

Abstract
Cancer cells are often characterized by abnormalities in DNA damage response including defects in cell cycle checkpoints and/or DNA repair. Synthetic lethality between DNA damage repair (DDR) pathways has provided a paradigm for cancer therapy by targeting DDR. The successful example is that cancer cells with BRCA1/2 mutations are sensitized to poly(adenosine diphosphate [ADP]‐ribose)polymerase (PARP) inhibitors. Beyond the narrow scope of defects in the BRCA pathway, “BRCAness” provides more opportunities for synthetic lethality strategy. In human pancreatic cancer, frequent mutations were found in cell cycle and DDR genes, including P16, P73, APC, MLH1, ATM, PALB2, and MGMT. Combined DDR inhibitors and chemotherapeutic agents are under preclinical or clinical trials. Promoter region methylation was found frequently in cell cycle and DDR genes. Epigenetics joins the Knudson's “hit” theory and “BRCAness.” Aberrant epigenetic changes in cell cycle or DDR regulators may serve as a new avenue for synthetic lethality strategy in pancreatic cancer.
Keywords: cell cycle, DNA damage repair, epigenetics, pancreatic cancer, synthetic lethality
Beyond the narrow scope of defects in the BRCA pathway, ‘BRCAness’ provides more opportunities for synthetic lethality strategy. Combined DDR inhibitors and chemotherapeutic agents are under preclinical or clinical trials. Aberrant epigenetic changes in cell cycle or DDR regulators may serve as a new avenue for synthetic Lethality strategy in pancreatic cancer.
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ATM, and Rad3‐related
- ATRi
ATR inhibitors
- BER
base excision repair
- CDKs
cyclin‐dependent protein kinases
- DDR
DNA damage repair
- DSB
double‐strand break
- EMT
epithelia‐mesenchymal transition
- HR
homologous recombination
- IR
ionizing radiation
- NHEJ
non‐homologous end‐joining
- OS
overall survival
- PARP
poly (adenosine diphosphate [ADP]‐ribose) polymerase
- PARPi
PARP inhibitors
- PC
Pancreatic cancer
- PDAC
pancreatic ductal adenocarcinoma
- PFS
progression free survival
- ROS
reactive oxygen species
- SSB
single‐strand breaks
- UV
ultraviolet
1. INTRODUCTION
Naturally occurring DNA damage accounts for up to 200 000 lesions per cell per day, and is caused by various intrinsic and extrinsic factors. 1 , 2 Extrinsic or environmental sources of DNA damage include UV light, IR and exposure to genotoxic agents. For example, UV light from the sun can induce up to 100 000 DNA lesions per cell per day. 2 , 3 Intrinsic factors mostly stem from metabolic by‐products, such as ROS. Mammalian cells have evolved some signaling pathways and mechanisms to engage the DDR. Mildly damaged cells may repair the DNA lesions and recover. However, if the damage is irreparable, it will trigger the cell death response to eliminate damaged cells. Aberrant genetic and epigenetic changes of DDR regulators may cause human diseases. A plethora of studies illustrates that DDR is inactivated in the early stage of carcinogenesis. 4 If DNA damage is not correctly repaired or stays unrepaired, DNA lesions will be induced, including mutations, deletions, insertions, and others. Cancer is regarded as a disease of accumulating driver mutations. 5 For cancer treatment, the purpose of chemotherapeutic agents is to induce DNA damage. It is a major challenge of toxic effect and chemo‐resistance in the clinic. Defects of DDR in cancer cells increase their sensitivity to DNA damage agents. 4 The concept of synthetic lethality originates from studies in Drosophila modal systems in which a combination of mutations in 2 or more separate genes leads to cell death. 6 There is application of synthetic lethality approaches in dysfunction of BRCA1 and BRCA2 sensitized cells to poly(adenosine diphosphate[ADP]‐ribose)polymerase (PARP) inhibitors in various cancers. 7 , 8 The tumor‐specific genetic defects serve as therapeutic markers to target human tumor‐induced tumor cell death without damaging normal cells. Beyond BRCA1/2 mutations, there are more aberrant genetic or epigenetic changes in human tumors. The key genes involved in cell cycle checkpoints and DDR are actionable under synthetic lethality. Accumulation of aberrant epigenetic changes, including DNA damage signaling, plays an important role in cancer development. 9 , 10
Pancreatic cancer is a very malignant disease, the overall 5‐y survival time is <10%. 11 Pancreatic cancer‐related death is increasing and by 2030 is predicted to be the second leading cause of cancer‐related death. 12 Although c. 10% cases of PC have a family history, the genetic basis for familial aggregation has not been identified in most cases. 13 , 14 Despite the vast genetic information gathered from PC genomic sequencing, biomarker‐based tailored therapies account for <1% of the total patient population. Targeting the DDR in tumors with defective DNA repair is a successful clinical example, but c. 4%‐7% of patients with PC have germline BRCA1/2 mutation. 15 Discovering more DDR‐related markers will promote the development of novel synthetic lethality strategies in pancreatic cancer. This review is mainly focused on aberrant DDR in genetics and epigenetics, the microenvironment and immune system are not discussed.
2. CELL CYCLE AND DDR
Loss of the normal controls of cellular replication is a fundamental defect in cancer. Thus, understanding the mechanisms of cell cycle control may lead to the development of novel strategies for cancer treatment. The cell cycle consists of the G1, S, G2, and M phases. Cyclins and CDKs are important regulatory components that are required for cell cycle progression. Human cells possess 20 CDKs and 29 cyclins. 16 G1‐phase cells are preparing for DNA synthesis. DNA is replicated during the S phase and cells enter into G2 phase for a period of growth. Then, cells enter the M phase and DNA is divided equally into 2 daughter cells. Most non‐dividing cells exit the cell cycle at the G1 phase into quiescence (G0 phase). Progression from 1 phase to another in the cell cycle is tightly regulated by CDKs. The integrity of the cell cycle is governed by surveillance mechanisms known as checkpoints, including G1/S, intra‐S, G2/M and mitotic checkpoints (M) in mammalian cells. 17 These checkpoints are important for DNA repair, replication, and spindle assembly. Cells rely on these checkpoints to prevent them from progressing into a new phase before they complete their current phase. In response to DNA damage, cell cycle progression is blocked by these checkpoints, giving cells time to repair damage before the next cell phase.
In early G1 phase, CDK4 and/or CDK6 are activated by cyclin D and phosphorylated retinoblastoma protein (Rb), releasing E2F transcription factors and resulting in the activation of E2F responsive genes for cell cycle progression. In the late G1 phase, CDK2 is activated by binding to cyclin E and completes the phosphorylation of Rb, leading to further activation of E2F‐mediated transcription. Then, cells pass through the boundary of the G1/S checkpoint and enter S phase. In S phase, CDK2 plays an important role by binding to cyclin A. During G2/M transition, CDK1, cyclin A, and cyclin B are required for completing mitosis. 18
One of the most harmful forms of DNA damage is the DSB. There are 2 major pathways to repair DSBs: NHEJ and HR. NHEJ takes part in DDR throughout the entire cell cycle, and it is preferably repairing during G0, G1, and early S phase. 19 HR is generally restricted to the late S and G2 phase, as it usually uses the intact sister chromatin as a template for synthesis‐dependent repair in mitotic cells. HR is regarded as an error‐free pathway. Interestingly, DSBs in vertebrate cells are predominantly repaired by NHEJ rather than HR. 20 DSBs are sensed by the heterotrimeric Mre11/Rad50/NBS1 (MRN) complex. 21 The MRN complex is recruited to DSBs and serves as an activation platform for the DNA damage checkpoint kinase, ATM. ATM is directly involved in DSBs repair by HR. 22 DSBs also activate another DNA damage responsive kinase DNA‐PK to initiate NHEJ signaling. NHEJ is more error‐prone, as the ends of DNA breaks are directly ligated without the need for a homologous template. When cells encounter SSBs or replication errors, ATR (ATM and Rad3‐related), another kinase, is activated. ATM is activated in response to DSBs, and ATR acts in response to SSBs. However, as cross‐talk between these pathways, both ATM and ATR are involved in repair of DSBs.
3. DDR INHIBITORS
A well recognized sensor of DNA damage is the protein PARP, which is best known for its role in DNA BER and repair of DNA SSB. Here, 30 y ago, small molecule nicotinamide analogs were shown to inhibit PARylation and to enhance the cytotoxicity of dimethyl sulfate, a DNA‐damaging agent. 23 More modified PARP inhibitors (PARPi) have been developed, including olaparib and niraparib. All these PARPi interact with the binding site of the PARP enzyme cofactor, β‐nicotinamide adenine dinucleotide (β‐NAD+), in the catalytic domain of PARP1 and PARP2. 24 Both BRCA1 and BRCA2 proteins are critical for the repair of DSBs by HR. In HR‐deficient cells, including defects in BRCA1, BRCA2, or other pathway components, the preferentially used mechanism is NHEJ. This may lead to DNA deletions or mutations. 25 , 26
In 2005, 2 groups described the synthetic lethal interaction between PARP inhibition and BRCA1 or BRCA2 mutation, and developed a novel treatment strategy for BRCA‐mutant tumors. 7 , 8 BRCA‐mutant tumor cells were more sensitive to PARPi than BRCA wild‐type cells, by as much as 1000 times. In addition to causing persistent SSBs by inhibiting PARP, it may create a DSB when SSBs encounter a replication fork, resulting in collapse of the fork. A recent study has suggested that some PARPi “trap” PARP1 on DNA, preventing auto‐PARylation and PARP1 release from the site of damage and therefore interfere with the catalytic cycle of PARP1. Thus, PARP1‐defective cells are resistant to PARPi. 24
Cancer cells often harbor a defect in DNA repair compared with normal cells. Therefore, cancer cells are more susceptible to DDR inhibition than normal cells. Faulty cell cycle checkpoint activation in cancer cells also results in replication stress and subsequent accumulation of DNA damage. DNA‐PKcs activity is essential for effective repair by classic NHEJ, which is the predominant DNA repair pathway of DSBs in human cells, occurring through all phases of the cell cycle. Classic NHEJ plays crucial roles in the repair of exogenous and endogenous DSBs. DNA‐PK inhibition sensitizes cells to replication‐independent DSB‐inducing agents such as topoisomerase 2 inhibitors. 27 Some novel DNA‐PK inhibitors have been developed, including MSC2490484A. 28 Similar to DNA‐PK, ATM promotes DNA DSB repair in cells and responds to DSBs generated throughout the cell cycle. The activation of CHK2, a well recognized substrate of ATM, is important for the G1/S checkpoint. AZD0156, an ATM inhibitor, is currently under clinical trials. 28 ATR is activated by replication protein A (RPA)‐bound ssDNA, which can arise as a result of stalled replication forks and also occurs following DNA end resection during the early stages of HR. 29 CHK1 is the best described substrate of ATR and, once activated by ATR, CHK1 inhibits CDK activity through phosphorylation of CDC25A. 30 CHK1 is a critical regulator of the G2/M and intra‐S cell cycle checkpoints. 30 VX‐970, an ATR inhibitor, is reported to increase antitumor activity in combination with cisplatin in vivo. 31 MK8776 is one of the CHK1 inhibitors. CHK1 inhibitors have a strong synergy with antimetabolites (such as gemcitabine), which generate replication‐dependent DNA damage. 32 A synthetic lethal relationship has now been established between ATR and CHK1 inhibition, with combination blockade leading to replication fork arrest. 33 The WEE1 protein kinase also plays a critical role in the activation of the G2/M checkpoint. WEE1 is not directly regulated by DNA damage, but is required for physiologic cell cycle progression. WEE1 inhibits CDK1 activity by phosphorylating CDK1 Tyr15, resulting in G2/M checkpoint activation. 34 A WEE1 inhibitor, AZD1775, has been shown to have cytotoxic effects in preclinical models. 35 As shown in Table 1, more DDR inhibitors have been studied in different cancers.
TABLE 1.
DDR inhibitors
| DDR pathway | Genes | DDR inhibitors | Application | Ref |
|---|---|---|---|---|
| BER | PARP | Olaparib (AD2281) | Germline BRCA‐mutated metastatic pancreatic cancer (NCT02184195) | (97) |
| Combination with irinotecan, cisplatin, and mitomycin C in pancreatic cancer (NCT01296763) | (98) | |||
| Combination with gemcitabine in pancreatic cancer (NCT00515866) | ||||
| Metastatic pancreatic cancer with a BRCAness somatic profile (NCT04348045) | ||||
| Veliparib (ABT‐888) | Combination with gemcitabine, cisplatin pancreatic cancer with BRCA1/2, PALB2 mutation (NCT01585805) | |||
| Combination with folinic acid, fluorouracil and irinotecan (FOLFIRI) in pancreatic cancer (NCT02890355) | ||||
| Combination with modified FOLFOX6 in pancreatic cancer (NCT01489865) | ||||
| Combination with gemcitabine and intensity modulated radiation therapy in pancreatic cancer (NCT01908478) | ||||
| Niraparib (MK‐4827) | Pancreatic cancer with BRCA1, BRCA2, PALB2, CHEK2 or ATM mutations (NCT03601923) | |||
| Pancreatic cancer after previous chemotherapy (NCT03553004) | ||||
| Talazoparib (BMN 673) | Advanced or recurrent solid tumors, including pancreatic cancer with BRCA mutations (NCT01286987) | (99) | ||
| Rucaparib (AG‐014699) | BRCA1, BRCA2 or PALB2 mutated pancreatic cancer (NCT03140670) | |||
| Pancreatic cancer with BRCA mutations (NCT02042378) | ||||
| APE1 | TRC102 | Combination with pemetrexed in advanced solid tumors (NCT00692159) | (100) | |
| NER | RPA | HAMNO | HAMNO was shown to act synergistically with etoposide to kill cancer cells (UMSCC38 and UMSCC11B) in vitro and slow tumor growth in vivo | (101) |
| TDRL‐505 | TDRL‐505 prevented cell cycle progression and acted synergistically with cisplatin in non–small‐cell lung cancer (NSCLC) cells | (102) | ||
| MCI13E | MCI13E showed chemotherapeutic promise in ovarian and lung cancer cell lines when used in combination with cisplatin, or as a single agent | (43) | ||
| XPA | NERI01(AB‐00026258) | NERI01 significantly sensitized human colon cancer cells to UV irradiation | (103) | |
| XPF | F06/NERI02(NSC130813) | F06 was shown to act synergistically with cisplatin and mitomycin C in NSCLC cells and human colon cancer cells | (104) | |
| HR | Mre11 | Mirin | Mirin abolished the G2/M checkpoint and homology‐dependent repair in mammalian cells (U2OS, TOSA4) | (105) |
| RAD51 | IBR2 | IBR2 was shown to overcome imatinib resistance in chronic myeloid leukemia (CML) cells and murine models | (106) | |
| NHEJ | DNA‐PK | M3814 | Advanced solid tumors (NCT02516813) | |
| Locally advanced rectal cancer (NCT03770689) | ||||
| VX‐984 | Advanced solid tumors (NCT02644278) | |||
| CC‐115 | Castration‐resistant prostate cancer (NCT02833883) | |||
| DDR checkpoints | ATM | AZD0156 | Monotherapy or combination with other anticancer treatment in advanced solid tumors (NCT02588105) | |
| AZD1390 | Combination with radiation therapy in brain cancer (NCT03423628) | |||
| KU‐60019 | PTEN‐deficient breast cancer cells are sensitive to KU‐60019 when combined with cisplatin | (107) | ||
| TP53 mutant glioblastoma (GBM) cells are sensitive to KU‐60019 under the ionizing radiation | (108) | |||
| Combination with sunitinib, pazopanib, temsirolimus in human renal tumors (NCT03571438) | ||||
| Combination with avelumab in DDR‐deficient advanced solid tumors (NCT04266912) | ||||
| ATR | VX‐970 (M6620) | Combination with Irinotecan in TP53‐mutant gastric or gastroesophageal junction cancer (NCT03641313) | ||
| Advanced solid tumors (NCT03718091) | ||||
| AZD6738 | Combined AZD6738 and olaparib potentiates genome instability and cell death in ATM‐deficient cancer cells | (109) | ||
| Combination with olaparib in advanced solid tumors including ATM‐deficient/proficient gastric cancer, lung cancer, breast cancer (NCT02264678) | ||||
| Combination with olaparib in pancreatic cancer, renal cancer, urothelial cancer (NCT03682289) | ||||
| Combination with olaparib in gynecological cancers with ARID1A loss (NCT04065269) | ||||
| BAY1895344 | Advanced solid tumors and lymphoma (NCT03188965) | |||
| Combination with niraparib in DDR‐deficient advanced solid tumors (NCT04267939) | ||||
| CHK1 | Prexasertib (LY2606368) | Prexasertib synergizes with olaparib in triple‐negative breast cancer cells | (110) | |
| Small cell lung cancer (NCT02735980) | ||||
| BRCA1/2 mutation associated breast cancer, ovarian cancer and prostate cancer (NCT02203513) | ||||
| Combination with olaparib in advanced solid tumors (NCT03057145) | ||||
| Rabusertib (LY2603618) | Combination with gemcitabine in pancreatic cancer (NCT00839332) | (111) | ||
| Combination with pemetrexed/cisplatin in non–small‐cell lung cancer (NCT01139775) | (112) | |||
| CHK1/2 | AZD7762 | Monotherapy or combination with gemcitabine in advanced solid tumors (NCT00413686) | ||
| WEE1 | AZD1775 | Combination with gemcitabine and radiation therapy in pancreatic cancer (NCT02037230) | (113) | |
| Combination with gemcitabine and nab‐paclitaxel in pancreatic cancer (NCT02194829) | ||||
| Small cell lung cancer with MYC family (MYC, MYCN, MYCL) amplification or CDKN2A mutation combined with p53 mutation (NCT02688907) | ||||
| Combination with paclitaxel, in gastric cancer harboring p53 mutation (NCT02448329) | ||||
| Combination with carboplatin in p53 mutated ovarian cancer (NCT01164995) |
4. COMBINED DDR INHIBITORS AND CHEMO‐RADIOTHERAPIES
The major effect of chemotherapy and radiotherapy is to induce DNA damage. Activated DDR may induce resistance to chemo‐radiotherapy. Therefore, it is reasonable to combine DNA damage checkpoint protein inhibitors and chemo‐radiotherapy to block this resistance. Targeting the compensatory DDR pathway may sensitize cancer cells that harbor a defect in DDR signaling to chemo‐radiotherapy. ATM inhibition has been demonstrated to sensitize cells to IR and DNA DSB‐inducing agents, including etoposide, camptothecin, and doxorubicin. 36
The loss of G1 checkpoint control is almost ubiquitous in cancer, making cancer cells more reliant on the S and G2/M checkpoints. Inhibition of ATR sensitizes cancer cells to various DNA‐damaging anticancer agents, such as combined VX‐970 with cisplatin or gemcitabine. 28 ATRi sensitizes PARPi to BRCA wild‐type breast cancer cells. 37 The combination of ATR inhibitors (ATRi) with IR is another promising strategy. 38 Prexasertib is a small ATP‐competitive selective inhibitor of CHK1 and CHK2. The combination of prexasertib with antimetabolites, PARPi or platinum for solid tumors has been included in preclinical studies. 39 Combinations of a Wee1 inhibitor (MK1775) and cisplatin, paclitaxel, and other agents are undergoing testing. 40 The combination of CDK4/6 inhibitors with endocrine therapy exhibited much more effective results in breast cancer compared with endocrine alone. LB100, a CDC25 inhibitor, sensitized cancer cells to chemo‐radiotherapy. 41 NU7026 is a novel DNA‐PK inhibitor and has been shown to inhibit NHEJ. 42 In combination with topoisomerase II poisons, NU7026 significantly sensitized K562 cells to radiation and etopside. 42 The nucleotide excision repair (NER) pathway is an incredibly versatile pathway and remedies DNA bulky adduct damage. The RPA protein is involved in essential interactions with DNA to support NER catalyzed repair of bulky adduct DNA damage. MCI13E, a RPA inhibitor, decreased cell viability, induced apoptosis, and in lung cancer showed a synergistic interaction with cisplatin. 43 KU‐55933, an ATM inhibitor, sensitized cancer cells to IR, camptothecin, doxorubicin, and etoposide. 44
5. THE RATIONALE OF COMBINING DDR INHIBITORS AND CHEMO‐RADIOTHERAPIES IN HUMAN CANCERS WITH DDR DEFECTS
The ability of cancer cells to repair DNA damage induced by chemo‐radiotherapies has reduced therapeutic efficiency. Deficiency of DDR is one of the characteristics of human cancer cells. This deficiency increases the sensitivity of cancer cells to chemo‐radiotherapies. Conversely, defects in one of the DDR pathways may sensitize cancer cells to inhibitors of the compensatory DDR signaling. BRCA1/2 mutation cancer cells sensitization to PARPi is a paradigm. In conventional chemotherapy, both cancer cells and normal cells are killed indiscriminately, while combining synthetic lethality with chemotherapy overcomes the limitation of chemotherapy by specifically killing cancer cells. Thus, it is rational to obtain more specific and stronger therapeutic effects by a combination of DDR protein inhibitors and chemo‐radiotherapies in cancer cells with DDR defects caused by mutation or epigenetics. PALB2 and HDAC2 have been reported to have a synthetic lethal interaction with PARPi. 45 , 46 Mateo et al 47 reported that sensitization of metastatic, castration‐resistant prostate cancer to olaparib is associated with PALB2 or HDAC2 defects. Combining carboplatin and the Wee1 inhibitor, AZD1775 increased the sensitivity of patients with platinum‐resistant ovarian cancer with the TP53‐mutant, with a partial response rate of 38% and a complete response rate of 5% in clinic. 48 SLFN11 is silenced by frequently methylation in human cancers. 49 , 50 Using a treatment combining olaparib and VE‐821, an ATR inhibitor, the sensitivity was increased in SLFN11 defect cancer cells. 51 DDR genes have multiple roles in the promotion of cancer cell growth via accumulation of driver mutations and abnormal epigenetic changes, generation of tumor heterogeneity and evasion of apoptosis. 9 , 10 It is important to identify the predictive biomarkers of response and/or resistance to DDR inhibitors. 52 “BRCAness” means a HR defect phenotype beyond the narrow scope of BRCA1 or BRCA2 mutations. 53 E‐cadherin has been predicted in preclinical models to be a sensitive marker for platinum and PARPi, and has been selected for a clinical trial. 54 Preclinical studies have shown that actionable onco‐proteins can directly or indirectly regulate DDR and cell cycle checkpoint pathways by driving HR gene expression. 52 For example, the RAS, PI3K and androgen receptor signaling pathways can promote HR repair. 55 , 56 Thus, targeting of these onco‐proteins together with DDR pathway components may lead to synthetic lethality in other DDR inhibitor resistant cells. 57 , 58 Inhibition of PI3K in BRCA1/2 wild‐type breast cancer cells suppressed BRCA1/2 expression and induced sensitivity to PARPi. 59 A phase I trial, combining PI3K inhibitor buparlisib with olaparib in ovarian and breast cancer, is being undertaken. 60 Another example is the combined inhibition of MYC and PARP inhibitor in HR defect cancers. 52 In addition to oncogene activation, epigenetic silencing of tumor suppressor expression may play similar roles in the regulation of DDR and the cell cycle. 9 However, epigenetic‐based synthetic lethality has not been extensively studied. 4 , 10
6. PANCREATIC CANCER‐RELATED SIGNALING AND DEFECTS OF DDR REGULATORS
Normal pancreatic ductal epithelium cells progress to PanIN‐1, PanIN‐2, PanIN‐3 to pancreatic ductal adenocarcinoma (PDAC), there is accumulation of mutations in KRAS, TP53, CDKN2A, SMAD4, RNF43, and GNAS. 61 More than 90% of PDACs harbor a KRAS driver mutation. KRAS may trigger proliferation in PC, but its presence in PanIN1 lesions shows that it alone is not sufficient to support a malignant phenotype. The signaling network in PC cells is very complex and involves KRAS. KRAS has 5‐8 downstream signaling pathways, including MAPK, ERK, PI3K, JNK, P53, and P16. Thus, inhibition of the KRAS pathway has a limited impact on PC cells. 62 GNAS is frequently mutated in intraductal papillary mucinous neoplasm (IPMN) and PDAC. Activated GNAS signaling, along with KRAS mutation, induces IPMN. 63 Epidermal growth factor receptors (EGFRs) (EGFR also known as HER) are known to be activators of KRAS, PI3K, and STATs. These pathways promote cancer cell growth, invasion, and prepare the cell for the next phase of cell cycle progression. 64 HER2 is expressed in 30%‐40% of PDAC. In patients with HER2 overexpression, combined trastuzumab and capecitabine does not result in improved PFS and OS. 65 P53 has been identified to be mutated in 40%‐75% of PDAC. 66 Germline P53 mutations result in Li‐Fraumeni syndrome. Somatic P53 mutations occur at the late stage of tumor development, unlike early KRAS oncogene activation. 66 P53 induces cell cycle arrest at the G1 phase by inhibiting CDK2 through upregulation of P21. P53 is also involved in DDR. 67 , 68 The complex roles of P53 include cross‐talk with other signaling pathways and synthetic lethality strategies based on P53 mutations and need to be further explored. TGF‐β signaling is a very complex signaling network and the role of the TGF‐β pathway in cellular homeostasis varies in cancer cells. TGF‐β signaling may act as a tumor suppressor to induce cell differentiation, and can also promote EMT in cancer cells. 69 , 70 Thus, development of synthetic lethality strategies based on a SMAD4 mutation is more complex. The CDKNA gene encodes 2 cell cycle regulators, P16 and P14. CDKN2A is mutated in more than 50% of PC. 71 CDKN2A is also frequently methylated in pancreatic early lesion and cancer tissue samples. 72 RNF43 is mutated in 10% of PDAC, and RNF43 is regarded as a tumor suppressor through inhibiting Wnt signaling to involve the cell cycle. 73 Driver mutations were reported to take part in PC initiation and progression, including KDM6A, RBM10, MLL3, TGFBR2, ARID1A, STK11, PRSS1, and SF3B1. 74 , 75 , 76 These aberrant changes of cell cycle regulators open new windows for synthetic lethality in PC.
It is estimated that c. 10% of all cases of PC have a hereditary component, involving DDR pathways and cell cycle regulatory genes. 77 The major known family PC susceptibilities include BRCA2, ATM, PALB2, CDKN2A, PRSS1, MLH1, MSH2, STK11, and P53. 78 The most common germline mutations are BRCA2 (1.4%‐7.4%) and ATM (1.2%) gene mutations in PC. 79 , 80 The lifetime risk of BRCA2 germline mutation carriers is approximately 4.9% for PC. 81 A recent study has suggested that germline BRCA mutations together with germline PALB2 mutations accounted for 7%. Inclusion of somatic mutations in BRCA1, BRCA2 and PALB2 doubled that number to 14% of patients, all of which were associated with an unstable genome or a BRCA mutational signature. 82 However, an unstable genome or BRCA mutational signature was present in 24% of patients, suggesting that DDR deficiency occurs in up to 24% of PDACs. 82 In addition, a significant proportion of patients with PDAC harbor heterozygous mutations in DDR pathways, with unknown functional consequences. The benefit of targeting heterozygous somatic or germline mutations with synthetic lethality strategies is yet to be determined and is complicated by our lack of knowledge on the functional consequences of many observed mutations in DDR genes. There is no consensus on whether the loss of the second allele is required to predict therapeutic sensitivity for the majority genes involved in DDR for present clinical trials. BRCA1 and BRCA2 germline carriers are known to respond to platinum and PARP inhibitor in multiple tumor types, including PDAC. 53 Novel targeted DDR agents, such as ATR and ATM inhibitors, offer significant potential in early preclinical studies. However, there is an urgent need to investigate patient selection markers.
“BRCAness” may be caused by aberrant epigenetic changes. As shown in Figure 1, promoter region methylation silenced BRCA1/2 expression, and offered an opportunity for a synthetic lethality strategy. Unlike genomics, epigenetics is not extensively studied in PC. Aberrant epigenetic changes play important roles in PC, including key components of cancer‐related signaling and DDR genes. Some genes were found frequently by our group and others to be methylated in human PC and early lesions. During carcinogenesis from IMPN to invasive cancer, there is a progressive tendency for gene methylation of which cell cycle regulators and DDR genes, including P14, P15, P16, P73, CHFR, APC, MLH1, BRCA1, GSTpi, MGMT, RASSF1A, play an important part. 83 , 84 , 85 , 86 , 87 , 88 Many tumor suppressors are frequently methylated in pancreatic and other cancers, including SOX17, HIN‐1, DACT2, and NKD2. Epigenetic silencing of these genes activated different cancer‐related signaling pathways to promote cell cycle progression, and further caused replication stress. Beyond classical “BRCAness,” epigenetic silencing of tumor suppressor genes in different signaling pathways may provide new opportunities for cancer synthetic lethality therapy (Figure 2A). 89 , 90 , 91 , 92 In the same DDR signaling pathway, different components may play alternative roles, for example RAD52 and BRCA1/2 are alternative components in RAD51‐mediated HR. Thus, inhibition of RAD52 in BRCA1/2 defect cells may cause synthetic lethality (Figure 2B). 93 However, synthetic lethality based on aberrant epigenetic changes need to be studied extensively in human PC.
FIGURE 1.
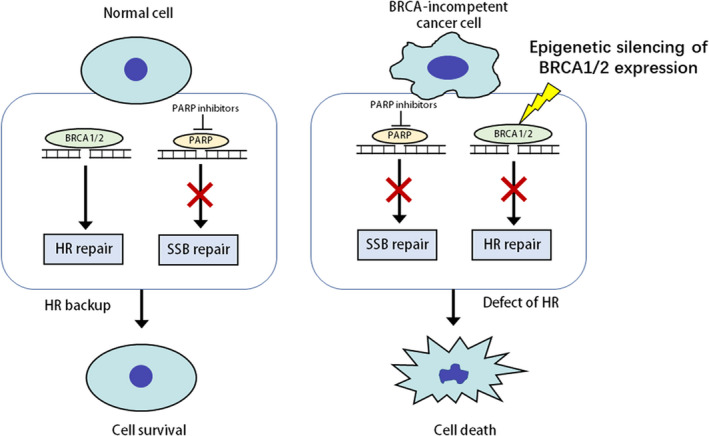
Epigenetic‐based synthetic lethality. HR, homologous recombination; SSB, single‐strand breaks.  : Defects of DDR
: Defects of DDR
FIGURE 2.
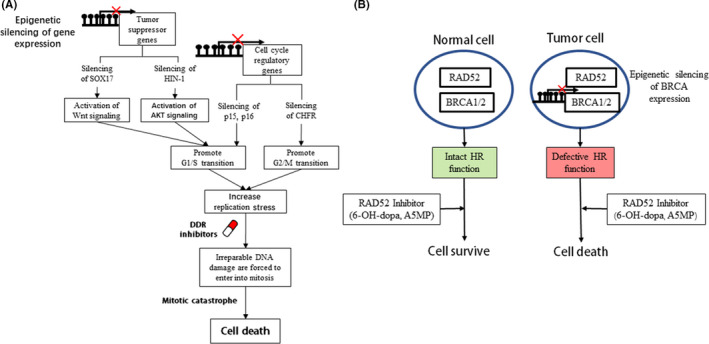
Synthetic lethality beyond BRCAness. A, Synthetic lethality induced by combining DDR inhibitors and epigenetic activation of cell cycle progression. B, Synthetic lethality induced by different HR inhibitors. CHFR: checkpoint with forkhead and ring finger domains; HIN‐1: high in normal‐1; p15, p16: cell cycle regulators; RAD52: homologous recombination repair gene; SOX17: SRY‐box‐transcription factor 17. ●: DNA methylation. : Silencing of gene expression
7. THE APPLICATION OF GENETIC AND EPIGENETIC MARKERS IN COMBINING DNA‐DAMAGING AGENTS AND DDR INHIBITORS IN HUMAN PC
It is well known that mutations may activate oncogene and inactivate tumor suppressor in PC. Epigenetics joins the Knudson's “hit” theory by silencing gene expression, including cell cycle regulators and DDR genes. DNA methylation is the most frequent epigenetic change in human cancers, including PC. 10 , 83 The epigenome is at the intersection of environment and genome, and genetics interacts with epigenetics. Disruption of a key epigenetic regulator by a mutation may lead to an altered transcriptome, including cell cycle regulators and DDR genes. DNMT3A is recurrently mutated in acute myeloid leukemia. 10 Histone modification modifiers were also found to be mutated in PC and precancerous lesions. These mutations may cause loss of, or increased, gene expression, and further influence drug sensitivity. 94 Miller et al 95 found that the bromodomain and extra‐terminal (BET) inhibitor JQ1 attenuated DSB repair and sensitized models of PC to PARPi. The exact mechanism of synthetic lethality needs to be further explored. Epigenetic silencing of DNA repair genes, such as MLH1, MGMT, and BRCA1, can lead to gene mutation and genomic instability in cancers, including PC. 87 In PC, the limited preclinical and clinical studies combining DNA‐damaging agents and DDR inhibitors have been based on BRCA1/2 mutation markers, and mainly focused on patients with germline mutation. 96 It is unclear if these patients had 1 allele or 2 allele mutations. The methylation status of BRCA1/2 and other DDR genes was not detected either. There is much room to improve treatment strategies of combined DNA‐damaging agents with DDR inhibitors.
8. PERSPECTIVE CONCLUSION AND CHALLENGES
Treatment of BRCA1/2 mutated cells with PARPi is a perfect model for synthetic lethality therapy in human cancer. However, mutations in BRCA1/2 are relatively rare, including in PC. Defects of any DDR regulator may serve as a synthetic lethality therapeutic marker, including for ATM, ATR, PALB2, MGMT, SLFN11, FANCC, and GSTpi. Several DDR inhibitors are undergoing testing in preclinical or clinical trials, however the application of these inhibitors in synthetic lethality needs to be extensively investigated. Clear pictures of the DDR pathway compensatory network remain to be produced. The rationale of “BRCAness” broadened the application of synthetic lethality therapy in human cancer. Some cancer‐related signaling pathways, including PI3K, AKT, Wnt and SMAD4, are involved in DDR by influencing the cell cycle. The efficiency of or deficiency in these cancer‐related signaling pathways regarding synthetic lethality has not been extensively studied. Almost all synthetic lethality studies have been based on gene mutations, even though DNA methylation occurs more frequently than gene mutations in human cancers, including in PC. Epigenetics has not been well studied in PC compared with other cancers. The exploration of synthetic lethality strategies based on epigenetic defects in PC is urgently needed. The interaction of genetics and epigenetics makes it more complex when aiming to determine the regimen of synthetic lethality based on available biomarkers. Techniques to discriminate single allele or double allele defects both through genetics and epigenetics are urgently need to be developed for clinical application. For precision medicine, evaluating genetic and epigenetic heterogeneity is necessary.
DISCLOSURE
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by grants from National Key Research and Development Program of China (2018YFA0208902, 2020YFC2002705); National Science Foundation of China (NSFC Nos. U1604281, 81672138); Beijing Science Foundation of China (BJSFC No. 7171008); National Key Scientific Instrument Special Program of China (Grant No. 2011YQ03013405).
Hu Y, Guo M. Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci. 2020;111:3111–3121. 10.1111/cas.14565
Funding information
National Key Scientific Instrument Special Program of China, (Grant/Award Number: “2011YQ03013405”) National Key Research and Development Program of China, (Grant/Award Number: “2018YFA0208902,” “2020YFC2002705”). National Natural Science Foundation of China, (Grant/Award Number: “81672138”) National Science Foundation of China, (Grant/Award Number: “U1604281”) Beijing Science Foundation of China, (Grant/Award Number: “7171008”).
REFERENCES
- 1. Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age‐dependent changes in base excision repair. Proc Natl Acad Sci USA. 2000;97(2):686‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matt S, Hofmann TG. The DNA damage‐induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. 2016;73(15):2829‐2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475‐1485. [DOI] [PubMed] [Google Scholar]
- 4. Gao D, Herman JG, Guo M. The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer. Oncotarget. 2016;7(24):37331‐37346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014;371(18):1725‐1735. [DOI] [PubMed] [Google Scholar]
- 7. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature. 2005;434(7035):913‐91. [DOI] [PubMed] [Google Scholar]
- 8. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 9. Guo M, Ren J, House MG, Qi Y, Brock MV, Herman JG. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res. 2006;12(15):4515‐4522. [DOI] [PubMed] [Google Scholar]
- 10. Guo M, Peng Y, Gao A, Du C, Herman JG. Epigenetic heterogeneity in cancer. Biomark Res. 2019;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7‐34. [DOI] [PubMed] [Google Scholar]
- 12. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913‐2921. [DOI] [PubMed] [Google Scholar]
- 13. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet (London, England). 2016;388(10039):73‐85. [DOI] [PubMed] [Google Scholar]
- 14. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371(11):1039‐1049. [DOI] [PubMed] [Google Scholar]
- 15. Gupta M, Iyer R, Fountzilas C. Poly(ADP‐Ribose) polymerase inhibitors in pancreatic cancer. A new treatment paradigms and future implications. Cancers. 2019;11(12):1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roskoski R Jr. Cyclin‐dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol Res. 2016;107:249‐275. [DOI] [PubMed] [Google Scholar]
- 17. Poon RY. Cell cycle control: a system of interlinking oscillators. Methods Mol Biol (Clifton, NJ). 2016;1342:3‐19. [DOI] [PubMed] [Google Scholar]
- 18. Satyanarayana A, Kaldis P. Mammalian cell‐cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28(33):2925‐2939. [DOI] [PubMed] [Google Scholar]
- 19. Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283(1):1‐5. [DOI] [PubMed] [Google Scholar]
- 20. Sonoda E, Takata M, Yamashita YM, Morrison C, Takeda S. Homologous DNA recombination in vertebrate cells. Proc Natl Acad Sci USA. 2001;98(15):8388‐8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carney JP, Maser RS, Olivares H, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double‐strand break repair to the cellular DNA damage response. Cell. 1998;93(3):477‐486. [DOI] [PubMed] [Google Scholar]
- 22. Goodarzi AA, Noon AT, Deckbar D, et al. ATM signaling facilitates repair of DNA double‐strand breaks associated with heterochromatin. Mol cell. 2008;31(2):167‐177. [DOI] [PubMed] [Google Scholar]
- 23. Purnell MR, Stone PR, Whish WJ. ADP‐ribosylation of nuclear proteins. Biochem Soc Trans. 1980;8(2):215‐227. [DOI] [PubMed] [Google Scholar]
- 24. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355(6330):1152‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moynahan ME, Cui TY, Jasin M. Homology‐directed dna repair, mitomycin‐c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61(12):4842‐4850. [PubMed] [Google Scholar]
- 26. Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology‐directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263‐272. [DOI] [PubMed] [Google Scholar]
- 27. Zhao Y, Thomas HD, Batey MA, et al. Preclinical evaluation of a potent novel DNA‐dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66(10):5354‐5362. [DOI] [PubMed] [Google Scholar]
- 28. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discovery. 2017;7(1):20‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR‐ and Cdc7‐dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11(1):203‐213. [DOI] [PubMed] [Google Scholar]
- 30. Stracker TH, Usui T, Petrini JH. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair. 2009;8(9):1047‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hall AB, Newsome D, Wang Y, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX‐970. Oncotarget. 2014;5(14):5674‐5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Daud AI, Ashworth MT, Strosberg J, et al. Phase I dose‐escalation trial of checkpoint kinase 1 inhibitor MK‐8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol: Off J Am Soc Clin Oncol. 2015;33(9):1060‐1066. [DOI] [PubMed] [Google Scholar]
- 33. Sanjiv K, Hagenkort A, Calderon‐Montano JM, et al. Cancer‐specific synthetic lethality between ATR and CHK1 kinase activities. Cell Rep. 2016;14(2):298‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aarts M, Sharpe R, Garcia‐Murillas I, et al. Forced mitotic entry of S‐phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discovery. 2012;2(6):524‐539. [DOI] [PubMed] [Google Scholar]
- 35. Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle (Georgetown, Tex). 2013;12(19):3159‐3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia‐telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24):9152‐9159. [DOI] [PubMed] [Google Scholar]
- 37. Peasland A, Wang LZ, Rowling E, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011;105(3):372‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tu X, Kahila MM, Zhou Q, et al. ATR inhibition is a promising radiosensitizing strategy for triple‐negative breast cancer. Mol Cancer Ther. 2018;17(11):2462‐2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Angius G, Tomao S, Stati V, Vici P, Bianco V, Tomao F. Prexasertib, a checkpoint kinase inhibitor: from preclinical data to clinical development. Cancer Chemother Pharmacol. 2020;85(1):9‐20. [DOI] [PubMed] [Google Scholar]
- 40. Schmidt M, Rohe A, Platzer C, Najjar A, Erdmann F, Sippl W. Regulation of G2/M transition by inhibition of WEE1 and PKMYT1 kinases. Molecules (Basel, Switzerland). 2017;22(12):2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin ZP, Zhu YL, Ratner ES. Targeting cyclin‐dependent kinases for treatment of gynecologic cancers. Front Oncol. 2018;8:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Willmore E, de Caux S, Sunter NJ, et al. A novel DNA‐dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103(12):4659‐4665. [DOI] [PubMed] [Google Scholar]
- 43. Neher TM, Bodenmiller D, Fitch RW, Jalal SI, Turchi JJ. Novel irreversible small molecule inhibitors of replication protein A display single‐agent activity and synergize with cisplatin. Mol Cancer Ther. 2011;10(10):1796‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gavande NS, VanderVere‐Carozza PS, Hinshaw HD, et al. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol Ther. 2016;160:65‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP‐ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109‐8115. [DOI] [PubMed] [Google Scholar]
- 46. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588‐5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mateo J, Carreira S, Sandhu S, et al. DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697‐1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leijen S, van Geel RM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53‐mutated ovarian cancer refractory or resistant to first‐line therapy within 3 months. J Clin Oncol: Off J Am Soc Clin Oncol. 2016;34(36):4354‐4361. [DOI] [PubMed] [Google Scholar]
- 49. He T, Zhang M, Zheng R, et al. Methylation of SLFN11 is a marker of poor prognosis and cisplatin resistance in colorectal cancer. Epigenomics. 2017;9(6):849‐862. [DOI] [PubMed] [Google Scholar]
- 50. Peng Y, Wang L, Wu L, Zhang L, Nie G, Guo M. Methylation of SLFN11 promotes gastric cancer growth and increases gastric cancer cell resistance to cisplatin. J Cancer. 2019;10(24):6124‐6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murai J, Feng Y, Yu GK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534‐76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pilie PG, Tang C, Mills GB, Yap TA. State‐of‐the‐art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16(2):81‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110‐120. [DOI] [PubMed] [Google Scholar]
- 54. Allison Stewart C, Tong P, Cardnell RJ, et al. Dynamic variations in epithelial‐to‐mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget. 2017;8(17):28575‐28587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang L, Zhang Y, Shan W, et al. Repression of BET activity sensitizes homologous recombination‐proficient cancers to PARP inhibition. Sci Trans Med. 2017;9(400):eaal1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun C, Yin J, Fang Y, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33(3):401‐416.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mo W, Liu Q, Lin CC, et al. mTOR inhibitors suppress homologous recombination repair and synergize with PARP inhibitors via regulating SUV39H1 in BRCA‐proficient triple‐negative breast cancer. Clin Cancer Res. 2016;22(7):1699‐1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun C, Fang Y, Yin J, et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Trans Med. 2017;9(392):eaal5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ibrahim YH, Garcia‐Garcia C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA‐proficient triple‐negative breast cancer to PARP inhibition. Cancer Discovery. 2012;2(11):1036‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Matulonis UA, Wulf GM, Barry WT, et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high‐grade serous ovarian and breast cancer. Ann Oncol: Off J Eur Soc Med Oncol. 2017;28(3):512‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matthaei H, Schulick RD, Hruban RH, Maitra A. Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2011;8(3):141‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Borazanci E, Dang CV, Robey RW, Bates SE, Chabot JA, Von Hoff DD. Pancreatic cancer: “A riddle wrapped in a mystery inside an enigma”. Clin Cancer Res. 2017;23(7):1629‐1637. [DOI] [PubMed] [Google Scholar]
- 63. Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Trans Med. 2011;3(92):92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424‐430. [DOI] [PubMed] [Google Scholar]
- 65. Harder J, Ihorst G, Heinemann V, et al. Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Br J Cancer. 2012;106(6):1033‐1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Casey G, Yamanaka Y, Friess H, et al. p53 mutations are common in pancreatic cancer and are absent in chronic pancreatitis. Cancer Lett. 1993;69(3):151‐160. [DOI] [PubMed] [Google Scholar]
- 67. Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53‐mediated G1 arrest in human cancer cells. Cancer Res. 1995;55(22):5187‐5190. [PubMed] [Google Scholar]
- 68. Bartek J, Lukas J. Mammalian G1‐ and S‐phase checkpoints in response to DNA damage. Curr Opin Cell Biol. 2001;13(6):738‐747. [DOI] [PubMed] [Google Scholar]
- 69. Massagué J. The TGF‐beta family of growth and differentiation factors. Cell. 1987;49(4):437‐438. [DOI] [PubMed] [Google Scholar]
- 70. Ellenrieder V, Hendler SF, Boeck W, et al. Transforming growth factor beta1 treatment leads to an epithelial‐mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal‐regulated kinase 2 activation. Cancer Res. 2001;61(10):4222‐4228. [PubMed] [Google Scholar]
- 71. Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nature Gen. 1994;8(1):27‐32. [DOI] [PubMed] [Google Scholar]
- 72. Goldstein AM, Fraser MC, Struewing JP, et al. Increased risk of pancreatic cancer in melanoma‐prone kindreds with p16INK4 mutations. N Engl J Med. 1995;333(15):970‐974. [DOI] [PubMed] [Google Scholar]
- 73. Jiang X, Hao HX, Growney JD, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA. 2013;110(31):12649‐12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89(6):442‐446. [DOI] [PubMed] [Google Scholar]
- 76. Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz‐Jeghers syndrome. Gastroenterology. 2000;119(6):1447‐1453. [DOI] [PubMed] [Google Scholar]
- 77. Hruban RH, Canto MI, Goggins M, Schulick R, Klein AP. Update on familial pancreatic cancer. Adv Surg. 2010;44:293‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wood LD, Yurgelun MB, Goggins MG. Genetics of familial and sporadic pancreatic cancer. Gastroenterology. 2019;156(7):2041‐2055. [DOI] [PubMed] [Google Scholar]
- 79. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol: Off J Am Soc Clin Oncol. 2017;35(30):3382‐3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Salo‐Mullen EE, O'Reilly EM, Kelsen DP, et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer. 2015;121(24):4382‐84388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ferrone CR, Levine DA, Tang LH, et al. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol: Off J Am Soc Clin Oncol. 2009;27(3):433‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. House MG, Herman JG, Guo MZ, et al. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003;238(3):423‐431; discussion 31‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Guo M, Jia Y, Yu Z, et al. Epigenetic changes associated with neoplasms of the exocrine and endocrine pancreas. Discovery Med. 2014;17(92):67‐73. [PMC free article] [PubMed] [Google Scholar]
- 85. Fu B, Guo M, Wang S, et al. Evaluation of GATA‐4 and GATA‐5 methylation profiles in human pancreatic cancers indicate promoter methylation patterns distinct from other human tumor types. Cancer Biol Ther. 2007;6(10):1546‐1552. [DOI] [PubMed] [Google Scholar]
- 86. Yi JM, Guzzetta AA, Bailey VJ, et al. Novel methylation biomarker panel for the early detection of pancreatic cancer. Clin Cancer Res. 2013;19(23):6544‐6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. House MG, Herman JG, Guo MZ, et al. Prognostic value of hMLH1 methylation and microsatellite instability in pancreatic endocrine neoplasms. Surgery. 2003;134(6):902‐908; discussion 9. [DOI] [PubMed] [Google Scholar]
- 88. Maekawa H, Ito T, Orita H, et al. Analysis of the methylation of CpG islands in the CDO1, TAC1 and CHFR genes in pancreatic ductal cancer. Oncol Lett. 2020;19(3):2197‐2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li JY, Han C, Zheng LL, Guo MZ. Epigenetic regulation of Wnt signaling pathway gene SRY‐related HMG‐box 17 in papillary thyroid carcinoma. Chin Med J. 2012;125(19):3526‐3531. [PubMed] [Google Scholar]
- 90. Yu Y, Yin D, Hoque MO, et al. AKT signaling pathway activated by HIN‐1 methylation in non‐small cell lung cancer. Tumour Biol: J Int Soc Oncodev Biol Med. 2012;33(2):307‐314. [DOI] [PubMed] [Google Scholar]
- 91. Zhang M, Linghu E, Zhan Q, et al. Methylation of DACT2 accelerates esophageal cancer development by activating Wnt signaling. Oncotarget. 2016;7(14):17957‐17969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cao B, Yang W, Jin Y, et al. Silencing NKD2 by promoter region hypermethylation promotes esophageal cancer progression by activating Wnt signaling. J Thoracic Oncol: Off Publ Int Assoc Study Lung Cancer. 2016;11(11):1912‐1926. [DOI] [PubMed] [Google Scholar]
- 93. Feng Z, Scott SP, Bussen W, et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. 2011;108(2):686‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hung YH, Hsu MC, Chen LT, Hung WC, Pan MR. Alteration of epigenetic modifiers in pancreatic cancer and its clinical implication. J Clin Med. 2019;8(6):903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Miller AL, Fehling SC, Garcia PL, et al. The BET inhibitor JQ1 attenuates double‐strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine. 2019;44:419‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lohse I, Borgida A, Cao P, et al. BRCA1 and BRCA2 mutations sensitize to chemotherapy in patient‐derived pancreatic cancer xenografts. Br J Cancer. 2015;113(3):425‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA‐mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yarchoan M, Myzak MC, Johnson BA 3rd, et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget. 2017;8(27):44073‐44081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. de Bono J, Ramanathan RK, Mina L, et al. Phase I, dose‐escalation, two‐part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discovery. 2017;7(6):620‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gordon MS, Rosen LS, Mendelson D, et al. A phase 1 study of TRC102, an inhibitor of base excision repair, and pemetrexed in patients with advanced solid tumors. Invest New Drugs. 2013;31(3):714‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Glanzer JG, Liu S, Wang L, Mosel A, Peng A, Oakley GG. RPA inhibition increases replication stress and suppresses tumor growth. Cancer Res. 2014;74(18):5165‐5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shuck SC, Turchi JJ. Targeted inhibition of Replication Protein A reveals cytotoxic activity, synergy with chemotherapeutic DNA‐damaging agents, and insight into cellular function. Cancer Res. 2010;70(8):3189‐3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Barakat KH, Jordheim LP, Perez‐Pineiro R, Wishart D, Dumontet C, Tuszynski JA. Virtual screening and biological evaluation of inhibitors targeting the XPA‐ERCC1 interaction. PloS One. 2012;7(12):e51329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jordheim LP, Barakat KH, Heinrich‐Balard L, et al. Small molecule inhibitors of ERCC1‐XPF protein‐protein interaction synergize alkylating agents in cancer cells. Mol Pharmacol. 2013;84(1):12‐24. [DOI] [PubMed] [Google Scholar]
- 105. Dupré A, Boyer‐Chatenet L, Sattler RM, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11‐Rad50‐Nbs1 complex. Nat Chem Biol. 2008;4(2):119‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhu J, Zhou L, Wu G, et al. A novel small molecule RAD51 inactivator overcomes imatinib‐resistance in chronic myeloid leukaemia. EMBO Mol Med. 2013;5(3):353‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Li K, Yan H, Guo W, et al. ATM inhibition induces synthetic lethality and enhances sensitivity of PTEN‐deficient breast cancer cells to cisplatin. Exp Cell Res. 2018;366(1):24‐33. [DOI] [PubMed] [Google Scholar]
- 108. Biddlestone‐Thorpe L, Sajjad M, Rosenberg E, et al. ATM kinase inhibition preferentially sensitizes p53‐mutant glioma to ionizing radiation. Clin Cancer Res. 2013;19(12):3189‐3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lloyd RL, Wijnhoven PWG, Ramos‐Montoya A, et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM‐deficient cancer cells. Oncogene. 2020;39(25):4869‐4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mani C, Jonnalagadda S, Lingareddy J, Awasthi S, Gmeiner WH, Palle K. Prexasertib treatment induces homologous recombination deficiency and synergizes with olaparib in triple‐negative breast cancer cells. Breast Cancer Res: BCR. 2019;21(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Laquente B, Lopez‐Martin J, Richards D, et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer. 2017;17(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wehler T, Thomas M, Schumann C, et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non‐small cell lung cancer. Lung Cancer (Amsterdam, Netherlands). 2017;108:212‐216. [DOI] [PubMed] [Google Scholar]
- 113. Cuneo KC, Morgan MA, Sahai V, et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol: Off J Am Soc Clin Oncol. 2019;37(29):2643‐2650. [DOI] [PMC free article] [PubMed] [Google Scholar]