

Abstract
Fibroblast growth factor receptor 4 (FGFR4) is known to induce cancer cell proliferation, invasion, and antiapoptosis through activation of RAS/RAF/ERK and PI3K/AKT pathways, which are also known as major molecular bases of colon cancer carcinogenesis related with epidermal growth factor receptor (EGFR) signaling. However, the interaction between FGFR4 and EGFR signaling in regard to colon cancer progression is unclear. Here, we investigated a potential cross‐talk between FGFR4 and EGFR, and the effect of anti‐EGFR therapy in colon cancer treatment. To explore the biological roles of FGFR4 in cancer progression, RNA sequencing was carried out using FGFR4 transfected colon cell lines. Gene ontology data showed the upregulation of genes related to EGFR signaling, and we identified that FGFR4 overexpression secretes EGFR ligands such as amphiregulin (AREG) with consequent activation of EGFR and ErbB3. This result was also shown in in vivo study and the cooperative interaction between EGFR and FGFR4 promoted tumor growth. In addition, FGFR4 overexpression reduced cetuximab‐induced cytotoxicity and the combination of FGFR4 inhibitor (BLU9931) and cetuximab showed profound antitumor effect compared to cetuximab alone. Clinically, we found the positive correlation between FGFR4 and AREG expression in tumor tissue, but not in normal tissue, from colon cancer patients and these expressions were significantly correlated with poor overall survival in patients treated with cetuximab. Therefore, our results provide the novel mechanism of FGFR4 in connection with EGFR activation and the combination of FGFR4 inhibitor and cetuximab could be a promising therapeutic option to achieve the optimal response to anti‐EGFR therapy in colon cancer.
Keywords: AREG, cetuximab, colon cancer, EGFR, FGFR4
Our study has characterized the cross‐talk between fibroblast growth factor receptor 4 (FGFR4) and epidermal growth factor receptor (EGFR)/ErbB3 signaling by the contribution of EGFR ligands secreted from FGFR4. These findings provide experimental evidence for combined treatment with FGFR4 inhibitor and anti‐EGFR therapy in colon cancer.

1. INTRODUCTION
Although the overall survival time has been prolonged by virtue of targeted agents, colon cancer is still the second leading cause of cancer‐related mortality worldwide. 1 Comprehensive molecular studies revealed common pathways of colon carcinogenesis and retrospective analyses of clinical features and molecular findings proposed consensus molecular subtyping of colon cancers to predict survival outcomes. 2 Despite these efforts, targeted therapies as current standards for colon cancer are limited to antiangiogenic (anti‐vascular endothelial growth factor) and anti‐epidermal growth factor receptor Abs (anti‐EGFR/ErbB1/HER1) such as cetuximab. In particular, anti‐EGFR therapy is used only in patients with WT KRAS, NRAS, or BRAF genes to achieve an optimal response, and even if selected as biomarkers, a quarter of them do not respond to anti‐EGFR therapy. 3 , 4 , 5 Hence, mapping the resistance mechanisms associated with targeted therapies or discovering new targets is critical to improve cancer treatment.
Epidermal growth factor receptor is a transmembrane receptor tyrosine kinases (RTK) and a member of the erythroblastic leukemia viral oncogene homolog (ErbB) family, which includes ErbB1 (EGFR), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). 6 Epidermal growth factor receptor plays an important role in various cellular functions such as cell proliferation, differentiation, migration, and antiapoptosis of breast, head and neck, ovary, lung, and colon cancer cells. 7 Upon binding with ligands, EGFR forms homo‐ or heterodimers with MET, insulin‐like growth factor type 1 receptor (IGF‐1R), and platelet‐derived growth factor receptor (PDGFR), as well as with other members of ErbB receptors. 8 , 9 , 10 , 11 , 12 Epidermal growth factor receptor dimerization is a very important step in the activation of downstream signaling pathways such as RAS/RAF/ERK and PI3K/AKT and resistance to EGFR‐targeted therapies such as cetuximab and erlotinib. 13 , 14 , 15 Therefore, in order to overcome the resistance to these targeted therapies, we urgently need to further understand the relationship between EGFR and its dimerization partners.
Fibroblast growth factor receptors (FGFRs) consist of a family of 4 highly conserved transmembrane receptor tyrosine kinases, FGFR1‐4, and FGFR5 (also known as FGFRL1) that has the ability to bind fibroblast growth factor ligands but lacks an intracellular kinase domain. 16 Of these, FGFR4 has recently received a great deal of attention – the overexpression of FGFR4 has been associated with cancer metastasis, poor survival outcomes in stomach, lung, colon, and breast cancers. 17 , 18 , 19 , 20 , 21 Previously, we also reported that FGFR4 promoted colon cancer cell proliferation and epithelial‐mesenchymal transition showing poor prognosis. 22 In addition, FGFR4 can decrease response to chemotherapy, such as 5‐fluorouracil and oxaliplatin, by increasing caspase‐dependent apoptosis in colon cancer. 21
Like EGFR, FGFR4 forms homo‐ and heterodimers with other RTKs to activate. Recently, the direct interaction between EGFR and FGFR4 was reported in FGFR4‐ and EGFR‐expressing lung cancer cells. 23 However, the mechanism of cross‐talk between these 2 signalings in colon cancer have not been clearly established. In this study, we investigated the interactions between FGFR4 and EGFR signaling, which is a key pathway in colon cancer progression, and examined whether FGFR4 might play a role in the response of anti‐EGFR therapy in colon cancer treatment.
2. MATERIALS AND METHODS
2.1. Cell culture
Human colon cancer cell lines HT29, SW1116, HCT116, and SW480 were purchased from ATCC. SNU‐C4 cells were purchased from Korean Cell Line Bank. All cells were cultured according to the manufacturer’s recommended media and protocols.
2.2. Clinical colon cancer biospecimens
Patients who underwent surgical resection and patients who received chemotherapy with cetuximab were identified through an institutional database. The biospecimens were provided by the Biobank of Chonnam National University Hwasun Hospital, a member of the Korea Biobank Network, with informed consent. This study was approved by the Chonnam National University Hwasun Hospital Institutional Review Board (approval number: IRB CNUHH‐2018‐173) and undertaken in accordance with the Declaration of Helsinki. For study of FGFR4 or amphiregulin (AREG) mRNA expression, 150 colon tumor and normal tissue samples were used from resected colon cancer diagnosed as stage II or III. To evaluate the relationship between FGFR4 or AREG expression and anti‐EGFR therapy response, retrospective analysis was carried out in patients who received cetuximab combination therapy for metastatic colorectal cancer. Immunohistochemical stains for FGFR4 and AREG for these patients were examined using tissue at the time of diagnosis and survival outcome was calculated by overall survival (OS).
2.3. Process of RNA sequencing analysis
RNA sequencing (RNA‐seq) data in FASTQ format were generated by an Illumina sequencer with 100 bp read length in paired‐end mode. The quality of reads was checked with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Overrepresented sequences in the QC result and low‐quality reads under Q20 were eliminated by Cutadapt, an adapter trimming program. 24 After that, only high‐quality reads were aligned to the hg19 version of the human reference genome using STAR aligner with default options. 25 Gene expression levels were calculated in consideration of replications in 2 groups, control and HT29/FGFR4 cells, using Cuffdiff of Cufflinks. 26 Scatter plots were drawn using CummeRBund R package designed for visualization of Cufflinks results. 27 Gene ontology (GO) analysis was carried out using DAVID to find highly matched GO terms with differentially expressed genes (DEGs). 28 Gene Set Enrichment Analysis was used to evaluate statistical enrichment scores with gene expression levels in each functional gene cluster. 29
2.4. Quantitative RT‐PCR
Total RNA was isolated using Hybrid‐R (GeneAll Biotechnology). Reverse transcription and quantitative PCR (qPCR) were carried out as described previously. 30 The following primers were used: FGFR4, forward 5′‐ CGTTGATGACGATGTGCTTC‐3′ and reverse 5′‐GCTGGCTTAAGGATGGACAG‐3′; AREG, forward 5′‐ CCAAAACAAGACGGAAAGTGA‐3′ and reverse 5′‐AGGATCACAGCAGACATAAAG‐3′; betacellulin (BTC), forward 5′‐CTTCACTGTGTGGTGGCAGATG‐3′ and reverse 5′‐ATGCAGTAATGCTTGTATTGCTTGG‐3′; EGF, forward 5′‐CCACACCAAACAAGGAGGAG‐3′ and reverse 5′‐ATGAGAAGCCCCACGATGAC‐3′; epigen (EPGN), forward 5′‐TTTGGGAGTTCCAATATCAGC‐3′ and reverse 5′‐TGTGATTGGAGGTGTTACAGTCA‐3′; epiregulin (EREG), forward 5′‐TACTGCAGGTGTGAAGTGGG‐3′ and reverse 5′‐TGGAACCGACGACTGTGATA‐3′; heparin‐binding EGF‐like growth factor (HBEGF), forward 5′‐ CCACACCAAACAAGGAGGAG‐3′ and reverse 5′‐ATGAGAAGCCCCACGATGAC‐3′; neuregulin 1 (NRG1), forward 5′‐AACGAGAAAGATATCAATGG‐3′ and reverse 5′‐ ACTACAAGCAAACAGAGGG‐3′; transforming growth factor‐α (TGF‐α), forward 5′‐GAGTGCAGACCCGCCCGTGGC‐3′ and reverse 5′‐CCAGGAGGTCCGCATGCTCAC‐3′; and β‐actin, forward 5′‐AGAAAATCTGGCACCACACC‐3′ and reverse 5′‐AGGAAGGAAGGCTGGAAGAG‐3′.
2.5. Statistical analysis
Categorical variables were compared by the χ2 test or Fisher’s exact test, and continuous variables were compared using the independent‐samples t test. Overall survival was calculated from the diagnosis of disease to death from any cause, and patients who were alive at the last follow‐up were recorded at that time. Overall survival was calculated using the Kaplan‐Meier method, and comparisons were made using log‐rank tests. Statistical analysis was undertaken using the SPSS 12.0 software package. Differences with P values less than .05 were considered statistically significant.
2.6. Online supporting methods
The detailed procedures for FGFR4 construction and transfection, western blotting, human phospho‐RTK array, immunoprecipitation, preparation and treatment of conditioned medium (CM), AREG ELISA, xenograft studies, cell viability and apoptosis assays, and immunohistochemistry are described in Data S1.
3. RESULTS
3.1. Fibroblast growth factor receptor 4 activates PI3K/AKT and RAS/RAF/ERK pathways in colon cancer cells
To explore the biological roles of FGFR4 in cancer progression, an FGFR4 expression plasmid was constructed and stably transfected into HT29 colon cancer cell lines. The FGFR4 expression was successfully upregulated in HT29 cells with no effects on the expression of other FGFRs (Figure S1A). As most RTKs, including FGFRs, facilitate similar signal pathways, such as PI3K/AKT and RAS/RAF/ERK, we next analyzed the activation of these targets and FGFR‐specific substrate 2 (FRS2) in FGFR4‐transfected cells by western blotting (Figure S1B). The phosphorylation of FRS2 was significantly increased in FGFR4‐transfected cells compared to vector control cells. Furthermore, we found activation of downstream signaling targets, including pAKT and pERK, in FGFR4‐transfected cells. These results indicated that FGFR4 expression in colon cancer cells activates downstream signaling of PI3K/AKT and RAS/RAF/ERK, which are crucial pathways in colon carcinogenesis.
3.2. Epidermal growth factor receptor signaling is activated by FGFR4
To investigate the differences in comprehensive signaling gene expression by FGFR4 in colon cancer cells, gene expression profiling was carried out using RNA‐seq. We found changes in the expression of 1777 genes (980 upregulated and 797 downregulated) in FGFR4‐transfected HT29 cells compared to vector control cells (Figure 1A). Gene ontology analysis revealed that DEGs altered by the expression of FGFR4 were related to biological functions such as cell proliferation, macroautophagy, wound healing, and angiogenesis (Figure 1B). Intriguingly, the gene set of EGFR signaling also showed altered expression patterns in FGFR4‐transfected HT29 cells. Gene Set Enrichment Analysis revealed a significant enrichment of gene sets belonging to EGFR signaling (Figure 1C). The heatmap for detailed enriched genes on EGFR signaling are listed in Figure S2.
FIGURE 1.
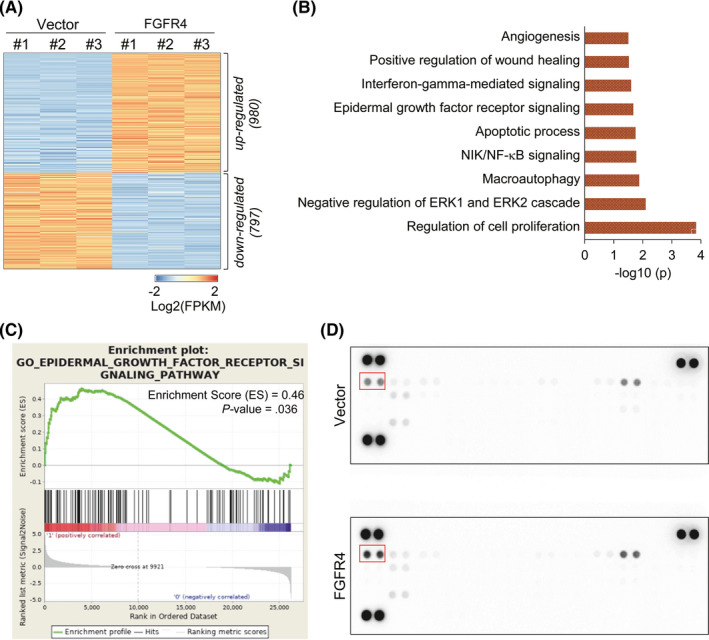
Epidermal growth factor receptor (EGFR) signaling is activated by fibroblast growth factor receptor 4 (FGFR4) in colon cancer cells. A, Heatmap of differentially expressed genes (DEGs) from RNA‐seq analysis of FGFR4‐transfected and vector control HT29 cells. DEGs were defined over 3 fragments per kilobase of transcript per million mapped reads (FPKM) at any group and fold change over 1.3. B, Gene ontology analysis of upregulated genes in FGFR4‐transfected cells. C, Gene Set Enrichment Analysis shown for genes related to EGFR signaling. P value and ES are shown on the graph. D, Effect of FGFR4 expression on the tyrosine phosphorylation of 49 different receptor tyrosine kinases (RTKs) was evaluated by phospho‐RTK array. The position of pEGFR is indicated with a red box
3.3. Fibroblast growth factor receptor 4 promotes phosphorylation of EGFR and ErbB3
To determine the effect of FGFR4 expression on activation of other RTKs, including EGFR, which showed a significant change in RNA‐seq results, phosphorylation of RTKs was measured using a human phospho‐RTK array. We found that the level of tyrosine phosphorylation of EGFR was highly increased in stable FGFR4‐transfected HT29 cells, compared to vector control cells (Figure 1D). To validate this result, the phosphorylation statuses and levels of ErbB family members were assessed by western blotting. The results showed not only pEGFR but also pErbB3 were significantly increased in stable FGFR4‐transfected HT29 cells (Figure 2A). In the RTK array, pErbB3 was not changed. It could be explained that many RTKs readily form homo‐ or heterodimers with other RTKs, and their dimerization is often indispensable in their kinase activities. Thus, we carried out immunoprecipitation with FGFR4 Ab to determine whether FGFR4 forms heterodimeric complexes with EGFR and/or ErbB3 and evaluated its involvement in their phosphorylation and transactivation in colon cancer cells. We found that FGFR4 did not bind to EGFR or ErbB3 directly, although levels of phosphorylated EGFR and ErbB3 were markedly increased in stable FGFR4‐transfected HT29 cells (Figure 2B). This result suggests that EGFR and ErbB3 could be activated by external stimulation, such as ligands. To assess the possibility that FGFR4 induces the expression and secretion of EGFR or ErbB3 ligands, parental colon cancer cell lines were treated with CM from stable FGFR4‐transfected (FGFR4‐CM) or vector control (VC‐CM) HT29 cells. After 1 hour of treatment, the phosphorylation of EGFR and ErbB3 was significantly elevated in FGFR4‐CM treated cells compared to serum‐free medium (SFM) and VC‐CM treated cells. In addition, the phosphorylation levels of their downstream effector molecules, such as ERK and AKT, were increased in FGFR4‐CM treated cells (Figure 2C). This result indicates that FGFR4 induced the expression of ErbB family ligands.
FIGURE 2.
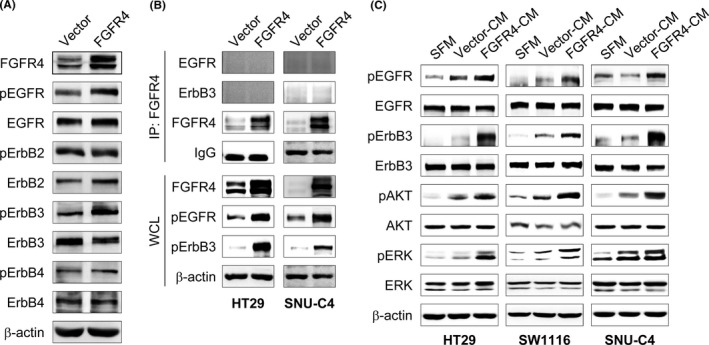
Fibroblast growth factor receptor 4 (FGFR4) promotes phosphorylation of epidermal growth factor receptor (EGFR) and erythroblastic leukemia viral oncogene homolog ErbB3. A, Phosphorylation of ErbB family members in stable FGFR4‐transfected HT29 cells were confirmed by western blotting. B, Whole‐cell lysates (WCL) were extracted from FGFR4‐transfected and vector control HT29 or SNU‐C4 cells and immunoprecipitated (IP) with FGFR4 Ab. The interaction was detected by western blotting with Abs of FGFR4 or ErbB family members. Immunoprecipitation with IgG was used as controls. C, Three parental colon cancer cells were incubated with conditioned medium (CM) obtained from FGFR4‐transfected (FGFR4‐CM) and vector control (Vector‐CM) HT29 cells. Serum‐free medium (SFM) was used as a negative control. After 1 h of incubation, cell lysates were prepared and subjected to western blotting with Abs to the indicated proteins. β‐Actin was used as a loading control
3.4. Fibroblast growth factor 4 induces AREG expression
To investigate which ligands of the ErbB family are associated with FGFR4 expression, we first reanalyzed the expression of 8 ErbB family ligands (AREG, BTC, EGF, EPGN, EREG, HBEGF, NRG1, and TGF‐α) using RNA‐seq data. We found that AREG, BTC, EREG, and HBEGF were markedly expressed in stable FGFR4‐transfected HT29 cells compared to vector control cells (Figure S3). Epidermal growth factor expression was decreased in stable FGFR4‐transfected HT29 cells, and EPGN and TGF‐α expressions were not changed. In addition, NRG1, which is known as Erb3 ligand, was not detected in these cells. Similar results were observed in RT‐qPCR analyses of ErbB family ligands (Figure 3A). To verify these RNA‐seq and RT‐qPCR results at the protein level, we examined the expression of elevated ErbB ligands by western blotting. Fibroblast growth factor receptor 4 signaling increased AREG, BTC, EREG, and HBEGF expressions and induced the phosphorylation of EGFR (Figure 3B). On the basis of the significant increase in both mRNA and protein levels of AREG compared to other ligands, we selected AREG for further studies. Using ELISA assay, we confirmed that FGFR4 signaling significantly induced AREG secretion (Figure 3C). Moreover, when cells were stimulated with FGFR4‐specific ligand FGF19, the expression of AREG was increased and the activation of EGFR, ErbB3, AKT, and ERK was also observed in FGF19 stimulated cells (Figure S4). These results show that FGFR4 activation results in AREG‐mediated EGFR activation.
FIGURE 3.
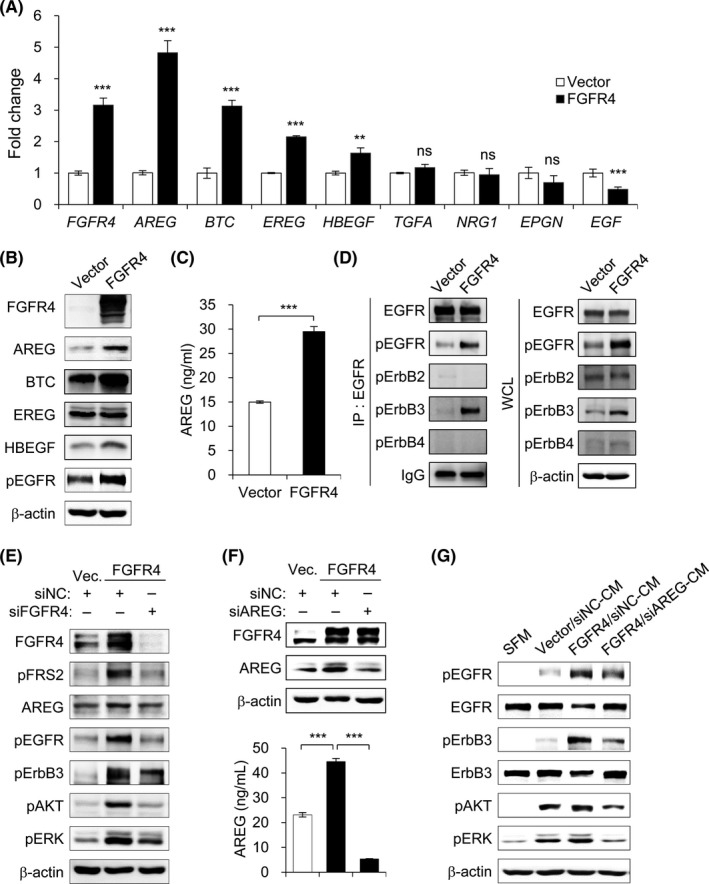
Fibroblast growth factor receptor 4 (FGFR4) induces amphiregulin (AREG) expression and then activates epidermal growth factor receptor (EGFR) signaling. A, mRNA expression levels of erythroblastic leukemia viral oncogene homolog (ErbB) family ligands were measured by RT‐quantitative PCR. Fold changes were calculated after normalization with β‐actin. B, Cell lysates of FGFR4‐transfected and vector control HT29 cells were prepared and subjected to western blotting with Abs to the indicated proteins. C, AREG level in culture supernatants of vector control and FGFR4‐transfected HT29 cells was measured by ELISA. D, Whole‐cell lysates (WCL) were extracted form FGFR4‐transfected and vector control HT29 cells and immunoprecipitated (IP) with EGFR Ab. The interaction was detected by western blotting with Abs for ErbB family members. Immunoprecipitation with IgG was used as controls. Expression levels of ErbB family members were confirmed by western blotting of WCL. E, Stable FGFR4‐transfected HT29 cells were treated with siFGFR4 or negative control (siNC). After 48 h incubation, cell lysates were prepared and subjected to western blotting with Abs to the indicated proteins. F, Stable FGFR4‐transfected HT29 cells were treated with siAREG or siNC. After 48 h incubation, AREG expression in cell lysates and culture supernatants were measured by western blotting and ELISA. G, Parental HT29 cells were incubated with serum‐free medium (SFM) or conditioned medium (CM) obtained from siNC (siNC‐CM) and siAREG treated (siAREG‐CM) HT29 cells. After 1 h of incubation, cell lysates were prepared and subjected to western blotting with Abs to the indicated proteins. β‐Actin was used as loading control. Data are presented as mean ± SD. **P < .01, ***P < .001. BTC, betacellulin; EGF, epidermal growth factor; EPGN, epigen; EREG, epiregulin; HBEGF, heparin‐binding EGF‐like growth factor; NRG1, neuregulin 1; ns, not significant; TGFA, transforming growth factor receptor‐α
Increase in EGFR ligands, such as AREG, induces the phosphorylation of EGFR and stimulates its homo‐ and/or heterodimerization with other ErbB receptors, resulting in the activation of downstream PI3K/AKT and RAS/RAF/ERK pathways. Neuregulin 1, an ErbB3 and ErbB4 ligand, did not increase in FGFR4‐transfected HT29 cells compared to vector control cells, despite the elevated phosphorylation of ErbB3. This suggests that ErbB3 might be simultaneously phosphorylated by binding with EGFR, which is activated by its ligands. To verify whether EGFR forms a heterodimeric complex with ErbB3 to induce its transphosphorylation and activation, immunoprecipitation assays were undertaken with EGFR Ab (Figure 3D). Phosphorylated EGFR and ErbB3 were significantly increased in the precipitates of FGFR4‐transfected HT29 cells compared to those of vector control cells, but ErbB2 and ErbB4 were not immunoprecipitated with EGFR from both HT29 cell lysates. These results suggest that FGFR4 activates EGFR and ErbB3 signaling through the induction of EGFR ligands.
3.5. Activation of EGFR abrogated by inhibition of FGFR4‐induced AREG expression
We further examined the effects of the lack of FGFR4 expression on EGFR ligand‐mediated EGFR and ErbB3 activation. We reduced FGFR4 expression in HT29 cells using FGFR4‐specific siRNAs (Figure 3E). Silencing the expression of FGFR4 led to a significant decrease in FRS2 phosphorylation, AREG expression, and EGFR and ErbB3 phosphorylation, with consequent decrease in activated downstream PI3K/AKT and RAS/RAF/ERK pathways. To examine whether the role of FGFR4 on the activation of EGFR and ErbB3 signaling is mediated by AREG, we treated FGFR4‐transfected and vector control HT29 cells with siAREG. The FGFR4 expression of FGFR4‐transfected HT29 cells was not altered after treatment with siAREG. We confirmed that the expression of AREG in cell lysate and culture media from FGFR4‐transfected HT29 cells was significantly decreased by the treatment with siAREG compared to negative control (siNC) (Figure 3F). Parental HT29 cells were also treated with CM from FGFR4‐transfected HT29 cells with siAREG (siAREG‐CM) or siNC (siNC‐CM). Phosphorylation of EGFR and ErbB3 was significantly elevated in FGFR4/siNC‐CM treated cells compared to SFM and vector/siNC‐CM treated cells. In addition, the previously observed increase in phosphorylation of their downstream effectors AKT and ERK was successfully maintained in FGFR4/siNC‐CM treated cells (Figure 3G). Conversely, knockdown of AREG in FGFR4‐transfected HT29 cells (FGFR4/siAREG‐CM) significantly decreased the levels of EGFR, ErbB3, AKT, and ERK phosphorylation. Together, these results confirm that the expression of EGFR ligands, such as AREG, is indispensable for FGFR4‐mediated EGFR activation.
3.6. Fibroblast growth factor receptor 4 promotes in vivo tumor growth of colon cancer cells
To determine the effect of FGFR4 in vivo, stable FGFR4‐transfected HCT116 cells were established and injected s.c. into nude mice. As expected, FGFR4 expression induced a marked promotion of tumor growth (Figure 4A). Following resection, tumors from stable FGFR4‐transfected xenografts weighed significantly more than the vector control counterparts (Figure 4B). To verify that FGFR4 promotes tumor growth by activating EGFR and ErbB3 signaling, we examined the expression of FGFR4 signaling molecules in xenograft tumor lysates. As expected, FRS2 phosphorylation and AREG expression were markedly elevated in stable FGFR4‐transfected xenograft tumors compared to vector control xenograft tumors. In addition, EGFR, ErbB3, AKT, and ERK phosphorylation was consequently increased in stable FGFR4‐transfected xenograft tumors (Figure 4C). Taken together, the results from these in vitro and in vivo studies showed that FGFR4 induced the expression of AREG to promote EGFR and ErbB3 activation, leading to cell proliferation in colon cancer (Figure 4D).
FIGURE 4.
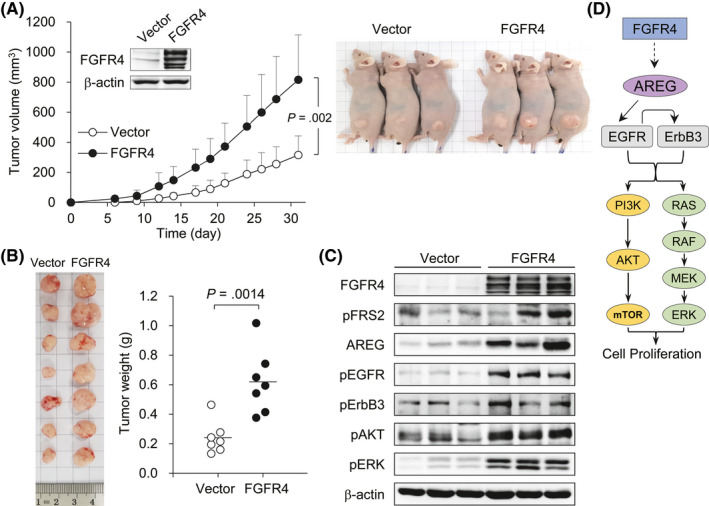
Fibroblast growth factor receptor 4 (FGFR4) induces in vivo growth of colon cancer cells. A, Stable FGFR4‐transfected (n = 7) or vector control (n = 7) HCT116 cells were s.c. injected into nude mice. After their injection, the lengths and widths of the xenograft tumors were measured individually every 2 or 3 days. Data are presented as mean ± SD. B, At day 31 from tumor injection, the mice were killed, and the implanted tumors were resected and weighed. Vertical lines indicate the mean values. C, Cell lysates of FGFR4‐transfected and vector control xenograft tumors were prepared and subjected to western blotting with Abs to the indicated proteins. β‐Actin was used as a loading control. D, A model of the boosting effect of FGFR4 on epidermal growth factor receptor (EGFR) and erythroblastic leukemia viral oncogene homolog (ErbB3) activation, leading to colon cancer cell proliferation. AREG, amphiregulin; FRS2, FGFR‐specific substrate 2
3.7. AREG expression is positively correlated with FGFR4 expression in colon cancer
As a next step, we validated our results in clinical samples to assess the observed correlation between FGFR4 and AREG in colon cancer. The levels of FGFR4 and AREG mRNA expression were analyzed in cancer and paired normal tissues from 150 resected stage II or III colon cancer specimens, using RT‐qPCR. The expression levels of FGFR4 and AREG mRNA in the cancer tissues were significantly higher than in normal tissues (Figure 5A). Furthermore, there was a significant positive correlation between the levels of FGFR4 and AREG mRNA expression in colon cancer tissues, but not in normal tissues (Figure 5B). This result suggests that FGFR4 expression is closely associated with AREG expression in colon cancer progression and could enhance EGFR signaling.
FIGURE 5.
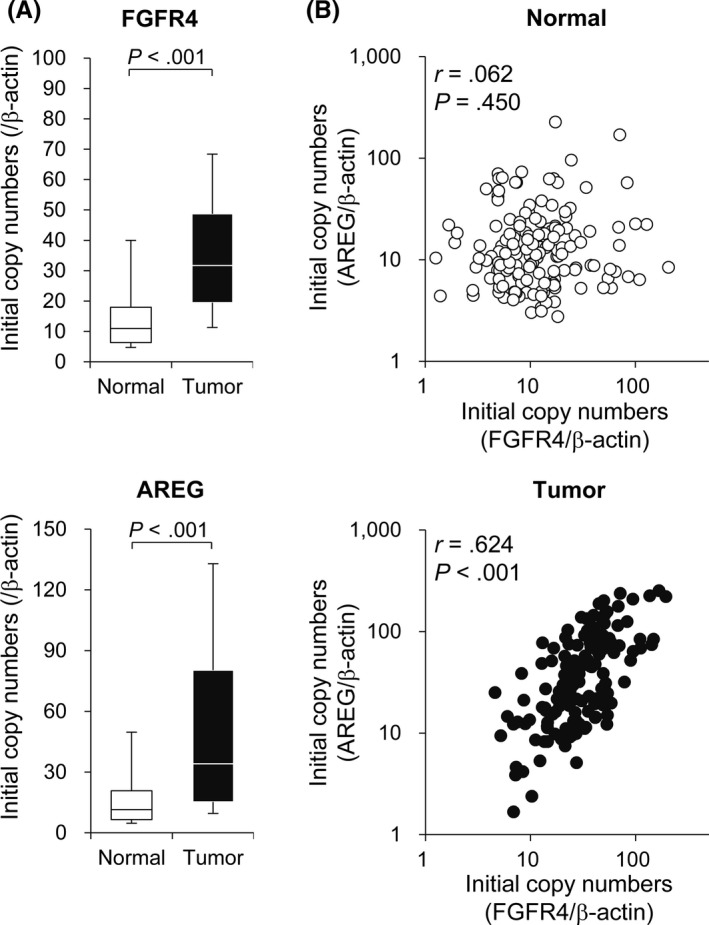
FGFR4 and AREG expression is positively correlated in human colon cancer. A, FGFR4 and AREG mRNA expression in colon cancer and adjacent normal tissues were analyzed by RT‐quantitative PCR. Initial copy numbers were calculated after normalization with β‐actin. In box graphs, the top and bottom of each box represent the first and third quartiles, and the solid line within the box represents the median value. Error bars represent 95% confidence intervals. B, Correlation analysis between FGFR4 and AREG expression in colon cancer and adjacent normal tissues was undertaken using Pearson’s correlation test. Statistical significance was assessed by Pearson’s correlation coefficient (r) and Student’s t test (P value)
3.8. Fibroblast growth factor receptor 4 attenuates response to cetuximab in colon cancer cells
Upregulation of EGFR activity promotes a multitude of carcinogenic processes, including cell proliferation, motility, adhesion, angiogenesis, and metastasis. Epidermal growth factor receptor has been a good therapeutic target, as previously mentioned, and the expression of EGFR ligands is closely associated with EGFR activation and response to EGFR inhibition. 31 Thus, we evaluated the effect of FGFR4 in cetuximab (anti‐EGFR Ab) response in colon cancer. The FGFR4‐transfected and vector control HT29, SW480, SW1116, and SNU‐C4 cells were treated with cetuximab and cell viability was measured to assess response to cetuximab, relative to FGFR4 expression in colon cancer cells. HT29 and SW480 cells are more sensitive to cetuximab than SNU‐C4 and SW116 cells. However, FGFR4 expression reduced cell cytotoxicity at each concentration of cetuximab compared to vector control cells regardless of cell lines (Figure 6A). Further, the cell cytotoxicity was increased dose‐dependently following treatments with the FGFR4‐specific inhibitor BLU9931 (Figure 6B) and additive effects were observed in cells treated with the combination of cetuximab and BLU9931 (Figure 6C). These findings were also represented by FACS analysis in FGFR4‐transfected SNU‐C4 cells treated with cetuximab and/or BLU9931 (Figure S5). Our results suggest that FGFR4 inhibitor has an antitumor effect and it can additively enhance the effect of cetuximab in colon cancer.
FIGURE 6.
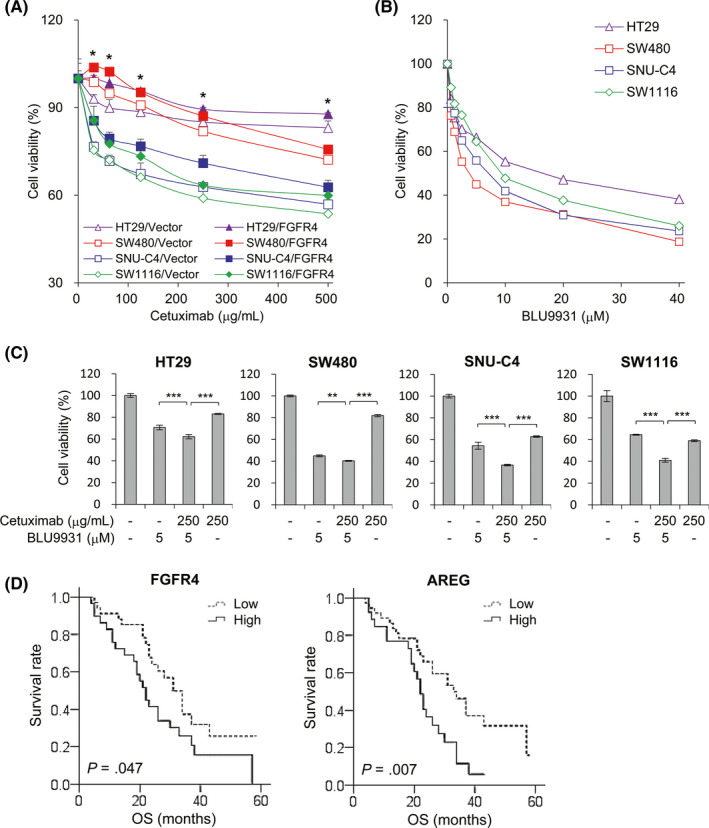
Fibroblast growth factor receptor 4 (FGFR4) increases cell viability against epidermal growth factor receptor inhibition in colon cancer cells. A, Colon cancer cells with FGFR4 expression or vector control plasmid were treated with different concentrations of cetuximab (range, 31‐500 μg/mL) for 48 h and cell viability were evaluated. *P < .05 compared to each vector control. B, Different concentrations of BLU9931 (range, 0.63‐40 μmol/L) were used to treat cancer cells for 48 h and cell viability were evaluated. C, Cells were treated with 250 μg/mL cetuximab, 5 μmol/L BLU9931, or their combination for 48 h and cell viability were evaluated. D, Kaplan‐Meier survival analysis for FGFR4 and amphiregulin (AREG) expression was carried out in 63 patients with metastatic colon cancer treated with cetuximab. Data are presented as mean ± SD. **P < .01, ***P < .001
To verify the role of FGFR4‐related AREG expression on cetuximab treatment in clinical samples, we examined the expression of FGFR4 and AREG in patients with metastatic colon cancer who received cetuximab combination chemotherapy as a first line of treatment. Immunohistochemical stains of specimen collected at the time of diagnosis were analyzed for the relationship between protein expression levels and OS outcomes (Figure S6). The expression of FGFR4 is significantly correlated with the expression of AREG (P = .035) and patients with higher expression of FGFR4 or AREG showed significantly poor OS compared to patients with low expression of these proteins (Figure 6D). Taken together, this work suggests that FGFR4 could be a biomarker for EGFR therapy and new druggable target for colon cancer. Thus, dual targeted therapy for FGFR4 and EGFR could be an effective approach for colon cancer treatment.
4. DISCUSSION
The EGFR signaling pathways, such as JAK/STAT, PI3K/AKT, and RAS/RAF/ERK, play key roles in tumor growth, drug response, and progression, promoting proliferation, survival, and invasion. 8 , 32 Many EGFR studies in cancer have focused on its aberrant expression and mutations, as well as heterodimerization with the other RTKs including of MET, IGF‐1R, PDGFR, FGFR4, and other ErbB receptors. 8 , 9 , 10 , 11 , 12 , 23 Fibroblast growth factor receptor 4 has been studied in a number of tumor types, including liver, prostate, breast, pancreas, colon, and gynecological cancer. It also plays vital roles in cancer cell proliferation, invasion, and antiapoptosis through FRS2‐ and phospholipase‐mediated activation of RAS/RAF/ERK and PI3K/AKT signaling pathways. 33 In this study, we newly elucidated the mechanism of FGFR4 that induces EGFR ligands, resulting in cooperative activation of EGFR signaling in colon cancer.
Recently it has been highlighted as a promising targeted therapy, especially in liver cancer by disrupting FGF19 (ligand of FGFR4)‐FGFR4 signaling pathway. 34 However, BLU9931 (selective FGFR4 inhibitor) and BGJ398 (pan‐FGFR inhibitor, data not shown) in colon cancer could not effectively block the downstream pathways of FGFR4, such as RAS/RAF/ERK and PI3K/ATK signals and AREG, although siFGFR4 treatment clearly showed the impaired downstream pathways. Therefore, further efforts are needed in the development of FGFR4 inhibitors that can sufficiently inhibit the FGFR4 downstream signals and the associated ligands. To date, studies focusing on FGFR4 in colon cancer have been limited; this is the first study to provide a rationale for combination treatment of FGFR4 and EGFR inhibitors in colon cancer.
In detail, this study showed that EGFR and ErbB3 are not activated directly by dimerization with FGFR4, but they are phosphorylated and activated depending on AREG secreted by FGFR4. It means that FGFR4 can stimulate EGFR externally regardless of the genetic status of KRAS, NRAS, BRAF, and PIK3CA, leading to colon cancer progression. In addition, FGFR4 induces cetuximab resistance, and the combination of FGFR4 inhibitor and cetuximab has shown an additive effect. Previously, the cooperative activation of EGFR and FGFR4 has been reported in lung adenocarcinoma. 23 Unlike our results, they showed the direct interaction between 2 proteins at the physical level using coimmunofluorescence and coimmunoprecipitation. The highlight of our study is that FGFR4 could be one of the intrinsic resistance mechanisms of anti‐EGFR therapy related to EGFR ligands that are involved in cancer progression and chemotherapy resistance. Indeed, other studies have shown that abundant expression of EGFR ligands, such as BTC and TGF‐α, can cause the downregulation of cell surface EGFR and reduce the binding ability of cetuximab, which could explain the associations with poorer response to cetuximab and shorter progression‐free survival. 35 Clinically, we showed the positive correlation between FGFR4 and AREG expression and these are associated with poor OS in cetuximab‐treated colon cancer patients. Thus, dual inhibition of FGFR4 and EGFR could be a promising treatment approach for colon cancer based on evidence that FGFR4 inhibitors not only block FGFR4, but can partially block EGFR, including ErbB3.
As previously mentioned in the Results, the levels of EGFR ligands are increased in FGFR4‐transfected colon cancer cells. One of these, AREG, is a pro‐oncogenic factor providing self‐sufficient growth and survival signals in lung, breast, pancreas, and colon cancer differentiation, proliferation, and invasion. 36 , 37 , 38 , 39 Expression of AREG has also been associated with resistance to conventional chemotherapeutic agents such as doxorubicin in hepatocellular carcinoma (HCC) and cisplatin in breast cancer. 40 , 41 Recently, it was reported that the sorafenib resistance in HCC showed aberrant activation of EGFR/Erb3 receptors as well as overexpression of EGFR ligands, including AREG. 42 These studies support our findings, stressing the importance of evaluating compensatory interactions of signaling pathways in targeted treatments.
The FGFR4 inhibitor BLU9931 can reduce colon cancer cell viability, either alone or in combination with cetuximab, and in the case of SW480, more profound cell cytotoxicity was observed compared to treatments with cetuximab alone. Based on our result, this combination strategy can be applied not only to colon cancer but also to other tumors if FGFR4 alteration is observed with EGFR activation, such as FGF19 gene amplification or FGFR4 Arg388 polymorphism. 33
To date, biomarkers for anti‐EGFR therapy in colon cancer have been restricted by known aberrant EGFR pathways, such as mutations in KRAS, NRAS, and BRAF. As reported in several clinical trials, EGFR status is not a reliable biomarker to select patients for cetuximab‐based therapy. 43 , 44 , 45 KRAS, NRAS, and BRAF status are examined in clinical practice before cetuximab use because their mutation constitutively activates the EGFR pathway and induces cetuximab resistance regardless of EGFR expression. Based on our study, FGFR4 can induce AREG expression and it dynamically activates the EGFR pathway. Thus, we identified FGFR4, together with AREG, as novel biomarkers for predicting cetuximab response for patients with metastatic colon cancer. Like EGFR, c‐Met is also known to be associated with anti‐EGFR resistance with heterodimerization of EGFR resulting in activation of similar signal pathways, such as PI3K/AKT and RAS/RAF/ERK. In the future, the role of FGFR4 in relation to other receptor tyrosine kinase family and its value as a biomarker to predict the clinical benefit should be validated in clinical trials.
Although multiple colon cancer cell lines have been used in a series of experiments, this study has some shortcomings. Only FGFR4 stable transfected HCT116 cells were used in the human colon cancer mouse model because other cell lines (HT29, SW116, SNU‐C4, and SW480, which were used in in vitro studies) did not grow in nude mice. As SCID mouse or mouse colon cancer cells were not available for this study, the functional roles of FGFR4 with EGFR ligands in colon cancer progression will be redetermined in SCID or nude mouse using multiple colon cancer cell lines.
To conclude, our study has characterized the cross‐talk between FGFR4 and EGFR/ErbB3 signaling by the contribution of EGFR ligands secreted from FGFR4. These findings provide experimental evidence for combined treatment with FGFR4 inhibitor and anti‐EGFR therapy in colon cancer.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Figures S1‐S6
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Myung‐Sook Park and Mi‐Ra Park for conducting the experimental studies. This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (NRF‐2017R1A2B4005003 and NRF‐2018R1A5A2024181) and a grant (HCRI17904‐21) from Chonnam National University Hwasun Hospital Institute for Biomedical Science.
Hong C-S, Sun E-G, Choi J-N, et al. Fibroblast growth factor receptor 4 increases epidermal growth factor receptor (EGFR) signaling by inducing amphiregulin expression and attenuates response to EGFR inhibitors in colon cancer. Cancer Sci. 2020;111:3268–3278. 10.1111/cas.14526
C.‐S. Hong and E.‐G. Sun contributed equally to this work.
Contributor Information
Ik‐Joo Chung, Email: ijchung@jnu.ac.kr.
Sang‐Hee Cho, Email: shcho@jnu.ac.kr.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Fontana E, Eason K, Cervantes A, et al. Context matters‐consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Ann Oncol. 2019;30:520‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Laurent‐Puig P, Lievre A, Blons H. Mutations and response to epidermal growth factor receptor inhibitors. Clin Cancer Res. 2009;15:1133‐1139. [DOI] [PubMed] [Google Scholar]
- 4. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol cell Biol. 2001;2:127‐137. [DOI] [PubMed] [Google Scholar]
- 5. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341‐354. [DOI] [PubMed] [Google Scholar]
- 6. Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65:1566‐1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maennling AE, Tur MK, Niebert M, et al. Molecular targeting therapy against EGFR family in breast cancer: progress and future potentials. Cancers. 2019;11:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saito Y, Haendeler J, Hojo Y, et al. Receptor heterodimerization: essential mechanism for platelet‐derived growth factor‐induced epidermal growth factor receptor transactivation. Mol Cell Biol. 2001;21:6387‐6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanizaki J, Okamoto I, Sakai K, et al. Differential roles of trans‐phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Brit J Cancer. 2011;105:807‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quintanal‐Villalonga A, Molina‐Pinelo S, Cirauqui C, et al. FGFR1 cooperates with EGFR in lung cancer oncogenesis, and their combined inhibition shows improved efficacy. J Thorac Oncol. 2019;14:641‐655. [DOI] [PubMed] [Google Scholar]
- 11. Morgillo F, Woo JK, Kim ES, et al. Heterodimerization of insulin‐like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006;66:10100‐10111. [DOI] [PubMed] [Google Scholar]
- 12. von der Heyde S, Bender C, Henjes F, et al. Boolean ErbB network reconstructions and perturbation simulations reveal individual drug response in different breast cancer cell lines. BMC Syst Biol. 2014;8:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sergina NV, Rausch M, Wang DH, et al. Escape from HER‐family tyrosine kinase inhibitor therapy by the kinase‐inactive HER3. Nature. 2007;445:437‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 15. Ali R, Wendt MK. The paradoxical functions of EGFR during breast cancer progression. Signal Transduct Target Ther. 2017;2:16042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sleeman M, Fraser J, McDonald M, et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271:171‐182. [DOI] [PubMed] [Google Scholar]
- 17. Spinola M, Leoni V, Pignatiello C, et al. Functional FGFR4 Gly388Arg polymorphism predicts prognosis in lung adenocarcinoma patients. J Clin Oncol. 2005;23:7307‐7311. [DOI] [PubMed] [Google Scholar]
- 18. Thussbas C, Nahrig J, Streit S, et al. FGFR4 Arg388 allele is associated with resistance to adjuvant therapy in primary breast cancer. J Clin Oncol. 2006;24:3747‐3755. [DOI] [PubMed] [Google Scholar]
- 19. Shen YY, Lu YC, Shen DP, et al. Fibroblast growth factor receptor 4 Gly388Arg polymorphism in chinese gastric cancer patients. World J Gastroentero. 2013;19:4568‐4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu T, Wang LN, Li W, et al. Downregulation of miR‐491‐5p promotes gastric cancer metastasis by regulating SNAIL and FGFR4. Cancer Sci. 2018;109:1393‐1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Turkington RC, Longley DB, Allen WL, et al. Fibroblast growth factor receptor 4 (FGFR4): a targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014;5:e1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho SH, Hong CS, Kim HN, et al. FGFR4 Arg388 is correlated with poor survival in resected colon cancer promoting epithelial to mesenchymal transition. Cancer Res Treat. 2017;49:766‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quintanal‐Villalonga A, Molina‐Pinelo S, Yague P, et al. FGFR4 increases EGFR oncogenic signaling in lung adenocarcinoma, and their combined inhibition is highly effective. Lung Cancer. 2019;131:112‐121. [DOI] [PubMed] [Google Scholar]
- 24. Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal. 2011;17:10‐12. [Google Scholar]
- 25. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goff LA, Trapnell C, Kelley D. CummeRbund: analysis, exploration, manipulation, and visualization of Cufflinks high‐throughput sequencing data. R package version 2280. 2019. [Google Scholar]
- 28. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat protoc. 2009;4:44‐57. [DOI] [PubMed] [Google Scholar]
- 29. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hong CS, Jeong O, Piao Z, et al. HOXB5 induces invasion and migration through direct transcriptional up‐regulation of β‐catenin in human gastric carcinoma. Biochem J. 2015;472:393‐403. [DOI] [PubMed] [Google Scholar]
- 31. Hatakeyama H, Cheng HX, Wirth P, et al. Regulation of heparin‐binding EGF‐like growth factor by miR‐212 and acquired cetuximab‐resistance in head and neck squamous cell carcinoma. PLoS One. 2010;5:e12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Parseghian CM, Napolitano S, Loree JM, et al. Mechanisms of innate and acquired resistance to anti‐EGFR therapy: a review of current knowledge with a focus on rechallenge therapies. Clin Cancer Res. 2019;25:6899‐6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang SY, Hao YL, Yuan Y, et al. Role of fibroblast growth factor receptor 4 in cancer. Cancer Sci. 2018;109:3024‐3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao LX, Wang XL, Tang YL, et al. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J Exp Clin Canc Res. 2017;36:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khelwatty S, Essapen S, Bagwan I, et al. The impact of co‐expression of wild‐type EGFR and its ligands determined by immunohistochemistry for response to treatment with cetuximab in patients with metastatic colorectal cancer. Oncotarget. 2017;8:7666‐7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Willmarth NE, Ethier SP. Autocrine and juxtacrine effects of amphiregulin on the proliferative, invasive, and migratory properties of normal and neoplastic human mammary epithelial cells. J Biol Chem. 2006;281:37728‐37737. [DOI] [PubMed] [Google Scholar]
- 37. Shao JY, Lee SB, Guo BHP, et al. Prostaglandin E‐2 stimulates the growth of colon cancer cells via induction of amphiregulin. Cancer Res. 2003;63:5218‐5223. [PubMed] [Google Scholar]
- 38. Panico L, D'Antonio A, Salvatore G, et al. Differential immunohistochemical detection of transforming growth factor α, amphiregulin and CRIPTO in human normal and malignant breast tissues. Int J Cancer. 1996;65:51‐56. [DOI] [PubMed] [Google Scholar]
- 39. Ebert M, Yokoyama M, Kobrin MS, et al. Induction and expression of amphiregulin in human pancreatic cancer. Cancer Res. 1994;54:3959‐3962. [PubMed] [Google Scholar]
- 40. Castillo J, Goni S, Latasa MU, et al. Amphiregulin induces the alternative splicing of p73 into its oncogenic isoform ΔEx2p73 in human hepatocellular tumors. Gastroenterology. 2009;137:1805‐1815. [DOI] [PubMed] [Google Scholar]
- 41. Eckstein N, Servan K, Girard L, et al. Epidermal growth factor receptor pathway analysis identifies amphiregulin as a key factor for cisplatin resistance of human breast cancer cells. J Biol Chem. 2008;283:739‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blivet‐Van Eggelpoel MJ, Chettouh H, Fartoux L, et al. Epidermal growth factor receptor and HER‐3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J Hepatol. 2012;57:108‐115. [DOI] [PubMed] [Google Scholar]
- 43. Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan‐refractory metastatic colorectal cancer. New Engl J Med. 2004;351:337‐345. [DOI] [PubMed] [Google Scholar]
- 44. Hebbar M, Di Fiore F, Conroy T, et al. Assessment of baseline clinical predictive factors of response to cetuximab‐irinotecan in patients with irinotecan‐refractory metastatic colorectal cancer. Oncology. 2007;73:185‐191. [DOI] [PubMed] [Google Scholar]
- 45. Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803‐1810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S6
Supplementary Material