

Abstract
Although next‐generation sequencing‐based panel testing is well practiced in the field of cancer medicine for the identification of target molecules in solid tumors, the clinical utility and clinical issues surrounding panel testing in hematological malignancies have yet to be fully evaluated. We conducted a multicenter prospective clinical sequencing study to verify the feasibility of a panel test for hematological tumors, including acute myeloid leukemia, acute lymphoblastic leukemia, multiple myeloma, and diffuse large B‐cell lymphoma. Out of 96 eligible patients, 79 patients (82%) showed potentially actionable findings, based on the clinical sequencing assays. We identified that genetic alterations with a strong clinical significance were found at a higher frequency in terms of diagnosis (n = 60; 63%) and prognosis (n = 61; 64%) than in terms of therapy (n = 8; 8%). Three patients who harbored a germline mutation in either DDX41 (n = 2) or BRCA2 (n = 1) were provided with genetic counseling. At 6 mo after sequencing, clinical actions based on the diagnostic (n = 5) or prognostic (n = 3) findings were reported, but no patients were enrolled in a clinical trial or received targeted therapies based on the sequencing results. These results suggest that panel testing for hematological malignancies would be feasible given the availability of useful diagnostic and prognostic information. This study is registered with the UMIN Clinical Trial Registry (UMIN000029879, multiple myeloma; UMIN000031343, adult acute myeloid leukemia; UMIN000033144, diffuse large B‐cell lymphoma; and UMIN000034243, childhood leukemia).
Keywords: clinical sequencing, feasibility study, hematological malignancy, panel testing, potentially actionable finding
This multicenter prospective study investigated feasibility of target capture‐based panel testing, focusing on hematological malignancies. Our results suggest that panel testing for hematological malignancies is feasible given the availability of useful diagnostic and prognostic information.

Abbreviations
- AITL
angioimmunoblastic T‐cell lymphoma
- ALL
acute lymphoblastic leukemia
- CNV
copy‐number variation
- CSeq
clinical sequencing
- DLBCL
diffuse large B‐cell lymphoma
- GCB
germinal center B‐cell
- ITD
internal tandem duplication
- JSH
Japanese Society of Hematology
- ML‐DS
myeloid leukemia associated with Down syndrome
- MLPA
multiplex ligation‐dependent probe amplification
- MM
multiple myeloma
- NGS
next‐generation sequencing
- PAF
potentially actionable finding
- PPA
positive percent agreement
- ROI
region of interest
- UPD
uniparental disomy
- VAF
variable allele frequency
- WHO
World Health Organization
1. INTRODUCTION
In the last decade, NGS technologies have revolutionized the understanding of the cancer genome through many genomic studies, which revealed recurrent driver mutations shared across different human cancers or that are specific to a certain type of cancer. 1 , 2 These studies also clarified the complexity of intra‐tumor and inter‐tumor clonal structures and the process of clonal evolution from benign to malignant states. 3 , 4 , 5 NGS technologies have been further applied toward clinical management for cancer patients, leading to molecular diagnosis, precise prognostic stratification, and identification of molecular target therapy. 6 , 7 , 8 Accurate, fast, and cost‐effective target‐enrichment NGS panel tests have accelerated a large‐scale practical implementation of precision medicine for patients with cancer. 9
Hematology has been the vanguard of genomic medicine since the 1980s, when Southern blot analysis was used to aid the diagnosis of lymphoma. Notably, identification of the BCR‐ABL1 and PML‐RARA fusion genes caused a paradigm shift in molecularly targeted therapies against CML 10 and acute promyelocytic leukemia, 11 and aided in making remarkable progress in long‐term outcomes. It is also apparent that genetic classification has become quite developed in the field of hematology, and many genetic subtypes are listed in among 2017 classifications by the WHO. 12 , 13 Recent comprehensive genomic studies further demonstrated the existence of patients with new genetic subtypes of morphologically diagnosed AML, 14 ALL, 15 and DLBCL. 16 Based on the disease subtypes or useful genetic markers, risk‐based stratified treatment has been practiced more commonly when managing patients with hematological malignancies, suggesting that genomic information is indispensable for clinical practice with these disorders.
Although relatively large numbers of reports have shown the utility of panel sequencing for solid tumors, 6 , 8 , 9 , 17 it remains unclear whether it is an effective approach with clinical benefit for hematological malignancies. In particular, few reports have discussed the value of panel testing in diagnostic and prognostic assessment, which are likely to provide useful information especially for patients with hematological malignancies. 7 , 18 , 19 Therefore, we developed a DNA‐panel testing method for hematological malignancies that can simultaneously detect various types of gene alterations including single‐nucleotide variants (SNVs), insertions/deletions (indels), CNVs, and immunoglobulin heavy chain locus (IGH) translocations. We then performed a prospective multicenter feasibility clinical sequencing (CSeq) study and assessed the clinical utility of the panel testing in a unified way across 96 patients with AML, ALL, MM, or DLBCL.
2. MATERIALS AND METHODS
2.1. Patients
To evaluate the feasibility of target capture‐based panel testing for hematological malignancies, adult patients with AML (n = 25), pediatric patients with AML or ALL (n = 25), adult patients with MM (n = 25), and adult patients with DLBCL (n = 25) were enrolled in the CSeq study. The patient samples were collected as ancillary studies, which were designated as the CS‐17‐CSeq arm of the CS‐17 study conducted by Japan Adult Leukemia Study Group for adult AML, the CSeq‐17 arm of the JPLSG‐CHM14 study conducted by the Japan Pediatric Leukemia/Lymphoma Study Group for pediatric leukemia, the MM‐15‐CSeq arm of the JSH‐MM‐15 prospective observational study conducted by JSH for MM, and the Lymphoma‐CSeq study for DLBCL. Most patients were registered for the study with an initial diagnosis, except for 13 relapsed pediatric patients. The studies were approved by the ethics committees at all participating institutions. Informed consent was obtained from all patients or guardians when children were enrolled. This study was conducted in accordance with the Declaration of Helsinki. Before genetic testing was conducted, patients were asked whether they wanted to receive a report describing any germline mutations that might be found.
2.2. Hybridization‐based targeted CSeq assay
To detect somatic SNVs, indels, CNVs, and IGH translocations with clinical or preclinical relevance in managing hematological malignancies, we designed a capture panel consisting of the entire coding regions of 330 genes [which included frequently mutated genes in hematological malignancies or targetable molecules in cancer (Table S1)], some IGH regions (Figure S1), and 1179 single‐nucleotide polymorphism baits. Tumor specimens were prepared from bone marrow (AML, ALL, and MM) or freshly resected tumor tissues (DLBCL), and subjected to DNA extraction. To enrich the tumor cells from patients with MM, CD138‐positive cells were selected before DNA extraction, using CD138 MicroBeads (Miltenyi Biotec). We used normal sample pairs (buccal mucosa or peripheral blood) as controls to discriminate between somatic and germline mutations. Sequencing libraries were prepared from 50‐200 ng DNA using the SureSelect XT reagent (Agilent Technologies) according to the manufacturer's instructions and subjected to NGS from both ends with the MiSeq or HiSeq2500 platform (Illumina).
2.3. Bioinformatics analysis
We considered the NGS to be successful if the average sequence depth in the tumor sample was above 300×, based on a previous report. 20 Using tumor cells and matched normal tissue, mutation calling was performed using the Genomon2 pipeline (https://genomon.readthedocs.io/ja/latest/), as previously described. 5 , 21 Putative somatic mutations with (i) a Fisher exact test P‐value of <.01, (ii) a VAF in the tumors of >.05, (iii) a sequencing depth in the tumor of ≥50 were adopted, and filtered by excluding (i) synonymous SNVs or noncoding variants and (ii) variants only present in unidirectional reads. The remaining variants were interrogated for evidence that they were present at significantly higher VAFs than expected for errors (P ≤ 10−3), for which the statistical significance was evaluated by empirical Bayesian mutation calling, as previously described. 22
To detect IGH translocations and tandem duplication, we used Genomon‐SV (https://github.com/Genomon‐Project/GenomonSV), 23 and searched for known variants by manual curation. Breakpoints of candidate alterations were inspected visually using the Integrative Genomics Viewer tool (http://software.broadinstitute.org/software/igv/). Candidate FLT3‐internal tandem duplication (FLT3‐ITD) calls were validated by PCR analysis, and the FLT3‐ITD allelic ratio was determined as previously reported. 24
CNVs were detected using the CNACS algorithm (https://github.com/papaemmelab/toil_cnacs). 5 , 21 Candidate focal CNVs (shorter than half of a chromosome arm, except for 17p deletions involving TP53) were manually reviewed and further filtered by removing the regions showing detection with <3 capture probes, as described previously. 21 Gene amplification was defined as increase in the number of copies of a gene to more than 4 copies, and hyperdiploidy was defined as the presence of >50 chromosomes.
2.4. Germline mutations
We defined reportable germline mutations in 22 genes, as follows: 16 genes (APC, BRCA1, BRCA2, MSH2, MSH6, NF2, PMS2, PTEN, RB1, RET, STK11, TP53, TSC1, TSC2, VHL, and WT1) in the panel were responsible for hereditary cancers for which American College of Medical Genetics and Genomics (ACMG) recommends reporting as incidental or secondary findings, 25 and 6 genes (ANKRD26, CEBPA, DDX41, ETV6, GATA2, and RUNX1) were associated with myeloid neoplasms with a germline predisposition that were proposed as a diagnostic category by the WHO in the 2017 classification scheme. 12 Germline variants in all 22 genes were analyzed as described above, except for determining Fisher exact test P‐values, which were omitted because a single analysis was performed. Putative variants were further filtered based on known variants listed in the 1000 Genomes Project (October 2014 release); the National Center for Biotechnology Information dbSNP database (build 131); Exome Aggregation Consortium (ExAC); the Human Genome Variation Database (April 2016 release); and 3.5KJPN (the ToMMo Japanese reference panel). 26
2.5. Analytical validation
We used 22 cell lines and 4 standard reference materials (HD728, HD731, HD753, and HD829; Horizon Discovery) for sensitivity analysis. For these specimens, the regions of interest (ROIs) for analytical validation were defined as follows: regions involving common somatic mutations that were verified by orthogonal methods including MLPA, information in the literature, the Catalog of Somatic Mutations in Cancer (version 87) 27 or Cancer Cell Line Encyclopedia 28 database, or manufacturer's validation data. As a result, the ROI comprised 89 genetic regions, and the PPA was evaluated for 26 specimens. For the analytical validation assays, the same analytical filter used for the germline mutation analysis was adopted, although the filter of tumor VAF >.05 was removed. For specificity analysis, 2 normal specimens from the Genome in a Bottle Consortium (RM8391 and RM8393) were used, and the sequencing specificity was determined for the same ROI, as verified by performing MLPA or searching the literature. 29
2.6. Analysis of clonality
To analyze the clonality of lymphoid malignancies, we designed a capture‐based NGS panel consisting of coding regions for IGH, IGK, IGL, TRA, TRB, TRD, and TRG (M. Sanada, Y. Iijima, manuscript in preparation). The clonality was assessed using the Vidjil pipeline. 30 Candidate clonal rearrangements were validated by PCR analysis.
2.7. Molecular tumor board for hematological malignancies
The multicenter molecular tumor board was composed of multidisciplinary members, and meetings were held once or twice a month to interpret the sequencing results of all patients, with the goal of identifying PAF. The tumor board included a hematological specialist, pathologists, genome researchers, bioinformaticians, medical geneticists, and genetic counselors. Before the molecular tumor board was established, clinically important information, such as age, sex, diagnoses, and leukemia‐associated translocation, was collected. Board members discussed analytical validity, clinical validity, and the clinical utility of the sequencing results. Based on the significance of clinical decision making, we categorized genetic alterations into 4 levels (Level A to Level D) according to standard guidelines for evidence‐based categorization of somatic variants. 31 In the process of curating of genetic alterations, professional guidelines, or crucial reports (Table S2) were used as a reference. The clinical utility was assessed regardless of the patients’ disease stage, clinical history, and accessibility to clinical trials for unapproved drugs. Candidate germline mutations were also reviewed by board members, and decisions were made as to whether to present the results to the patients.
2.8. Definition of PAF
For the purposes of this study, we defined a PAF as any genomic finding obtained by the CSeq assay that was capable of providing (a) a disease subtype or change in diagnosis (evidence level A or B), 31 (b) a risk category (evidence level A or B), or (c) a targetable molecular aberration (evidence level C or above).
3. RESULTS
3.1. Analytical validation of the CSeq assay
We first evaluated the analytical performance of the CSeq assay. The sensitivity of our assay, as determined from the PPA of the ROI, was 95.6% for SNVs (65/68), 100% for short indels (10/10), 100% for large indels (7/7), 100% for CNVs (13/13), 72.2% for IGH translocations (13/18), and 93.3% overall for all variants combined (113/121) (Tables S3 and 1). The PPA for variants with a VAF of approximately 5% – the threshold set for SNVs and indels – was 93.3% (42/45, Table S3). These results were considered acceptable, except for the sensitivity of detecting IGH translocations. Specificity analysis was performed for 2 normal samples, and the CSeq assay showed no false‐positive variant calls in the ROIs (Table 1).
TABLE 1.
Summary of analytical validation of CSeq assay
| Mutation type | Sensitivity | Specificity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Orthogonal methods | No. of analytical variants | True positive (TP) | False negative (FN) | PPA (%) TP/TP + FN | Sample | Orthogonal methods | No. of analytical regions | True negative (TN) | False positive (FP) | Specificity (%) TN/TN + FP | |
| SNV | Cell line, Standard material | Database (CCLE, COSMIC) | 68 | 65 | 3 | 95.6 | RM8391, RM8393 | Literature | 106 | 106 | 0 | 100 |
| Short indel | 10 | 10 | 0 | 100 | 20 | 20 | 0 | 100 | ||||
| Large indel | 7 | 7 | 0 | 100 | 8 | 8 | 0 | 100 | ||||
| CNV | MLPA | 18 | 18 | 0 | 100 | MLPA | 26 | 26 | 0 | 100 | ||
| Translocation | Cell line | Literature | 18 | 13 | 5 | 72.2 | Literature | 18 | 18 | 0 | 100 | |
Abbreviations: CCLE, Cancer Cell Line Encyclopedia; COSMIC, Catalog of Somatic Mutations in Cancer; MLPA, multiplex ligation‐dependent probe amplification; SNV, single nucleotide variant.
3.2. Feasibility of the CSeq assay
From November 2017 to April 2019, 100 patients with hematological malignancies (0 y old to 87 y old) were enrolled in this study. Four patients were excluded from further analysis due to misdiagnosis (n = 3) or patient death before registration (n = 1). We performed target capture sequencing for DNA isolated from the remaining 96 patients (Table 2). The average sequence depth in each tumor sample was 597× (range, 357×‐837×), and all cases showed a depth above the set threshold (300×), making the sequencing success rate 100% (Figure S2).
TABLE 2.
Clinical information of the patients enrolled in CSeq study
| Characteristics | No. (n = 96) |
|---|---|
| Diagnosis | |
| Acute myeloid leukemia | 30 |
| CBFB‐MYH11 | 2 |
| RUNX1‐RUNX1T1 | 1 |
| Other | 27 |
| Acute lymphoblastic leukemia | 17 |
| B‐cell | 15 |
| BCR‐ABL1 | 2 |
| KMT2A‐AFF1 | 1 |
| KMT2A‐MLLT3 | 1 |
| TCF3‐HLF | 1 |
| TCF3‐PBX1 | 1 |
| Other | 9 |
| T‐cell | 2 |
| Multiple myeloma | 24 |
| Diffuse large B‐cell lymphoma | 25 |
| non‐GCB | 13 |
| GCB | 12 |
| Age, y | |
| 0‐4 | 9 |
| 5‐14 | 11 |
| 15‐39 | 8 |
| 40‐64 | 27 |
| 65‐79 | 30 |
| ≥80 | 11 |
| Gender | |
| Male | 54 |
| Female | 42 |
| Disease status | |
| Primary | 83 |
| Relapse | 13 |
The median turnaround time, defined as the interval between the date of sample shipping and the date of returning the analysis report, was 41 d (range, 21‐80 d), which was 1 or 2 wk longer than our anticipated timeline. The primary reason for the delay was the waiting period for the next molecular tumor board meeting, which usually took place approximately 2‐3 wk after completion of the bioinformatic analysis.
Disease‐specific mutational landscapes for AML, ALL, MM, and DLBCL are presented in Figure 1 in terms of the major driver mutations, and a full listing of the observed gene mutations is provided in Table S4. At least 1 genetic alteration was identified in 93 of the 96 (96.9%) cases. The common driver alterations found with each disease were as follows: NPM1 mutations (n = 7) and Del (7q) (n = 7) in AML, CDKN2A deletion (n = 7) and IKZF1 deletion (n = 7) in ALL, Del(13q) (n = 12) and hyperdiploidy (n = 10) in MM, and CD79A/B mutations (n = 7) and MYD88 mutations (n = 7) in DLBCL. In addition, the CSeq assay also detected FLT3‐ITD (n = 5) and IGH translocations including IGH‐CCND1 (n = 4), IGH‐BCL6 (n = 4), IGH‐NSD2 (n = 3), and IGH‐MYC (n = 3).
FIGURE 1.

Major driver mutations detected by the CSeq assay. The figure shows the major driver mutations involved in the pathogenesis of hematological malignancies, including AML, ALL, MM, and DLBCL. Clinical information and clinical interpretations assessed by the molecular tumor board are described. PAF, potentially actionable findings
One of the purposes of this study was to estimate the prevalence of patients with PAF. The CSeq assay identified PAF in 26 of 30 patients (86.7%) with AML, 11 of 17 patients with ALL (64.7%), 20 of 24 patients (83.3%) with MM, and 22 of 25 patients (88.0%) with DLBCL (Figure 1). Thus, a total 79 of 96 patients with hematological malignancies (82.2%) had PAF, demonstrating the high clinical efficacy of this assay.
3.3. Clinical utility of the CSeq assay in drug selection
We identified actionable alterations leading to drug selection in 44 cases, most of which were considered as being of preclinical significance (n = 36, evidence level C) (Figure 2 and Table S5). These alterations (n ≥ 3) and potential modes of targeted therapy (evidence level C) included RAS pathway mutations (n = 13, BRAF and MEK inhibition), CD79A/79B mutations (n = 7, PKC inhibition), MYD88 mutations (n = 4, BTK inhibition), IGH‐CCND1 (n = 4, BCL2 inhibition), TP53 mutations (n = 4, DNMT1 inhibition), and CREBBP mutations (n = 3, HDAC inhibition). Two types of alterations, namely FLT3‐ITD (n = 5, FLT3 inhibition) and IDH1/2 mutations (n = 5, IDH1/2 inhibition) were identified with a clinical evidence of level A in 8 patients with AML (Table S5).
FIGURE 2.

Clinical utility of the CSeq assay, as assessed by the evidence‐based categorization system. The frequencies of patients with mutations having clinical implications (evaluated using the evidence‐based categorization system) are shown
3.4. Clinical diagnostic utility of the CSeq assay
Based on information from diagnostic guidelines and some crucial reports (Table S1), we tried to divide morphologically diagnosed disease into molecular subtypes after discussing the validity at the molecular tumor board meeting. Through our analysis, AML, 1 of the diseases with the most advanced molecular diagnosis, could be subclassified into 6 subtypes 12 , 14 , 32 : AML with NPM1 mutations (n = 7), AML with mutated chromatin, RNA‐splicing genes, or both (n = 5), AML with TP53 mutations, chromosome aneuploidy, or both (n = 4), AML with biallelic CEBPA mutations (n = 3), myeloid leukemia associated with Down syndrome (ML‐DS, n = 2), and myeloid neoplasms with DDX41 germline mutations (n = 2) (Figure 3 and Table S4).
FIGURE 3.
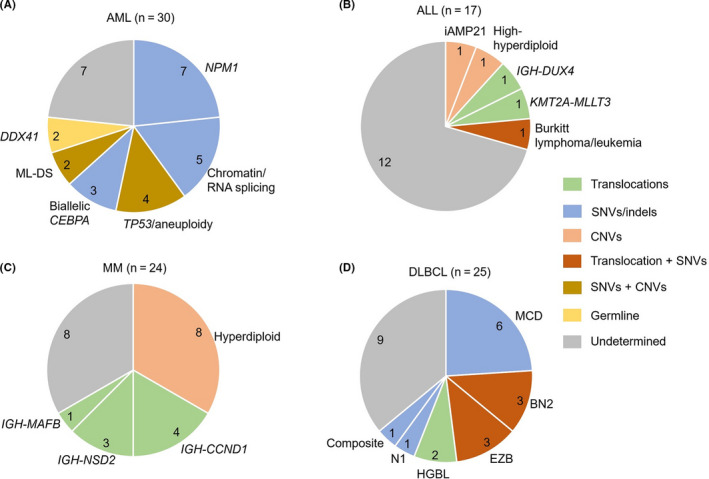
Subtype classification based on the CSeq assay results. Pie charts showing the molecular subtypes in patients with AML, ALL, MM, and DLBCL, based on the results of the CSeq assay. The mutation types of key genetic alterations are color‐coded. NPM1, AML with NPM1 mutation; Chromatin/RNA‐splicing, AML with mutated chromatin, RNA‐splicing genes, or both; TP53, AML with TP53 mutations, chromosomal aneuploidy, or both; Biallelic CEBPA, AML with biallelic CEBPA mutations; ML‐DS, myeloid leukemia associated with Down syndrome; DDX41, Myeloid neoplasms with germline DDX41 mutation; HGBL, high‐grade B‐cell lymphoma
We also analyzed the diagnostic utility of CSeq analysis for patients with ALL, MM, and DLBCL. CSeq analysis revealed subtype‐defining events in 4 cases with B‐ALL (iAMP21; n = 1, IGH‐DUX4; n = 1, high‐hyperdiploidy; n = 1, and KMT2A‐MLLT3; n = 1). For patients with MM, 4 groups including hyperdiploidy (n = 8) and IGH‐CCND1 (n = 4), IGH‐NSD2 (n = 3), and IGH‐MAFB (n = 1) translocations were identified, based on our analysis of CNVs and IGH translocations. We also classified DLBCL into 5 subtypes 16 ; the MCD type (n = 6), the BN2 type (n = 3), the EZB type (n = 3), the high‐grade B‐cell lymphoma type (n = 2), and the N1 type (n = 1) (Figure 3 and Table S4). These results for DLBCL were almost consistent with pathological findings in cell of different origins independently analyzed by a pathologist, since 5 of 6 MCD types were non‐GCB (germinal center B‐cell) types and 2 of 3 EZB type were GCB types (Table S4). 16
Interestingly, the CSeq assay leads to diagnostic changes in 4 cases. In 1 case with morphologically DLBCL, we identified both RHOA G17V and IDH2 R172W hot‐spot mutations, which is strongly suggestive of angioimmunoblastic T‐cell lymphoma (AITL). 33 Confirmation of clonality involving both T‐cell receptor and immunoglobulin production in tumors enabled us to diagnose this case as composite lymphoma (AITL and DLBCL; Figure S3). 34 In 1 case initially diagnosed as B‐ALL and 2 cases initially diagnosed as DLBCL, identification of key diagnostic alterations lead to a new diagnosis of Burkitt lymphoma/leukemia, high‐grade B‐cell lymphoma with MYC and BCL6 rearrangements, and high‐grade B‐cell lymphoma, not otherwise specified, respectively, according to the WHO’s 2017 classification scheme (Figure 3 and Tables S4, S6). 13
3.5. Clinical utility of the CSeq assay in prognosis
The CSeq assay provided prognostic information with strong clinical evidence (level A or B) for 24 patients with AML (80.0%), 8 patients with ALL (47.0%), 19 patients with MM (79.1%), and 10 patients with DLBCL (40.0%) (Figure 2 and Table S4). The most frequent genetic alterations associated with prognosis in each disease were NPM1 mutations in AML (n = 7, favorable risk or intermediate risk), IKZF1 loss in ALL (n = 7, adverse risk), hyperdiploidy in MM (n = 8, standard risk), and TP53 mutations/del (17p) in DLBCL (n = 7, adverse risk).
3.6. Cancer‐related germline mutations
We identified 2 AML patients who harbored both a DDX41 germline mutation (A500fs) and a DDX41 somatic mutation (R525H), and another AML patient harboring a deleterious BRCA2 germline mutation, and these results were validated by Sanger sequencing (Table 3). The former patient was diagnosed as having myeloid leukemia with DDX41 mutations (evidence level A) and the latter was diagnosed as having a risk for developing hereditary breast and ovarian cancer (evidence level A). All 3 patients with germline mutations received genetic counseling for future cancer risks (Table 3). No germline mutations were identified in patients with ALL, MM, and DLBCL.
TABLE 3.
Summary of the sequencing results with strong influence on clinical actions
| ID | Diagnosis | Clinical stage | Therapy options without sequencing | Actionable findings | Clinical interpretation | Clinical actions |
|---|---|---|---|---|---|---|
| CSeq‐01 | AML | Primary | No genetic counseling | DDX41 p.A500fs | Myeloid neoplasms with germline DDX41 mutation (Diagnosis) | Genetic counseling for DDX41 mutations |
| CSeq‐09 | AML | Primary | No genetic counseling | DDX41 p.A500fs | Myeloid neoplasms with germline DDX41 mutation (Diagnosis) | Genetic counseling for DDX41 mutations |
| CSeq‐10 | AML | Primary | Allogeneic stem cell transplantation at first CR | Biallelic CEBPA mutations | Favorable risk (Prognosis) | Continuation of cytotoxic chemotherapy without allogeneic stem cell transplantation |
| CSeq‐11 | AML | Primary | Consolidation therapy containing multi‐agent chemotherapy | Biallelic CEBPA mutations | Favorable risk (Prognosis) | Change to consolidation therapy containing high‐dose cytarabine alone |
| CSeq‐14 | AML | Primary | No genetic counseling | BRCA2 p.S1882X | Risk for developing hereditary breast and ovarian cancer (Diagnosis) | Genetic counseling for BRCA2 mutations |
| CSeq‐73 | DLBCL | Primary | R‐CHOP 6 cycles | TP53 p.E271K | Adverse risk (Prognosis) | R‐CHOP 8 cycles |
| CSeq‐77 | DLBCL | Primary | R‐CHOP 8 cycles | MYD88 p.243N, CD79A p.190_202del | MCD type (Diagnosis) | Addition of radiation therapy for primary tumor site |
| CSeq‐92 | DLBCL | Primary | Treatment for DLBCL | RHOA p.G17V, IDH2 p.R172W | Composite lymphoma (Diagnosis) | Treatment for both DLBCL and AITL |
3.7. Sequencing results that strongly influenced clinical actions
At 6 mo after registration, all patients were prospectively surveyed regarding their clinical course, treatment regimen, and participation in a clinical trial. For the patients with targetable molecular aberrations, we assessed whether treatment according to the CSeq reports was delivered or not. Although 44 patients received sequencing results regarding target therapy (evidence level A: 8 cases; evidence level C: 36 cases), no patients enrolled in clinical trials or received therapies based on sequencing results, except for 1 patient with an FLT3‐ITD mutation that was already identified before registration (Table S5). Based on the prognostic information, 2 patients with AML underwent different treatment strategies; 1 patient selected chemotherapy rather than allogeneic stem cell transplantation; the other patient underwent a different treatment protocol (Table 3). Furthermore, although there is not enough clinical evidence in the management of DLBCL, the clinician also changed the treatment for 2 patients with DLBCL according to diagnostic or prognostic information; 1 patient underwent different treatment cycles, the other patient was given an additional radiation therapy. Patients diagnosed with composite lymphoma were treated and followed as having AITL, rather than DLBCL.
4. DISCUSSION
To assess the utility of genomic medicine in hematological malignancies, we performed a multicenter prospective study of capture‐based panel sequencing for patient with these disorders. This prospective study demonstrated the feasibility of the CSeq assay in that it showed: (i) a high incidence of cases with PAF as assessed by standard criteria, (ii) a permissible turnaround time, and (iii) sequencing results likely to have high specificity. However, despite the growing availability of genomic medicine, 2 major issues need to be resolved. First, manual curation of sequencing results in the context of diagnostic, prognostic, and therapeutic value requires substantial work by the curator and causes a long waiting period before the final reports are returned to the patients, as reflected by the turnaround times in this study. Second, as illustrated in Table S5, the clinical utility of CSeq assay for drug selection was limited. The primary reasons for not being able to act upon the therapeutic findings was limited access to unapproved drugs for patients with hematological malignancies in Japan, as well as the disease conditions of the patients, most of who were enrolled at the time of initial diagnosis and did not require further treatment beyond standard chemotherapy. Both problems might be major barriers or bottlenecks against implementing genomic medicine in patients with hematological malignancies. The application of artificial intelligence to genomic curation, 35 , 36 establishing systems to share enormous genomic and clinical data sets for cancer patients, 37 and developing easily available target therapeutics could be helpful in solving this problem.
This feasibility study clearly showed that the percentage of patients with genetic alterations considered to have strong clinical significance (level A or level B) in terms of disease diagnosis (63%) or prognosis (64%) was higher than that considered in terms of therapy (8%) (Figure 2). Most actionable findings in the therapeutic category were assigned as preclinical evidence (level C). Consistent with these findings, our follow‐up survey revealed that the clinical actions taken by 8 patients were mainly based on the diagnostic or prognostic information shown in Table 3. These results suggested that the current CSeq assay may be a useful tool for precision diagnosis and accurate disease prognosis, and, therefore, may be performed at the initial diagnosis. Appropriate timing of the panel test would depend on the cancer type, as the panel tests for solid tumors are usually performed at refractory or relapse stage to detect the target molecule.
The CSeq assay identified 3 cases with germline mutations. Interestingly, 2 of 3 patients harbored recurrent DDX41 somatic and germline mutations, which might indicate that the percentage of patients with DDX41 germline mutations is higher than expected. 38 , 39 Based on previous reports describing patients with donor cell leukemia that harbored DDX41 germline mutations, 40 , 41 the CSeq assay reported here suggested that patients may have a familial hereditary predisposition to leukemia and that it might be better not to select sibling donors for allogeneic stem cell transplantation, although the biological and clinical significance of this germline mutation is still fully unknown. Our results thus show that germline mutations and somatic mutations can potentially impact patient management if clinicians carefully consider core ethical issues regarding the germline mutations.
The CSeq assay offers the advantage of detecting various types of gene alterations with a single DNA‐sequencing platform including SNVs, indels, CNVs and IGH translocations, which can efficiently identify gene‐mutation profiles and classify the molecular subtypes across hematological diseases. However, this assay has several limitations. First, CSeq assay did not detect gene fusions because the CSeq assay does not include RNA‐based sequencing, a suitable method for detection of gene fusions. As shown by the low molecular‐diagnostic yield of the CSeq assay for patients with ALL (Figure 3), this limitation would be especially true for ALL, as a wide variety of recurrent chromosome rearrangements define different disease subtypes for ALL. 15 , 42 , 43 Second, this assay is less sensitive in detecting IGH translocations (Table 1), which was probably caused by insufficient disposition of IGH capture probes or a mapping failure due to tandemly repeated sequences in the IGH regions. Combining this assay with RNA sequencing as well as existing laboratory tests would provide more excellent sequencing performance and improve clinical decision making for patients with hematological malignancies. Third, this assay is not suitable for evaluating clonal hematopoiesis of indeterminate potential (CHIP), a risk factor of hematological malignancies and cardiovascular diseases, 3 , 44 because we used buccal mucosa as a control specimen for the patients with AML or ALL. Lastly, DLBCL tumor specimens were extracted not from formalin‐fixed paraffin‐embedded tissues but from freshly resected tumor tissues to ensure the sensitivity and specificity of this genomic analysis. This may differ from the actual clinical practice.
In conclusion, the CSeq assay enables detection of somatic and germline mutations in patients with hematological malignancies, which makes it a useful diagnostic and prognostic testing tool. Our findings suggest that using the panel test for hematological malignancies would be feasible, but further optimization of NGS analysis and developing system that allows easy access to unapproved drugs may improve treatment outcomes for patients with these disorders.
DISCLOSURE
NA received honoraria from SRL, Inc and Nippon Shinyaku Co., Ltd., and research funds from Chugai Pharmaceutical Co., Ltd. KU received honoraria from Novartis Pharma KK, research funds from Astellas Pharma Inc, Alexion Pharmaceuticals Inc, AbbVie Inc, Gilead Sciences Inc, SymBio Pharmaceuticals Limited, Daiichi Sankyo Co., Ltd., Sumitomo Dainippon Pharma Co., Ltd., Chugai Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Novartis Pharma KK, Bristol‐Myers Squibb KK, Takeda Pharmaceutical Co., Ltd., Amgen Astellas BioPharma KK, and Nippon Shinyaku Co., Ltd. MK received research funds from Daiichi Sankyo Co., Ltd. MR received honoraria from Janssen Pharmaceutical KK, research funds from Celgene Corporation, Daiichi Sankyo Co., Ltd., and Bristol‐Myers Squibb KK, and scholarships from Takeda Pharmaceutical Co., Ltd., Sanofi KK, Ono Pharmaceutical Co., Ltd., and Chugai Pharmaceutical Co., Ltd. HH received honoraria from Janssen Pharmaceutical KK, Celgene Corporation, Takeda Pharmaceutical Co., Ltd., and Ono Pharmaceutical Co., Ltd., research funds from Celgene Corporation, Takeda Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd, Kyowa Kirin Co., Ltd., Sanofi KK, Bayer Yakuhin, Ltd., and Astellas Pharma Inc TI received honoraria from Takeda Pharmaceutical Co., Ltd., Celgene Corporation, Ono Pharmaceutical Co., Ltd., and Janssen Pharmaceutical KK. HS received honoraria from Takeda Pharmaceutical Co., Ltd., Novartis Pharma KK, Celgene Corporation, Janssen Pharmaceutical KK, Chugai Pharmaceutical Co., Ltd., and Kyowa Kirin Co., Ltd., research funds from Janssen Pharmaceutical KK, Ono Pharmaceutical Co., Ltd., Celgene Corporation, Novartis Pharma KK, Sanofi KK, AstraZeneca KK, AbbVie Inc, and Chugai Pharmaceutical Co., Ltd., and scholarships from Astellas Pharma Inc, Teijin Pharma Limited, Shionogi Pharma Co., Ltd., Eisai Co., Ltd., Sanofi KK, Taiho Pharmaceutical Co. Ltd., and Nippon Shinyaku Co., Ltd. MA received honoraria from Daiichi Sankyo Co., Ltd., Celgene Corporation, Takeda Pharmaceutical Co., Ltd., and Janssen Pharmaceutical KK, research grants from Bristol‐Myers Squibb KK, Ono Pharmaceutical Co., Ltd., Celgene Corporation, and Takeda Pharmaceutical Co., Ltd., and scholarships from Chugai Pharmaceutical Co., Ltd., Kyowa Kirin Co., Ltd., MSD KK, Ono Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Novartis Pharma KK, Astellas Pharma Inc, Teijin Pharma Limited, Pfizer Japan Inc, Takeda Pharmaceutical Co., Ltd. MN received honoraria from Eisai Co., Ltd. and research funds from Eisai Co., Ltd. and Sumitomo Dainippon Pharma Co., Ltd. KO received research funds from Daiichi Sankyo Co., Ltd. HG received honoraria from Amgen Inc HK received consultant fee from Astellas Pharma Inc, Amgen Astellas BioPharma KK, and Daiichi Sankyo Co., Ltd., honoraria from Bristol‐Myers Squibb KK, Astellas Pharma Inc, and Novartis Pharma KK, research funds from Chugai Pharmaceutical Co., Ltd., Kyowa Kirin Co., Ltd., Zenyaku Kogyo Co., Ltd., FUJIFILM Corporation, Daiichi Sankyo Co., Ltd. Co., Ltd., Astellas Pharma Inc., Otsuka Pharmaceutical Co., Ltd., Nippon Shinyaku Co., Ltd., Eisai Co., Ltd., Pfizer Japan Inc, Takeda Pharmaceutical Co., Ltd., Novartis Pharma KK, Sumitomo Dainippon Pharma Co., Ltd., Sanofi KK, Perseus Proteomics Inc, and Celgene Corporation. YM received honoraria from Novartis Pharma KK, Celgene Corporation, Sumitomo Dainippon Pharma Co., Ltd., Nippon Shinyaku Co., Ltd., Chugai Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Astellas Pharma Inc, and Kyowa Kirin Co., Ltd., research funds from Sumitomo Dainippon Pharma Co., Ltd., and scholarships from Pfizer Japan Inc, Takeda Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Nippon Shinyaku Co., Ltd. SO is stockholder in Asahi Genomics Co., Ltd., received consulting fees from KAN Research Institute, INC., and Chordia Therapeutics Inc, research funds from KAN Research Institute, Inc, Chordia Therapeutics Inc, Sumitomo Dainippon Pharma Co., Ltd., Otsuka Pharmaceutical Co., Ltd., and Eisai Co., Ltd., and accepted researchers from Chordia Therapeutics. HY received research funds from Astellas Pharma Inc SI received honoraria from Takeda Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd, Celgene Corporation, Janssen Pharmaceutical KK, Bristol‐Myers Squibb KK, Daiichi Sankyo Co., Ltd., and Sanofi KK, research funds from Takeda Pharmaceutical Co., Ltd., Janssen Pharmaceutical KK, AbbVie Inc, Bristol‐Myers Squibb KK, MSD KK, and scholarships from Takeda Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Kyowa Kirin Co., Ltd., and Sanofi KK AT received honoraria from Chugai Pharmaceutical Co., Ltd. and research funds from Chugai Pharmaceutical Co., Ltd., Kyowa Kirin Co., Ltd., Takeda Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co. Ltd., Ono Pharmaceutical Co., Ltd., and Perseus Proteomics Inc. Other authors have no conflict of interest.
Supporting information
Fig S1
{kind=link}
Fig S2
{kind=link}
Fig S3
{kind=link}
Tables S1‐S6
ACKNOWLEDGMENTS
This work was supported by AMED under Grant Number JP18kk0205005h003 (to KH) and JP20cm0106501h0005 (to SO); JSPS KAKENHI Grant Numbers JP26221308 and JP19H05656 (to SO). The authors are grateful to Koichi Akashi and Itaru Matsumura at the JSH (who approved the MM‐15‐CSeq study) and all physicians participating in the CS‐17‐CSeq study, the CSeq‐17 study, the MM‐15‐CSeq study, and the Lymphoma‐CSeq study, for their cooperation. We thank Toshiki Saito, Nobutaka Kiyokawa, and Hirokazu Nagai, who promoted developing a program for an integrated database of clinical and genomic information. We also appreciate Tadashi Kumamoto and Hideki Muramatsu for sharing their insights at a molecular tumor board meeting, and Tomomi Ishida, Mika Fuyama, Kanako Okada, and Masumi Hosaka for providing technical assistance.
Yasuda T, Sanada M, Nishijima D, et al. Clinical utility of target capture-based panel sequencing in hematological malignancies: A multicenter feasibility study. Cancer Sci. 2020;111:3367–3378. 10.1111/cas.14552
REFERENCES
- 1. Bailey MH, Tokheim C, Porta‐Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371‐385 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23:227‐238 e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaiswal S, Fontanillas P, Flannick J, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47:458‐468. [DOI] [PubMed] [Google Scholar]
- 5. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harris MH, DuBois SG, Glade Bender JL, et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the Individualized Cancer Therapy (iCat) study. JAMA Oncol. 2016;2:608‐615. [DOI] [PubMed] [Google Scholar]
- 7. Mody RJ, Wu Y‐M, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314:913‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sunami K, Ichikawa H, Kubo T, et al. Feasibility and utility of a panel testing for 114 cancer‐associated genes in a clinical setting: a hospital‐based study. Cancer Sci. 2019;110:1480‐1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR‐ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031‐1037. [DOI] [PubMed] [Google Scholar]
- 11. Huang ME, Ye YC, Chen SR, et al. Use of all‐trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567‐572. [DOI] [PubMed] [Google Scholar]
- 12. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391‐2405. [DOI] [PubMed] [Google Scholar]
- 13. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gu Z, Churchman ML, Roberts KG, et al. PAX5‐driven subtypes of B‐progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B‐cell lymphoma. N Engl J Med. 2018;378:1396‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meric‐Bernstam F, Brusco L, Shaw K, et al. Feasibility of large‐scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33:2753‐2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He J, Abdel‐Wahab O, Nahas MK, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004‐3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oberg JA, Glade Bender JL, Sulis ML, et al. Implementation of next generation sequencing into pediatric hematology‐oncology practice: moving beyond actionable alterations. Genome Med. 2016;8:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. White BS, Lanc I, O’Neal J, et al. A multiple myeloma‐specific capture sequencing platform discovers novel translocations and frequent, risk‐associated point mutations in IGLL5. Blood Cancer J. 2018;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watatani Y, Sato Y, Miyoshi H, et al. Molecular heterogeneity in peripheral T‐cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33:2867‐2883. [DOI] [PubMed] [Google Scholar]
- 22. Shiraishi Y, Sato Y, Chiba K, et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res. 2013;41:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiba K, Shiraishi Y, Nagata Y, et al. Genomon ITDetector: a tool for somatic internal tandem duplication detection from cancer genome sequencing data. Bioinformatics. 2015;31:116‐118. [DOI] [PubMed] [Google Scholar]
- 24. Zwaan CM, Meshinchi S, Radich JP, et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood. 2003;102:2387‐2394. [DOI] [PubMed] [Google Scholar]
- 25. Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2. 0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249‐255. [DOI] [PubMed] [Google Scholar]
- 26. Tadaka S, Katsuoka F, Ueki M, et al. 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum Genome Var. 2019;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011;39:D945‐D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barretina J, Caponigro G, Stransky N, et al. The cancer cell line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zook JM, McDaniel J, Olson ND, et al. An open resource for accurately benchmarking small variant and reference calls. Nat Biotechnol. 2019;37:561‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giraud M, Salson M, Duez M, et al. Fast multiclonal clusterization of V(D)J recombinations from high‐throughput sequencing. BMC Genom. 2014;15:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakata‐Yanagimoto M, Enami T, Yoshida K, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171‐175. [DOI] [PubMed] [Google Scholar]
- 34. Suefuji N, Niino D, Arakawa F, et al. Clinicopathological analysis of a composite lymphoma containing both T‐ and B‐cell lymphomas. Pathol Int. 2012;62:690‐698. [DOI] [PubMed] [Google Scholar]
- 35. Itahashi K, Kondo S, Kubo T, et al. Evaluating clinical genome sequence analysis by Watson for genomics. Front Med. 2018;5:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sundaram L, Gao H, Padigepati SR, et al. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet. 2018;50:1161‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jensen MA, Ferretti V, Grossman RL, Staudt LM. The NCI genomic data commons as an engine for precision medicine. Blood. 2017;130:453‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheah JJC, Hahn CN, Hiwase DK, Scott HS, Brown AL. Myeloid neoplasms with germline DDX41 mutation. Int J Hematol. 2017;106:163‐174. [DOI] [PubMed] [Google Scholar]
- 39. Polprasert C, Schulze I, Sekeres M, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer Cell. 2015;27:658‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berger G, van den Berg E, Sikkema‐Raddatz B, et al. Re‐emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia. 2017;31:520‐522. [DOI] [PubMed] [Google Scholar]
- 41. Kobayashi S, Kobayashi A, Osawa Y, et al. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia. 2017;31:1020‐1022. [DOI] [PubMed] [Google Scholar]
- 42. Roberts KG, Li Y, Payne‐Turner D, et al. Targetable kinase‐activating lesions in Ph‐like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yasuda T, Tsuzuki S, Kawazu M, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569‐574. [DOI] [PubMed] [Google Scholar]
- 44. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Tables S1‐S6