one of the RTKs, and YAP in head and neck squamous cell carcinoma (HNSCC) remains unknown. In this study, the crosstalk between AXL and YAP was thoroughly investigated in vitro and in vivo. We determined that there was a positive correlation between AXL and YAP in the HNSCC tissue samples and the Cancer Genome Atlas (TCGA) dataset, and high co‐expression was associated with poor prognosis. Inhibiting YAP decreased AXL expression in HNSCC cells, while YAP overexpression increased AXL. Moreover, ectopic expression of AXL reversed tumor suppressor phenotypes mediated by YAP silencing. This reversal effect was also confirmed in vivo. In addition, AXL overexpression and Gas6, a ligand of AXL, stimulated YAP dephosphorylation, nuclear translocation, and target gene transcription. AXL inhibition decreased YAP dephosphorylation and nuclear translocation. Mechanistically, Gas6 induced a competitive binding to phosphorylated signal transducers and activators of transcription 3 (STAT3) with large tumor suppressor kinase 1 (LATS1) and inhibited the Hippo pathway. This study revealed a novel non‐transcriptional effect of STAT3 in Gas6/AXL‐induced YAP activity, suggesting that STAT3 acted as a critical “molecular switch” during the mutual promotion between AXL and YAP, which might be a promising therapeutic target in HNSCC.
Keywords: AXL, head and neck squamous cell carcinoma, signal transducers and activators of transcription 3, signaling mutual regulation, yes‐associated protein
Gas6/AXL‐induced STAT3 activation and then competitively bound to LATS1, which blocked Hippo inhibitory regulation on YAP and induced YAP nuclear translocation and transcriptional regulation. STAT3 activation is a novel regulation mediating the crosstalk between Gas6/AXL and Hippo/YAP in carcinogenesis and might be an intriguing therapeutic target in HNSCC.
Abbreviations
- CCN2
cellular communication network factor 2
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- HNSCC
head and neck squamous cell carcinoma
- LATS1
large tumor suppressor kinase 1
- rhGas6
recombinant human growth arrest‐specific protein 6
- rhIL‐6
recombinant human interleukin‐6
- RTK
receptor tyrosine kinase
- STAT3
signal transducers and activators of transcription 3
- TCGA
The Cancer Genome Atlas
- TEAD
TEA domain‐containing sequence‐specific transcription factor
- YAP
Yes‐associated protein
1. INTRODUCTION
HNSCC is a common and aggressive malignant cancer with 40%‐50% mortality in advanced patients. 1 RTKs, including EGFR and human epidermal growth factor receptor 2 (HER2), provide growth signaling to maintain the essential cell behaviors that have become the most popular therapeutic targets in various tumors. 2 Regrettably, cetuximab, an EGFR monoclonal antibody, showed unsatisfactory outcomes as a first‐line targeted drug in HNSCC with a response rate of only 13% in single‐agent application. 3 , 4 , 5 Thus, extensive investigation of the RTK regulatory network during cancer progression in HNSCC is an impending need.
Hippo‐YAP plays a critical role in tumor growth, metastasis, and chemoresistance. 6 , 7 Hippo cascade restricts YAP activity by phosphorylating large tumor suppressor kinase 1 (LATS1) and MOB1, leading to YAP phosphorylation, cytoplasmic retention, and consequently degradation. 8 When the Hippo pathway is inhibited, YAP accumulating in nucleus interacts with the TEA domain‐containing sequence‐specific transcription factors (TEADs) and initiates target gene transcription. Abnormally elevated levels of YAP and its pro‐oncogenic transcriptional regulation have attracted much attention. 9 , 10 Upstream signals that regulate the Hippo‐YAP pathway in HNSCC, however, remain poorly understood.
Recent studies have observed a strong relationship between RTKs and the Hippo pathway. Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) phosphorylates HER3 and regulates the Hippo‐YAP pathway. 11 EGF treatment dissociates the Hippo core complex, resulting in YAP nuclear translocation. 12 AXL, a member of the RTK family, plays a critical role in HNSCC. 13 If AXL regulates YAP signaling and its details of its mechanism in HNSCC are still unknown.
In this study, we demonstrated that AXL upregulation reversed tumor suppressor phenotypes by YAP silencing in HNSCC. Gas6/AXL inhibited the Hippo pathway and then induced YAP nuclear localization and transcriptional regulation. Furthermore, signal transducers and activators of transcription 3 (STAT3) activation was a key mediator in the regulation of Gas6/AXL on Hippo‐YAP. Our findings provided an insight into the mutual regulatory mechanism of AXL and YAP in HNSCC. STAT3 activation in this crosstalk was revealed for the first time and was suggested as a potential therapeutic target in HNSCC.
2. MATERIALS AND METHODS
2.1. Patients and specimens
In total, 63 HNSCC tumor tissue specimens were collected from January 2009 to December 2010 at the Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (Shanghai, China). Informed consent was provided by all patients. This study was approved by Ethics Committee of the Ninth People's Hospital. Demographic characteristics are listed in Table S1.
2.2. Immunohistochemistry and immunofluorescence assay
Immunohistochemistry (IHC) was performed with a tissue microarray (TMA). Protein levels were evaluated by 2 clinical pathologists using histochemistry score (H‐score) criteria. 14 H‐score = Σ (staining intensity percentage of stained cells). Immunofluorescence assay was performed according to protocols. Antibodies used are as following: AXL (AF154) (R&D Systems), Ki67 (IR626) (DAKO), LATS1 (17049‐1‐AP) (Proteintech).
2.3. Cell culture
Cal27, SCC9, and SCC25 cell lines were purchased from American Type Culture Collection (ATCC). HN4, HN6, and HN30 cell lines were kindly provided by the University of Maryland Dental School, USA. SCC7 cell line was kindly provided by Prof. Liu in Suzhou University, China. Cal27, HN4, HN6, HN30, and human embryonic kidney (HEK) 293T were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco) with 10% FBS (Gibco) and 1% penicillin/streptomycin (Gibco). DMEM/nutrient mixture F12 (Gibco) medium was used for SCC9 and SCC25. Recombinant human growth arrest‐specific protein 6 (rhGas6) and interleukin‐6 (rhIL‐6) were purchased from R&D Systems and Proteintech, respectively. Verteporfin, BGB324, cryptotanshinone (Selleck), AKTi‐1/2, and SCH772984 were purchased from MedChemExpress.
2.4. Cell transfection
Small interfering RNAs (siRNAs) were synthesized by RiboBio (Table S2). Plasmids and lentiviruses were synthesized by Genechema and Genomeditech. Cell transfection was performed using the Lipofectamine® 3000 Transfection Kit (Invitrogen).
2.5. Total mRNA extraction and quantitative Real‐time PCR
Total mRNA was extracted using TRIzol (Takara) and cDNA was synthesized with PrimerScript™ RT reagent Kit (Takara). Primers were synthesized by Sangon Biotech (Table S3).
2.6. Western blot and immunoprecipitation (IP) analysis
Total protein was extracted with SDS lysis buffer (Beyotime Biotechnology). Nuclear and cytoplasmic proteins were prepared as described previously. 15 Cells for IP were lysed with RIPA lysis buffer (Beyotime Biotechnology). Protein A/G Magnetic Beads were purchased from Bimake. Antibodies are listed as follows: YAP (#14074), p‐YAP (Ser127) (#13008), AXL (#8661), p‐AXL (Tyr702) (#5724), STAT3 (#9139), p‐STAT3 (Tyr705) (#9145), AKT (#4691), p‐AKT (Ser473) (#4060), ERK (#4695), p‐ERK1/2 (Thr202/Tyr204) (#4370), MMP2 (#40994), MMP9 (#13667), MST1 (#3682), p‐MST1/2 (Thr183/Thr180) (#49332), LATS1 (#3477), p‐LATS1 (Thr1079) (#8654), LATS2 (#5888), MOB1 (#13730), p‐MOB1 (Thr35) (#8699) (Cell Signaling Technology, CST), MST2 (12097‐1‐AP) (Proteintech).
2.7. RNA sequencing and data analysis
Sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit. RNA sequencing was performed on Illumina NovaSeq platform. Differential expression analysis and Gene Ontology (GO) enrichment analysis were performed with a fold change (FC) > 2.0 and Padj < .05.
2.8. Dual‐luciferase assay
The binding site between AXL and TEAD1 was predicted in JASPER dataset. AXL WT‐promoter, AXL 572‐583 MUT‐promoter, and cellular communication network factor 2 (CCN2)‐promoter were constructed by Genomeditech. Promoter activities were detected using the Dual‐Luciferase Reporter Gene Assay Kit (Beyotime Biotechnology).
2.9. Cell proliferation and invasion assay
Cell Counting Kit‐8 (CCK‐8; Dojindo), 5‐ethynyl‐2′‐deoxyuridine (EdU) (Cell‐Light EdU In Vitro Kit, RiboBio), and transwell assays were performed according to our previous study. 16
2.10. Animal studies
Lentivirus transfected CAL27 (1.5 × 106 cells per injection) and SCC7 cells (1.5 × 105 cells per injection) were subcutaneously injected into BALB/c and C3H mice, respectively. Hematoxylin and eosin stain (HE), IHC and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Beyotime Biotechnology) were performed according to manufacturer's protocols.
2.11. Statistical analysis
Statistical analysis and diagramming were performed using SPSS Version 25.0, R Studio and GraphPad Prism version 6.0. X‐Tile software version 3.6.1 (Yale University School of Medicine) was used to determine mRNA cut‐off values. 17 Student t test or one‐way ANOVA were used to analyze for 2 or more variables. Data were presented as the mean ± SD of 3 independent experiments. P < .05 was regarded as statistically significant difference.
3. RESULTS
3.1. A positive correlation between YAP and AXL is observed and indicates poor prognosis in HNSCC patients
Immunohistochemistry was performed to measure YAP and AXL protein expressions in the TMA of 63 HNSCC patients (Table S1). YAP was mainly localized in the cytoplasm and nucleus, while AXL was predominantly on plasma membranes (Figure 1A). Protein expression was divided into low, moderate, or high based on H‐score criteria. We further analyzed the relationship between YAP and AXL. A positive correlation was observed between total YAP and AXL protein expression (Figure 1B). Given that YAP activation requires nuclear translocation, we also observed a positive correlation between nuclear YAP and AXL protein expression (Figure 1C). According to the HNSCC dataset from TCGA database including 466 HNSCC samples, a positive correlation between YAP and AXL mRNA expression was also observed, thus supporting our IHC results (Figure 1D). Clinical prognosis analysis was performed based on YAP and AXL mRNA according to TCGA database (YAP cut‐off value: −0.33; AXL cut‐off value: 1.24; Figure S1). High YAP or AXL expression was associated with poor prognosis (Figure 1E,F and Tables 1 and 2). Furthermore, patients with high expression of YAP combined with AXL had worse prognosis than patients with respective high expression of the proteins (Figure 1G and Table 2). These results demonstrated that YAP and AXL were positively correlated and indicated poor prognosis in HNSCC.
FIGURE 1.
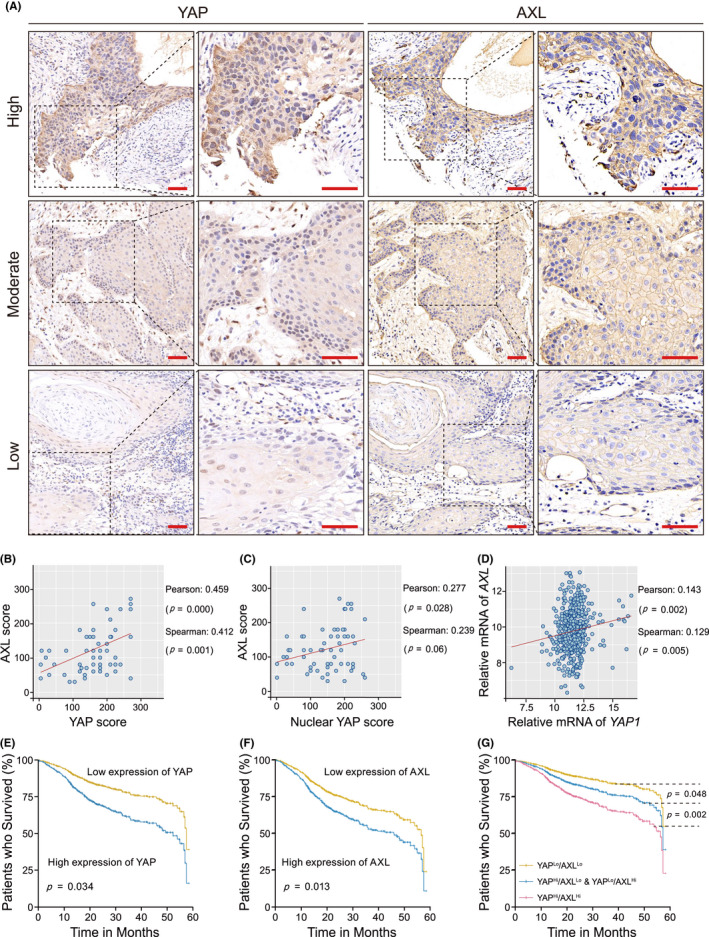
A positive correlation between Yes‐associated protein (YAP) and AXL is observed and indicates poor prognosis in head and neck squamous cell carcinoma (HNSCC) patients. A, YAP and AXL protein expression was detected using tissue microarray assay (TMA) including 63 HNSCC patients. Representative images of high, moderate, and low expression of YAP and AXL are shown. Scale bars: 50 μm. B, A correlation between total YAP and AXL protein expression were analyzed in HNSCC TMA. C, A correlation between nuclear YAP and AXL protein expression was analyzed in HNSCC TMA. D, A correlation between YAP and AXL mRNA expression in the HNSCC dataset including 466 samples from the Cancer Genome Atlas (TCGA) database (https://xenabrowser.net) was performed. E, F, Five‐year overall survival analysis was performed based on YAP or AXL expression in the HNSCC dataset. G, Five‐year overall survival analysis was performed according to the combined YAP and AXL expression in the HNSCC dataset. P‐value was according to Student t test
TABLE 1.
Univariate Cox regression models for estimating the overall survival
| Characteristics | HR | 95% CI | P‐value |
|---|---|---|---|
| Overall survival | |||
| Univariate analysis | |||
| Age | 1.021 | 1.009‐1.034 | .001 |
| Gender (male vs female) | 0.692 | 0.514‐0.932 | .016 |
| Alcohol history (Alcohol vs none Alcohol) | 0.775 | 0.577‐1.042 | .092 |
| Pathologic stage | |||
| II vs I | 2.018 | 0.728‐5.591 | .177 |
| III vs I | 3.271 | 1.208‐8.861 | .020 |
| YAP expression (high vs low) | 1.346 | 0.819‐2.214 | .241 |
| AXL expression (high vs low) | 1.755 | 1.265‐2.436 | .001 |
| YAP/AXL expression | |||
| YAPHi/AXLLo & YAPLo/AXLHi vs YAPLo/ AXLLo | 1.338 | 0.785‐2.281 | .284 |
| YAPHi/ AXLHi vs YAPHi/AXLLo & YAPLo/AXLHi | 1.965 | 1.325‐2.913 | .001 |
P‐values in bold print indicate statistical significance
Abbreviations: CI, confidence interval, HR, hazard ratio.
TABLE 2.
Multivariate Cox regression models for estimating the overall survival
| Characteristics | HR | 95% CI | P‐value |
|---|---|---|---|
| Overall survival | |||
| Multivariate analysis of YAP expression (Figure 1E) | |||
| Age | 1.025 | 1.011‐1.040 | .000 |
| Pathologic stage | |||
| II vs I | 2.156 | 0.778‐5.979 | .140 |
| III vs I | 3.917 | 1.440‐10.656 | .007 |
| YAP expression (high vs low) | 1.940 | 1.050‐3.583 | .034 |
| Multivariate analysis of AXL expression (Figure 1F) | |||
| Age | 1.025 | 1.011‐1.039 | .000 |
| Pathologic stage | |||
| II vs I | 2.147 | 0.774‐5.952 | .142 |
| III vs I | 3.621 | 1.331‐9.852 | .012 |
| AXL expression (high vs low) | 1.571 | 1.100‐2.244 | .013 |
| Multivariate analysis of combined YAP and AXL expression (Figure 1G) | |||
| Age | 1.025 | 1.012‐1.040 | .000 |
| Pathologic stage | |||
| II vs I | 2.165 | 0.781‐6.002 | .138 |
| III vs I | 3.792 | 1.393‐10.320 | .009 |
| YAP/AXL expression | |||
| YAPHi/AXLLo & YAPLo/AXLHi vs YAPLo/ AXLLo | 1.923 | 1.006‐3.676 | .048 |
| YAPHi/ AXLHi vs YAPHi/AXLLo & YAPLo/AXLHi | 2.013 | 1.281‐3.163 | .002 |
P‐values in bold print indicate statistical significance.
Abbreviations: CI, confidence interval; HR, hazard ratio.
3.2. YAP positively regulates AXL expression in HNSCC
We examined YAP and AXL expressions in 6 HNSCC cell lines and normal oral mucosal epithelial cells. Most HNSCC cell lines have upregulated YAP and AXL expression (Figure 2A). HN4 and HN30, with high expression levels of YAP, were selected for follow‐up experiments. AXL mRNA and protein levels were significantly attenuated after YAP silencing (Figures 2B and S2). The percentage of YAP‐ or AXL‐positive cells was decreased after si‐YAP transfection, as shown by immunofluorescence analysis (Figure 2C). To further explore YAP target genes, RNA sequencing analysis was performed after si‐YAP transfection. Here, 192 significantly altered genes were identified in the si‐YAP group, including 109 downregulated genes and 83 upregulated genes (Document S1). AXL was one of the downregulated genes in the si‐YAP group (Figure 2D). Other YAP target genes, such as AMOTL2 and AJUBA, were also downregulated. Moreover, verteporfin, an inhibitor of YAP, had substantially decreased AXL expression in a dose‐dependent manner (Figure 2E). Furthermore, ectopic expression of YAP increased AXL expression (Figure 2F). Verteporfin was used to explore the effect of YAP on AXL downstream signaling. Previous studies have found that Janus kinase (JAK)/STAT3, phosphoinositide 3‐kinase (PI3K)/AKT, and MEK/ERK signaling were main downstream pathways mediating AXL regulation in tumors. 18 , 19 We observed that p‐STAT3 and p‐AKT were reduced by verteporfin (Figure 2G). There was no obvious change in p‐ERK (data not shown). To verify AXL as a target gene of YAP, AXL‐promoter activity was measured after cotransfection of the YAP plasmid. We observed that YAP overexpression activated wild‐type AXL transcriptional initiation, not AXL mutant (572‐583) (Figure 2H,I). Our results suggested that YAP positively regulated AXL expression in HNSCC cells.
FIGURE 2.
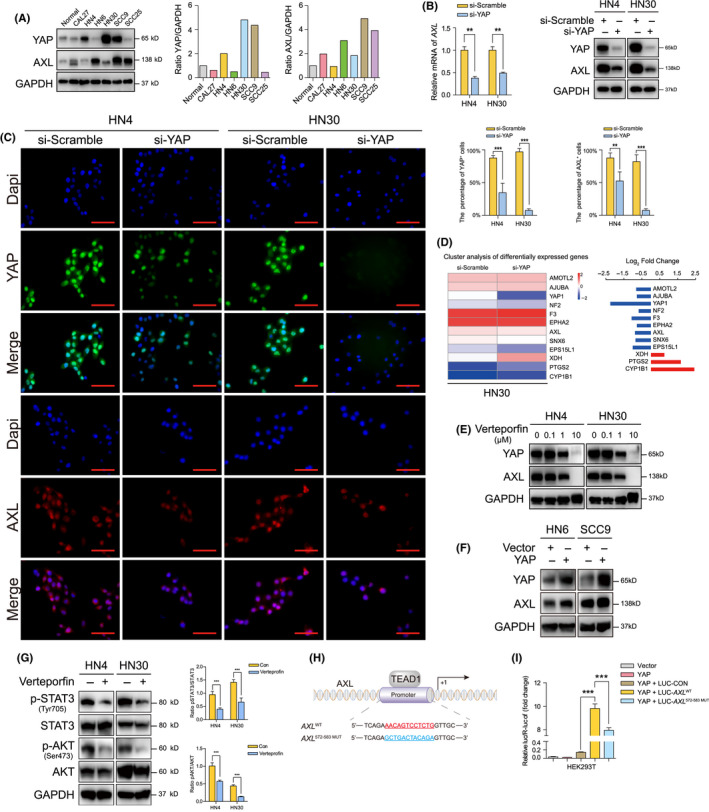
Yes‐associated protein (YAP) positively regulates AXL expression in head and neck squamous cell carcinoma (HNSCC) cells. A, YAP and AXL protein expression were detected in HNSCC cell lines and normal oral mucosal epithelial cell. B, The expression of AXL and YAP was determined after transfection with si‐YAP for 48 h. C, The percentage of AXL‐ and YAP‐positive cells was determined using immunofluorescence transfected with si‐YAP for 24 h. Scale bars: 50 μm. D, RNA sequencing analysis of mRNA extracted after transfection with si‐YAP for 48 h. Significantly altered genes associated with regulation of Hippo signaling and tyrosine kinase activity and corresponding log2 fold change are listed in the heat map. (blue, downregulation; red, upregulation). E, AXL expression was detected after YAP inhibitor verteporfin for 24 h. F, AXL expression was determined after ectopic expression of YAP for 48 h. G, The main downstream molecules of AXL were detected after treatment with 1 μmol/L verteporfin for 24 h. H, The binding site of TEAD1 in AXL‐promoter was predicted and mutant AXL‐promoter was designed. I, Wild‐type and mutant AXL‐promoter activities were detected after cotransfection with YAP plasmid for 24 h. *P < .05, **P < .01, ***P < .001, based on Student t test
3.3. AXL reverses tumor suppressor phenotypes mediated by YAP silencing in vitro and in vivo
To determine the involvement of AXL in YAP oncogenic function in HNSCC progression, salvage experiments using cotransfection with si‐YAP and AXL plasmid were performed. The inhibited proliferation mediated by YAP silencing was reversed by AXL overexpression according to EdU and CCK‐8 assays (Figures 3A,B and S3). Moreover, colony formation was inhibited after YAP silencing and increased after AXL cotransfection (Figure 3C). Decreases in migration and invasion mediated by YAP knockdown were also reversed after AXL overexpression according to wound healing and transwell assays (Figures 3D,E,F, S4, and S5). Moreover, 2 invasion‐associated proteins, matrix metallopeptidase 2 (MMP2) and MMP9, 20 were decreased after YAP silencing and increased by AXL overexpression (Figure 3G). These in vitro results demonstrated that AXL mediated the oncogenic regulation of YAP on HNSCC.
FIGURE 3.
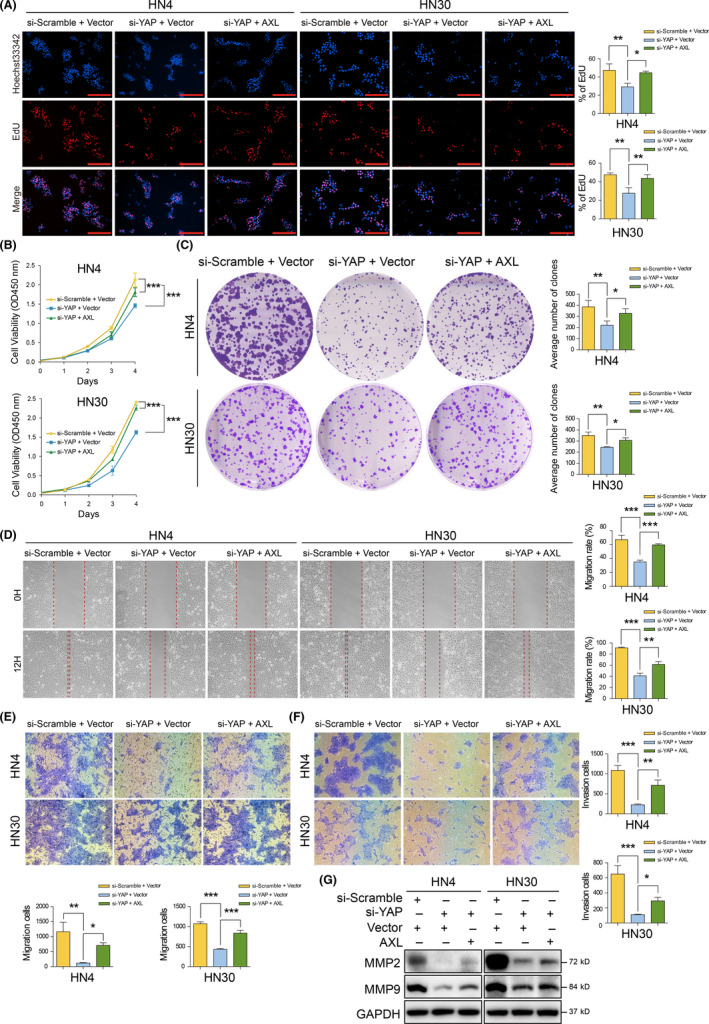
AXL reverses tumor suppressor phenotypes mediated by Yes‐associated protein (YAP) silencing in vitro. A, B, EdU and CCK‐8 assay was performed after cotransfection with si‐YAP and the AXL plasmid for 24 h. Scale bars: 200 μm. C, Colony formation assay was performed after cotransfection with si‐YAP and AXL plasmid. D, E, Cell migration was assessed with wound healing and transwell assay after YAP silencing and AXL overexpression for 24 h. F, Cell invasion was performed using transwell assay with coated Matrigel cotransfection with si‐YAP and AXL plasmid. G, MMP2 and MMP9 were detected after cotransfection with si‐YAP and AXL plasmid for 48 h. *P < .05, **P < .01, ***P < .001, according to one‐way ANOVA
To confirm in vitro results, xenograft tumor models were established using the lentivirus system in immunodeficient mice (Figure 4A). Our data showed that all 12 tumors were formed in Vector group, 4/12 tumors were observed in YAP knockdown group, and 6/12 tumors were formed after AXL overexpression (Figure 4B). These results indicated that YAP played a critical role during HNSCC carcinogenesis. AXL overexpression rescued the oncogenic inhibition of YAP knockdown. Moreover, tumor volume and tumor weight decreased in YAP silencing group were reversed by AXL overexpression (Figure 4C,D). YAP and AXL expressions in tumors were further confirmed using IHC (Figure 4E). Ki‐67 and TUNEL assays revealed that AXL ectopic expression could reverse tumor suppressor phenotypes mediated by YAP silencing (Figure 4E,F). We also explored the effect of YAP and AXL in vivo using immunocompromised mice (Figure 5A,B). The results showed that YAP knockdown inhibited tumor growth, which could be reversed by AXL overexpression (Figure 5C–E). Representative HE images are shown in Figure 5F. These in vivo results further supported that AXL acted as a downstream molecule of YAP in HNSCC progression.
FIGURE 4.
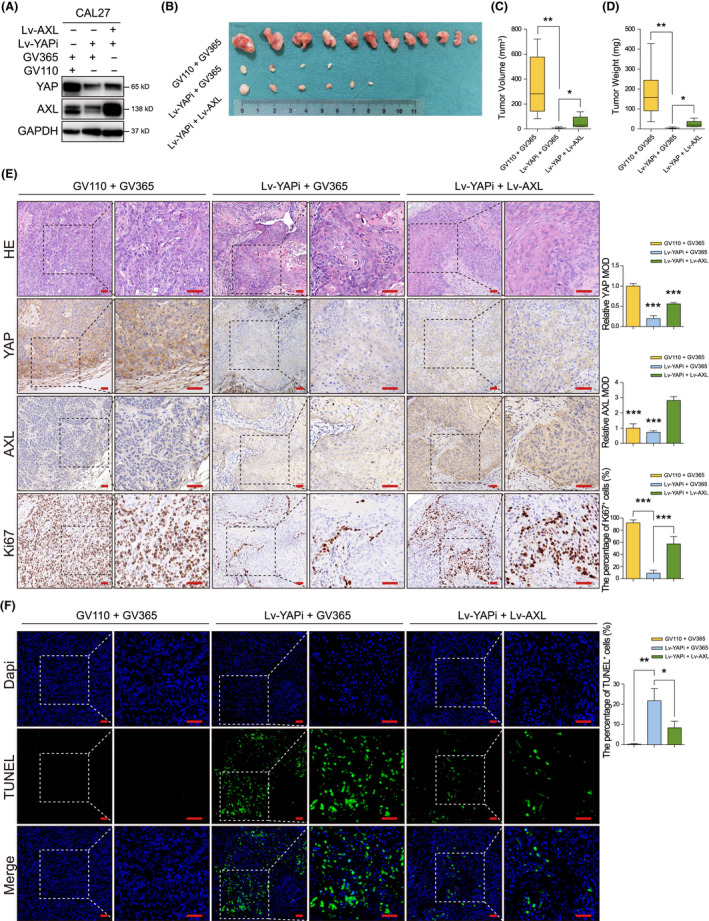
AXL reverses tumor growth inhibition mediated by Yes‐associated protein (YAP) silencing in vivo using immunodeficient mice. A, YAP and AXL were detected in CAL27 after cotransfection with Lv‐YAPi and Lv‐AXL. B, The tumors derived from BALB/c mice are shown. GV110 and GV365 were blank vector controls for Lv‐YAPi and Lv‐AXL, respectively. C, D, Tumor volume and tumor weight were measured after the mice were sacrificed. E, Representative images of HE, YAP, AXL, and Ki‐67 staining by IHC are shown. Scale bars: 50 μm. F, TUNEL staining was performed to measure cell apoptosis. Scale bars: 20 μm. *P < .05, **P < .01, ***P < .001, according to Student t test
FIGURE 5.
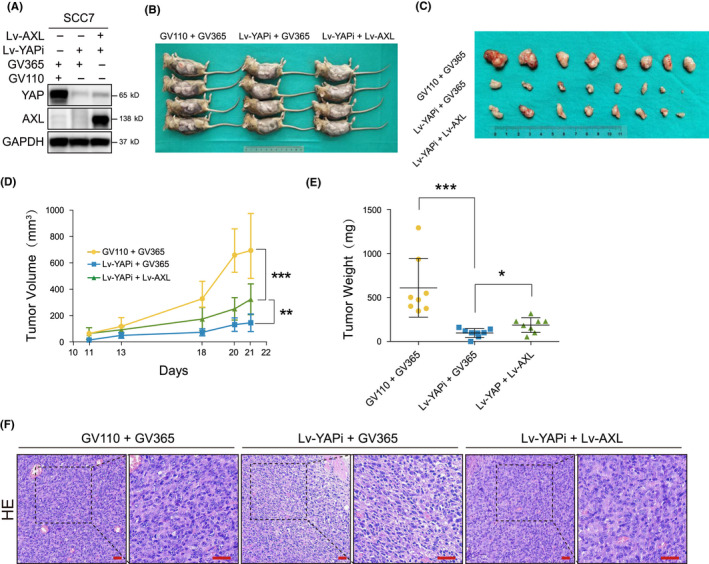
AXL reverses tumor growth inhibition mediated by Yes‐associated protein (YAP) silencing in vivo using immunocompromised mice. A, YAP and AXL were detected in SCC7 after cotransfection with Lv‐YAPi and Lv‐AXL. B, The xenograft models using C3H mice are shown. C, The tumors derived from C3H mice are shown. D, Tumor volume was measured during the experiment. E, Tumor weight was measured after the mice were sacrificed. F, Representative HE images are shown. Scale bars: 50 μm
3.4. AXL promotes YAP nuclear translocation and transcriptional regulation via dephosphorylation
To test whether AXL can act as an upstream signaling component and inversely regulate YAP activity, AXL plasmids, and its ligand, Gas6, were used to activate AXL activity. BGB324, an AXL inhibitor, was used to inhibit AXL activity. We observed that p‐YAP (Ser127) was significantly decreased after AXL overexpression or rhGas6 stimulation in a dose‐dependent manner (Figure 6A,B). Conversely, BGB324 significantly increased the levels of p‐YAP in a dose‐dependent manner (Figure 6C). To further explore whether AXL can affect YAP cellular location, immunofluorescence assay was performed. According to previous studies, lysophosphatidic acid (LPA) from serum can also induce YAP nuclear translocation via G‐protein‐coupled receptor (GPCR) signaling, regardless of Gas6/AXL. 21 High serum concentration in culture medium led to a high baseline of nuclear YAP expression, which would affect the observation on YAP nuclear translocation. Therefore, before the treatment of rhGas6, cells were pre‐starved with 2% FBS to ensure enough YAP in cytoplasm. While BGB324 was directly added with 10% FBS to ensure most YAP accumulating in nucleus in the baseline. Our results observed that Gas6 stimulation substantially promoted YAP nuclear translocation (Figure 6D) while BGB324 inhibited YAP nuclear translocation (Figure 6E). Cytoplasmic and nuclear proteins were separated for semiquantitative analysis for YAP distribution, nuclear YAP was enhanced with increasing dose of rhGas6 (Figure 6F), while BGB324 decreased YAP in the nucleus (Figure 6G). We also observed that si‐AXL inhibited YAP nuclear translocation induced by rhGas6 (Figures 6H and S6A). Moreover, mRNA expression of CCN2 and AREG, 2 well known target genes of YAP, 22 , 23 were increased after AXL overexpression (Figure 6I). To testify the effect of AXL in YAP transcriptional regulation, we detected CCN2‐promoter activity using dual‐luciferase assay. YAP knockdown decreased CCN2‐promoter activity induced by Gas6 (Figure 6J). The Hippo pathway has an inhibitory regulation on YAP activity. Activated LATS1 and MOB1 directly promoted YAP phosphorylation (mainly at Ser127), leading to YAP cytoplasmic retention and degradation. YAPS127A, an activated mutant, 24 was constructed to avoid LATS1 inhibition and enhance CCN2‐promoter activity (Figure S7). Our result showed that Gas6 increased CCN2‐promoter activities in Vector groups and YAPWT groups. However, Gas6 could not increase CCN2‐promoter activity in the presence of YAPS127A, which indicated that Gas6/AXL activated YAP via LATS1‐phosphorylated site at Ser127 (Figure 6K). We further found that Gas6 stimulation and AXL overexpression inhibited activities of MST1/2, LATS1, and MOB1, as well as AXL knockdown inhibited MST1/2 activity (Figures 6L and S8). These results suggested that Gas6/AXL‐induced YAP dephosphorylation and nuclear translocation via inhibiting the Hippo pathway in HNSCC.
FIGURE 6.
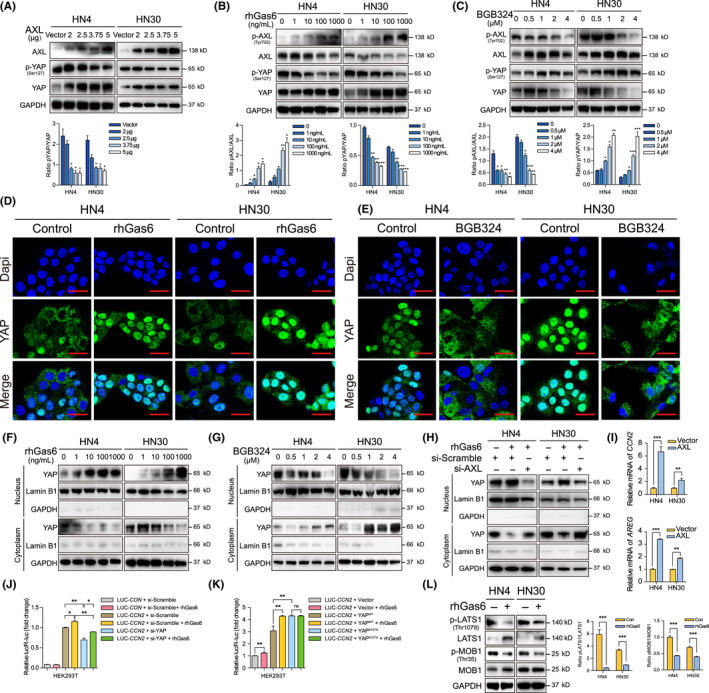
AXL promotes Yes‐associated protein (YAP) nuclear translocation and transcriptional regulation via dephosphorylation. A, P‐YAP was detected after AXL plasmid transfection for 48 h. B, P‐YAP was detected after treatment with 2% FBS for 12 h and rhGas6 for 1 h. C, After AXL inhibitor BGB324 treatment for 24 h, p‐YAP expression was assessed. D, Cells were treated with 2% FBS for 12 h and 1000 ng/mL rhGas6 for 1 h. Confocal immunofluorescence assay was used to visualize YAP localization. Scale bars: 20 μm. E, The localization of YAP was assessed after treatment with 2 μmol/L BGB324 for 24 h. Scale bar: 20 μm. F, Nuclear and cytoplasmic YAP proteins were separated and analyzed after treatment with 2% FBS for 12 h and 1000 ng/mL rhGas6 for 1 h. G, YAP expression in nucleus and cytoplasm were analyzed after 2 μmol/L BGB324 treatment for 24 h. H, Nuclear and cytoplasmic YAP were determined after si‐AXL transfection with 2% FBS for 48 h and 1000 ng/mL rhGas6 treatment for 1 h. I, CCN2 and AREG relative mRNA expression was measured after AXL plasmid transfection for 24 h. J, CCN2‐promoter activities were detected after cotransfection of si‐YAP with 2% FBS for 24 h and 1000 ng/mL rhGas6 treatment for 1 h. K, CCN2‐promoter activities were analyzed after cotransfection of YAPWT or YAPS127A plasmid with 2% FBS for 24 h and 1000 ng/mL rhGas6 treatment for 1 h. L, P‐LATS1 and p‐MOB1 were determined after treatment with 2% FBS for 12 h and 1000 ng/mL rhGas6 for 1 h. *P < .05, **P < .01, ***P < .001, according to Student t test
3.5. STAT3 activation played a critical role in the crosstalk between Gas6/AXL and the Hippo‐YAP pathway
To explore how Gas6/AXL promotes YAP activity, several candidates that could inhibit AXL were selected. A positive correlation between STAT3 and YAP mRNA expression was observed in the HNSCC dataset from TCGA database (Figure 7A). Our previous results demonstrated that the YAP inhibitor significantly attenuated STAT3 and AKT phosphorylation (Figure 2G). To explore how Gas6/AXL regulated the Hippo‐YAP pathway, 3 inhibitors of STAT3, AKT, and ERK were used. Only cryptotanshinone, an inhibitor blocking STAT3 phosphorylation at Tyr705, 25 significantly increased p‐YAP expression (Figure 7B), suggesting that STAT3 might be involved in YAP activation. We also testified that Gas6 induced STAT3 and AKT phosphorylation (Figures 7C and S9). We then used si‐STAT3 (Figure S6B) and cryptotanshinone to further explore STAT3 regulation on YAP activation. The data indicated that si‐STAT3 reversed p‐YAP expression, which was decreased by rhGas6 (Figure 7D). Cryptotanshinone also inhibited Gas6 promotion in YAP nuclear translocation (Figure 7E). Moreover, CCN2 and AREG mRNA expression was also attenuated by STAT3 knockdown (Figure 7F). To further explore the effect of STAT3 on the Hippo pathway, we observed that cryptotanshinone blocked Gas6 inhibition on p‐LATS1 and p‐MOB1 (Figure 7G). Overexpression of STAT3Δ705, lacking phosphorylation site at Tyr705, reversed the inhibition in LATS1/2 phosphorylation mediated by MST1 silencing (Figures S6C and S10). We then observed a co‐localization of STAT3 and LATS1 in cytoplasm (Figure S11). Immunoprecipitation experiments revealed that Gas6 induced binding to LATS1 with p‐STAT3, as well as inhibiting the interaction between total STAT3 and LATS1 (Figure 7H,I). In addition, Gas6 dissociated the interaction between LATS1 and other molecules in the Hippo pathway (Figure 7H). The affinity of p‐STAT3 for MST1 and MOB1 was also inhibited by Gas6 (Figure 7J). In addition, binding to LATS1 with p‐STAT3 was inhibited by STAT3Δ705 overexpression (Figure 7K). Moreover, we also found that IL‐6‐induced STAT3 activation promoted YAP nuclear translocation, which could be inhibited by STAT3 silencing (Figures 7L and S12). These data indicated that Gas6 induced a competitive binding to p‐STAT3 with LATS1. The interaction inhibited LATS1 activity, then blocked the Hippo inhibitory signal on YAP, and finally promoted YAP activity. In this study, a mutual regulation between Gas6/AXL and Hippo‐YAP was established in HNSCC progression. Gas6‐induced p‐STAT3 abolished the inhibition of YAP by the Hippo pathway, then promoted YAP dephosphorylation and nuclear translocation to initiate target gene transcription (Figure 7M).
FIGURE 7.
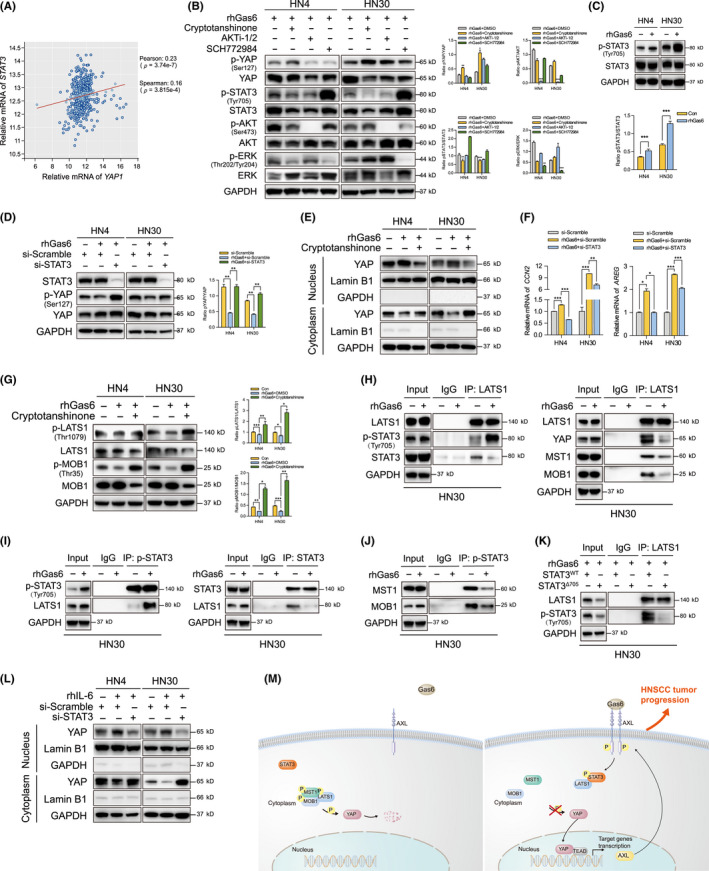
STAT3 activation is involved in the crosstalk between Gas6/AXL and the Hippo‐YAP pathway. A, A correlation between STAT3 and Yes‐associated protein (YAP) mRNA expression in the HNSCC dataset from TCGA database was analyzed. B, P‐YAP was detected after treatment with 5 μmol/L cryptotanshinone, AKTi‐1/2, or SCH772984 (STAT3, AKT or ERK inhibitors, respectively) with 2% FBS for 24 h and 1000 ng/mL rhGas6 for 1 h. C, P‐STAT3 was detected after treatment with 2% FBS for 12 h and 1000 ng/mL rhGas6 for 1 h. D, P‐YAP was measured after si‐STAT3 transfection and 2% FBS treatment for 48 h and 1000 ng/mL rhGas6 for 1 h. E, YAP expression in nucleus and cytoplasm were analyzed after treatment with 5 μmol/L cryptotanshinone with 2% FBS for 24 h and 1000 ng/mL rhGas6 for 1 h. F, Relative mRNA expression of CCN2 and AREG were analyzed after si‐STAT3 transfection and 2% FBS for 48 h and 1000 ng/mL rhGas6 for 1 h. G, P‐LATS1 and p‐MOB1 were assessed after treatment with cryptotanshinone and rhGas6 as described above. H, Immunoprecipitation assay was performed using anti‐LATS1 antibody after treatment with 2% FBS for 12 h and 1000 ng/mL rhGas6 for 1 h. I, J, After 2% FBS and rhGas6 treatment, IP assay was performed using antibodies against p‐STAT3 and STAT3. K, IP assay was performed after STAT3WT or STAT3Δ705 plasmid transfection and 2% FBS treatment for 48 h and 1000 ng/mL rhGas6 for 1 h. L, YAP expression in nucleus and cytoplasm were analyzed after treatment with 2% FBS and 100 ng/mL rhIL‐6 for 24 h. M, A schematic diagram illustrating that Gas6/AXL‐induced STAT3 activation blocked the Hippo inhibitory regulation on YAP by competitive binding to LATS1, which induced YAP dephosphorylation, nuclear translocation, and transcriptional regulation. *P < .05, **P < .01, ***P < .001, according to Student t test
4. DISCUSSION
In this study, we found that Gas6/AXL‐induced STAT3 activation blocked the inhibition by the Hippo pathway via competitive binding to LATS1, and subsequently activated YAP nuclear translocation and transcriptional regulation. This mutual promotion suggested that AXL and YAP jointly derived HNSCC progression. Our study firstly established a non‐transcriptional regulation function of STAT3, mediating the crosstalk between AXL and YAP, which was very useful for revealing HNSCC cancer mechanisms.
Upstream signals of the Hippo‐YAP pathway remain poorly understood. Hippo‐YAP was able to receive soluble factors, biomechanical signals, and metabolic products to regulate cell behavior. 21 , 26 , 27 , 28 Our study indicated that AXL was an upstream regulator that triggered YAP nuclear translocation and initiated transcription of YAP target genes, CCN2 and AREG. 22 , 23 Recent studies also observed that AXL stimulated YAP activity. 29 , 30 We found that YAP was indirectly dephosphorylated at Ser127 by AXL activation, which was different from YAP directly phosphorylation at tyrosine residues in Saab's study. 29 However, the complex regulatory mechanism mediating AXL effect remained obscure. Considering YAP as an effector of the Hippo pathway, we then inquired into the effect of AXL on LATS1 and MOB1, 2 direct inhibitory regulators of YAP. The results showed that Gas6 suppressed p‐LATS1 and p‐MOB1 to block their inhibitory regulation on YAP. We also observed that AXL inhibited p‐MST1 in a LATS1‐independent manner. We speculated that certain kinases, as adaptor proteins, bound to the AXL cellular domain. This anchoring might lead to membrane retention of upstream kinases and/or RASSFs which control MST phosphorylation. 31 Our results indicated that AXL could inhibit the Hippo pathway and induced YAP activity.
Given that YAP is a transcription factor driving oncogenic genes in tumors, 6 we found that AXL, as a target gene of YAP, rescued tumor suppressor phenotypes by YAP silencing in HNSCC growth and invasion, similar to the findings for hepatocellular carcinoma. 32 A previous study found that YAP was a key factor in embryonic development and tissue homeostasis, and that YAP activation was an early event in liver cancer development. 33 , 34 Our experiments in vivo also indicated that YAP exerted a vital effect on tumorigenesis and tumor growth. We further found that AXL was involved in this process to promote HNSCC progression. Therefore, our study revealed a crosstalk between Gas6/AXL and Hippo‐YAP. AXL, as a target gene of YAP, promoted YAP activation. Recent studies also reported the positive feedback loop between other signaling events and the Hippo pathway. 35 , 36 The interaction could reinforce a robust status of YAP, which promoted tumor progression via transcriptionally activating more oncogenes. Our results indicated that the mutual promotion between YAP and AXL, both as oncoproteins, further exacerbated tumor progression, accounting for our observation that HNSCC patients with co‐expression of YAP and AXL had a worse prognosis.
AXL is a critical RTK in tumor growth, invasion, and therapeutic resistance. 37 , 38 JAK/STAT3, PI3K/AKT and MEK/ERK signaling pathways have been reported as AXL main downstream functions. 18 , 19 Our inhibitors screening demonstrated that AXL‐induced YAP dephosphorylation in a STAT3‐dependent manner. JAK/STAT3 signaling participates in several physiological processes. 39 Cytoplasmic STAT3 is also involved in non‐transcriptional mechanisms. 40 Our results firstly demonstrated that STAT3 was a key determinant in AXL‐mediated YAP activation. A novel mechanism was revealed in inhibiting p‐LATS1 and binding to LATS1. We observed that p‐STAT3 competitively bound to LATS1 and could block Hippo inhibitory regulation on YAP, which ultimately promoted YAP activation.
In conclusion, Gas6/AXL‐induced STAT3 activation and then competitively bound to LATS1, which blocked Hippo inhibitory regulation on YAP and induced YAP nuclear translocation and transcriptional regulation. Our study suggested that a novel regulation of STAT3 activation mediated the crosstalk between Gas6/AXL and Hippo/YAP in carcinogenesis and might be an intriguing therapeutic target in HNSCC.
DISCLOSURE
The authors declare that they have no conflict of interest.
ETHICAL APPROVAL
This study was approved by Ethics Committee of the Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine.
Supporting information
Figures S1–S12
Tables S1–S3
Document S1
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (81902747 and 81430012) and the Shanghai Sailing Program (19YF1427000).
Li J, Shi C, Zhou R., et al. The crosstalk between AXL and YAP promotes tumor progression through STAT3 activation in head and neck squamous cell carcinoma. Cancer Sci. 2020;111:3222–3235. 10.1111/cas.14546
Jiayi Li and Chaoji Shi contributed equally to this work.
Contributor Information
Hailong Ma, Email: mahl21@sjtu.edu.cn.
Zhiyuan Zhang, Email: zhangzy1650@sh9hospital.org.
REFERENCES
- 1. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. 2018;18:269‐282. [DOI] [PubMed] [Google Scholar]
- 2. Casaletto JB, McClatchey AI. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer. 2012;12:387‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sacco AG, Cohen EE. Current treatment options for recurrent or metastatic head and neck squamous cell carcinoma. J Clin Oncol. 2015;33:3305‐3313. [DOI] [PubMed] [Google Scholar]
- 4. Adelstein D, Gillison ML, Pfister DG, et al. NCCN guidelines insights: head and neck cancers, version 2.2017. J Natl Compr Canc Netw. 2017;15(6):761‐770. [DOI] [PubMed] [Google Scholar]
- 5. Vermorken JB, Trigo J, Hitt R., et al. uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum‐based therapy. J Clin Oncol. 2007;25:2171‐2177. [DOI] [PubMed] [Google Scholar]
- 6. Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15:73‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zanconato F, Cordenonsi M, Piccolo S. YAP and TAZ: a signalling hub of the tumour microenvironment. Nat Rev Cancer. 2019;19:454‐464. [DOI] [PubMed] [Google Scholar]
- 8. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13:63‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Segrelles C, Paramio JM, Lorz C. The transcriptional co‐activator YAP: a new player in head and neck cancer. Oral Oncol. 2018;86:25‐32. [DOI] [PubMed] [Google Scholar]
- 11. Li C, Wang S, Xing Z, et al. A ROR1‐HER3‐lncRNA signalling axis modulates the Hippo‐YAP pathway to regulate bone metastasis. Nat Cell Biol. 2017;19:106‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3‐kinase and phosphoinositide‐dependent kinase‐1. Proc Natl Acad Sci U S A. 2013;110:2569‐2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brand TM, Iida M, Stein AP, et al. AXL is a logical molecular target in head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21:2601‐2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu B, Wu K, Wang X., et al. Periostin secreted by cancer‐associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 2018;9:1082. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15. Yang HL, Tsai YC, Korivi M., et al. Lucidone promotes the cutaneous wound healing process via activation of the PI3K/AKT, Wnt/beta‐catenin and NF‐kappaB Signaling Pathways. Biochim Biophys Acta Mol Cell Res. 1864;2017:151‐168. [DOI] [PubMed] [Google Scholar]
- 16. Jin S, Yang X, Li J, et al. p53‐targeted lincRNA‐p21 acts as a tumor suppressor by inhibiting JAK2/STAT3 signaling pathways in head and neck squamous cell carcinoma. Mol Cancer. 2019;18:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Camp RL, Dolled‐Filhart M, Rimm DL. X‐tile: a new bio‐informatics tool for biomarker assessment and outcome‐based cut‐point optimization. Clin Cancer Res. 2004;10:7252‐7259. [DOI] [PubMed] [Google Scholar]
- 18. Wu G, Ma Z, Hu W, et al. Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 2017;8(3):e2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer. 2017;116:415‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu FX, Zhao B, Panupinthu N, et al. Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell. 2012;150:780‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Y, Huang T, Cheng AS, et al. The TEAD family and its oncogenic role in promoting tumorigenesis Int J Mol Sci. 2016;17:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin KC, Park HW, Guan KL. Regulation of the Hippo Pathway Transcription Factor TEAD. Trends Biochem Sci. 2017;42:862‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J, Ye L, Li Q., et al. Synaptopodin‐2 suppresses metastasis of triple‐negative breast cancer via inhibition of YAP/TAZ activity. J Pathol. 2018;244:71‐83. [DOI] [PubMed] [Google Scholar]
- 25. Hu F, Zhao Y, Yu Y., et al. Docetaxel‐mediated autophagy promotes chemoresistance in castration‐resistant prostate cancer cells by inhibiting STAT3. Cancer Lett. 2018;416:24‐30. [DOI] [PubMed] [Google Scholar]
- 26. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20:888‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elosegui‐Artola A, Andreu I, Beedle AEM, et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell. 2017;171(6):1397‐1410. e1314. [DOI] [PubMed] [Google Scholar]
- 28. Santinon G, Pocaterra A, Dupont S. Control of YAP/TAZ Activity by Metabolic and Nutrient‐Sensing Pathways. Trends Cell Biol. 2016;26:289‐299. [DOI] [PubMed] [Google Scholar]
- 29. Saab S, Chang OS, Nagaoka K., et al. The potential role of YAP in Axl‐mediated resistance to EGFR tyrosine kinase inhibitors. Am J Cancer Res. 2019;9:2719‐2729. [PMC free article] [PubMed] [Google Scholar]
- 30. Azad T, Nouri K, Janse van Rensburg HJ., et al. A gain‐of‐functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene. 2020;39:334‐355. [DOI] [PubMed] [Google Scholar]
- 31. Ma S, Meng Z, Chen R., et al. The hippo pathway: biology and pathophysiology. Annu Rev Biochem. 2019;88:577‐604. [DOI] [PubMed] [Google Scholar]
- 32. Xu MZ, Chan SW, Liu AM., et al. AXL receptor kinase is a mediator of YAP‐dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2011;30:1229‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perra A, Kowalik MA, Ghiso E., et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J Hepatol. 2014;61:1088‐1096. [DOI] [PubMed] [Google Scholar]
- 35. Gill MK, Christova T, Zhang YY., et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun. 2018;9:3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weiler SME, Lutz T, Bissinger M., et al. TAZ target gene ITGAV regulates invasion and feeds back positively on YAP and TAZ in liver cancer cells. Cancer Lett. 2020;473:164‐175. [DOI] [PubMed] [Google Scholar]
- 37. Elkabets M, Pazarentzos E, Juric D., et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27:533‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dunne PD, McArt DG, Blayney JK., et al. AXL is a key regulator of inherent and chemotherapy‐induced invasion and predicts a poor clinical outcome in early‐stage colon cancer. Clin Cancer Res. 2014;20:164‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Graham DK, DeRyckere D, Davies KD., et al. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14:769‐785. [DOI] [PubMed] [Google Scholar]
- 40. Huynh J, Chand A, Gough D., et al. Therapeutically exploiting STAT3 activity in cancer ‐ using tissue repair as a road map. Nat Rev Cancer. 2019;19:82‐96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S12
Tables S1–S3
Document S1