

Abstract
A pediatric patient diagnosed initially with B-lymphoblastic leukemia (B-ALL) relapsed with lineage switch to acute myeloid leukemia (AML) after chimeric antigen receptor T-cell (CAR-T) therapy and hematopoietic stem cell transplant. A TCF3-ZNF384 fusion was identified at diagnosis, persisted through B-ALL relapse, and was also present in the AML relapse cell population. ZNF384-rearrangements define a molecular subtype of B-ALL characterized by a pro-B-cell immunophenotype; furthermore, ZNF384-rearrangements are prevalent in mixed-phenotype acute leukemias. Lineage switch following CAR-T therapy has been described in patients with KMT2A (mixed lineage leukemia) rearrangements, but not previously in any patient with ZNF384 fusion.
Keywords: AML, B-ALL, CAR-T, leukemia
1 |. INTRODUCTION
CD19-specific chimeric antigen receptor (CAR)–modified T-cell therapy is effective for relapsed and refractory CD19-positive B-cell malignancies;1 however, a significant number of patients still relapse. Some of these disease recurrences are associated with inadequate persistence of the CD19 chimeric antigen receptor T cell (CAR-T), or with emergence of leukemic clones that have lost expression of the targeted CD19 epitope due to mutations and splicing variants in the CD19 gene.2,3 A more rarely observed relapse mechanism post-CD19 immunotherapy is a myeloid lineage switch.3 This phenomenon has been described in late relapses in murine models bearing E2A-PBX1 leukemia,4 in Ph-positive B-cell acute lymphoblastic leukemia (BALL),5 and in patients with B-ALL harboring rearrangements of the KMT2A (mixed lineage leukemia) gene.3 Here, we describe a pediatric patient with a ZNF384-rearranged leukemia who was diagnosed initially with B-ALL, but who relapsed with a clonally related acute myeloid leukemia (AML) after the third CD19 CAR-T treatment and second hematopoietic stem cell transplant (HSCT).
2 |. RESULTS
The patient was initially diagnosed with B-ALL at 13 months of age. Presenting features were leukocyte count 300,000 cells/cu·mm and CNS2 (leukocyte count <5/cu·mm with blasts on cytospin in cerebrospinal fluid). The immunophenotype was remarkable for a lack of CD10 expression (Table 1). Karyotype analysis showed a deletion in the short arm of chromosome 12 as a sole anomaly. A chemotherapy regimen for high-risk ALL was initiated with vincristine, daunorubicin, dexamethasone, and peg-asparaginase. Bone marrow (BM) examination at the end of induction was positive for minimal residual disease (MRD) (0.039%). The patient achieved MRD-negative remission at the end of consolidation chemotherapy (cyclophosphamide, cytarabine, mercaptopurine, vincristine, peg-asparaginase), but developed an early BM relapse 3 months later. He had little response to salvage cytotoxic therapy with clofarabine, etoposide, and cyclophosphamide, and was referred for a CD19 CAR-T clinical trial using a second-generation anti-CD19, 4–1BB/CD3ζ vector (NCT02435849). The initial CAR-T therapy resulted in a short-lived MRD-negative remission (4-months), followed by re-emergence of MRD (0.09%). Since the residual blasts were still CD19-positive, he underwent a second CAR-T infusion with the same construct, without response (MRD 0.08%). The patient then proceeded to a HSCT from a matched unrelated donor, but was diagnosed with a CD19+ relapse 4 months after the procedure, and was then treated on a second CAR-T study using a CD28tm/4–1BBζ-T2A-EGFRt CAR vec tor (PLAT02 trial—NCT02028455). This resulted in an MRD-negative remission, and he subsequently underwent a second HSCT (unrelated cord blood). Chimeric antigen receptor T-cell DNA was detectable from infusion until beginning conditioning for the second HCST, about 9 weeks later. Approximately 8 months post-HSCT, detection of recipient chimerism prompted further evaluation. Surprisingly, the subsequent morphologic and immunophenotypic examination of a BM aspirate and biopsy was consistent with AML with minimal differentiation and 26% blasts (Table 1). The blasts expressed exclusively myeloid markers by flow cytometry (although Pax5 was weakly positive by immunohistochemistry) and no residual B-lymphoblasts were present. The karyotype was normal, however FISH testing showed a loss of the ETV6 (12p13) signal, consistent with the 12p deletion detected at diagnosis. FISH results thus suggested that the leukemia was clonally related to the pre-HSCT B-ALL, and the disease was interpreted as a relapse with lineage switch rather than a new primary leukemia or therapy-related myeloid neoplasm. The patient received palliative care and expired from progressive disease 4 months after the last relapse.
TABLE 1.
Pathology and genetic testing results for tumor samples
| Pathology | Karyotype | FISH | Chromosomal microarray analysis | DNA mutations (OncoKids) | Gene fusions (OncoKids and/or RT-PCR) | |
|---|---|---|---|---|---|---|
| Diagnosis | Immunophenotype (flow cytometry): positive for CD19, CD20 (partial), CD22, CD34, CD38, CD13, CD33 (partial), and HLA-DR; negative for CD10, MPO, andTdT | 46,XY,del(12)(p13) [3]/46,XY[27] | Negative for BCR/ABL1, ETV6/RUNX1, PBX1/TCF3 fusions and PDGFRB and KMT2A (mixed lineage leukemia) rearrangements | Not performed | Not performed | TCF3 Exon 11 (ENST00000262965)-ZNF384 Exon 2 (ENST00000396795) |
| B-ALL relapse, CD19- positive | Immunophenotype (flow cytometry): CD19 variable (dim to moderate), CD22 bright, CD10 variable (negative to dim), TdT, CD34, CD38, CD58, HLA-DR bright, CD123; negative for CD24, CD13, CD33, and MPO | 46,XY[1]//46,XX[19] (posttransplant chimerism) | Positive for a loss of ETV6 (12p13) signal in 20% of the cells | Not performed | Not performed | TCF3 Exon 11 (ENST00000262965)-ZNF384 Exon 2 (ENST00000396795) |
| AML relapse, lineage switch | Immunophenotype (flow cytometry): positive for CD13, CD33 (partial, dim), CD34, CD117, CD123,CD11b (partial), CD38 (moderate) and CD7; negative for CD19, CD10, CD20, CD24, MPO, TdT, and CD22 | 46,XY[20] | Positive for a loss of ETV6 (12p13) signal in 18% of the cells | Copy number loss in: 7q36.1q36.3 (10.8 Mb), 12p13.31p12.3 (9.8 Mb), 13p13.31p12.3 (86.5 Mb), 16 q12.2q23.1 (19.6 Mb), 18 p11.32p11.21 (11.6 Mb) and 21q21.3q22.11 (5 Mb) Copy number gain in 8q21.11q24.3 (71.4 Mb) | NM 004119 (FLT3) c.1788_1789ins GGCCCTGATTT CAGAGAA (p.Glu596_Tyr597insGlyPro AspPheArgGlu) | TCF3 Exon 11 (ENST00000262965)-ZNF384 Exon 2 (ENST00000396795) |
Chromosome microarray (CMA) analysis was performed on the AML sample to evaluate for submicroscopic copy number abnormalities. CMA results were remarkable for a 12p13 deletion with a breakpoint within the ZNF384 gene, suggesting the presence of a ZNF384 fusion (Table 1). Testing with a custom next-generation sequencing (NGS) based panel (OncoKids™) revealed the presence of a TCF3-ZNF384 fusion (Figure 1)6–9 and an internal tandem duplication in the FLT3 gene. To determine if the TCF3-ZNF384 fusion was present in the patient’s B-ALL, RT-PCR and Sanger sequencing were performed using RNA from an FFPE clot sample from the BM aspirate obtained at the time of the CD19-positive relapse after the first HSCT. This analysis confirmed the presence of the TCF3- ZNF384 fusion at the time the patient was still presenting with B-ALL (Figure 1), and subsequent testing of archival diagnostic material was also positive for the TCF3-ZNF384 fusion by RT-PCR.
FIGURE 1.
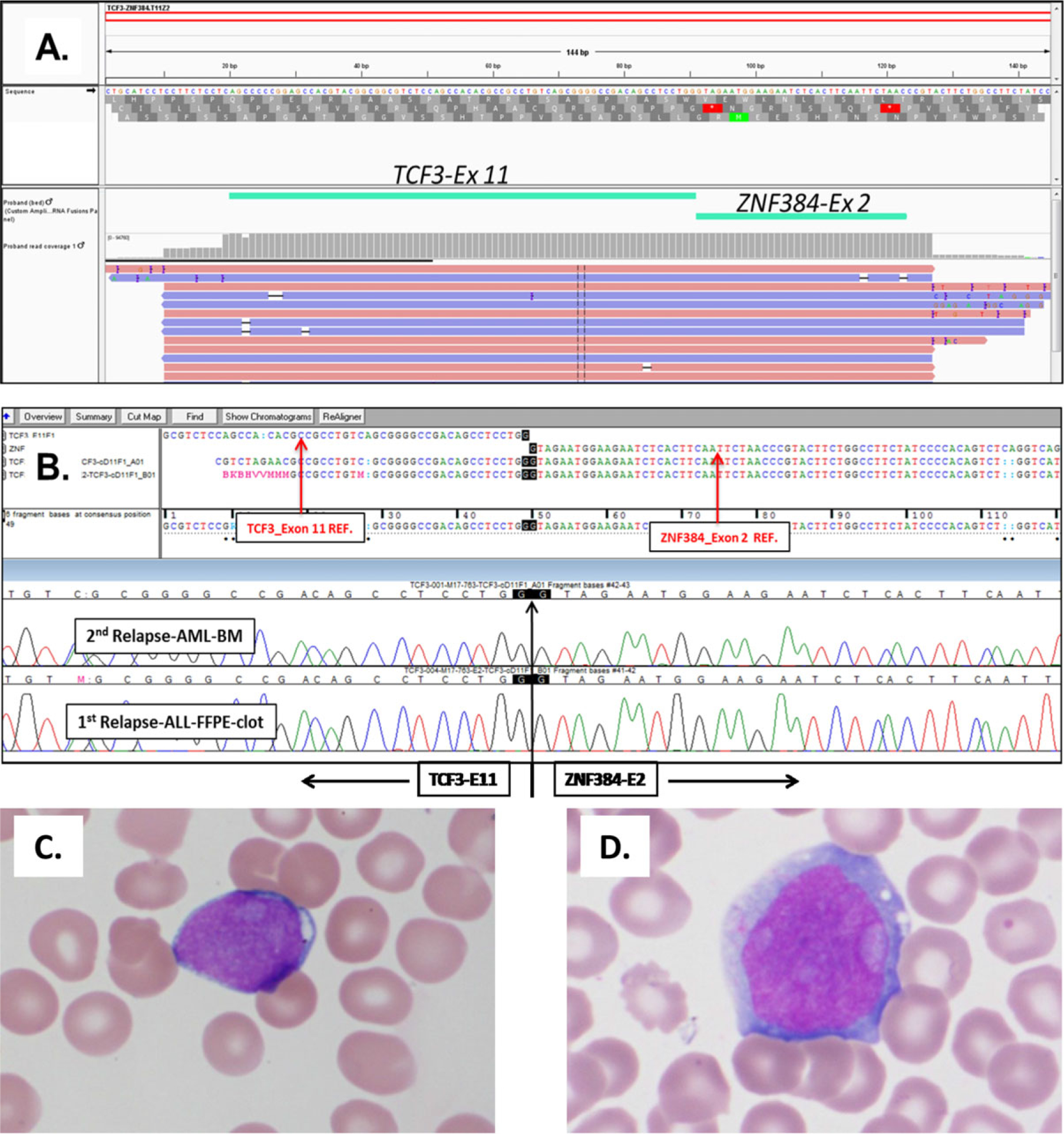
A TCF3-ZNF384 fusion is detected at diagnosis and following lineage switched relapse. Panel (A): A screenshot from the Integrated Genome Viewer showing the junction reads that support the presence of a TCF3-ZNF384 fusion. The fusion occurred between the TCF3 Exon 11 (ENST00000262965) and ZNF384 Exon 2 (ENST00000396795), and was supported by 94,759 reads (out of 1,775,839 total mapped reads). Panel (B): Confirmation of the TCF3-ZNF384 fusion by RT-PCR and Sanger sequencing in the AML sample at the time of relapse after the second HSCT (top chromatogram) and in the B-ALL sample at relapse after the first HSCT (bottom chromatogram). Panel (C): Wright Giemsa stain of BM aspirate at time of B-ALL relapse. Blasts were characterized by intermediate size, high N:C ratio, scant agranular vacuolated cytoplasm, and open nuclear chromatin. Panel (D): Wright Giemsa stain of BM aspirate at the time of AML relapse. Blasts were characterized by large size, open chromatin, and ample granular cytoplasm
3 |. METHODS
Karyotype analysis and FISH testing were performed following standard procedures. Nucleic acids were extracted using the Promega Maxwell RSC DNA and simplyRNA Blood Kit (BM DNA and RNA, respectively) and Agencourt FormaPure Kit (FFPE RNA). The Affymetrix CytoScan™ HD arrays (Thermofisher Scientific, Waltham, MA) were used for CMA analysis. Molecular testing of leukemia cells was done using the custom Children’s Hospital Los Angeles OncoKids™ panel, which is a PCR-enrichment-based (AmpliSeq) NGS assay designed to detect diagnostic, prognostic, and therapeutic markers in pediatric malignancies. Sequencing was performed using the 540 chip on the Ion Torrent S5 sequencing platform (Thermofisher), and data analysis was conducted by Ion Reporter (Thermofisher) and an in-house custom software suite. Confirmation of the abnormal fusion by RT-PCR was done using custom primers for TCF3 exon 11 and ZNF384 exon 2, with cDNA synthesis performed by SuperScript™ III Reverse Transcriptase (Thermofisher).
4 |. DISCUSSION
Abnormal gene fusions involving ZNF384 define a specific molecular subtype of B-ALL, characterized by a unique gene expression profile and distinctive immunophenotype with weak or absent CD10 and aberrant CD13 and/or CD33 expression.6–9 Additionally, ZNF384 fusions have been observed with a particularly high frequency in childhood acute leukemias with a mixed B/myeloid phenotype.10 Atleast eight different ZNF384 fusion partners have been reported to date.7–9 While as a group ALLs with ZNF384 fusions show standard risk features,7–9 some studies suggest that the cases with the TCF3-ZNF384 fusion may be characterized by higher cell counts, younger age at presentation, significantly poorer steroid response, and a higher frequency of relapse.11
The TCF3- ZNF384 fusion is generated by a reciprocal translocation between chromosomes 12 (ZNF384 in 12p13) and 19 (TCF3 in 19p13), which is cryptic by conventional cytogenetic analysis and typically remains undetected. However, as suggested by previous studies and illustrated by our patient’s aggressive clinical course, recognition of this abnormality at diagnosis may be of high prognostic importance.
In the current case, the TCF3-ZNF384 fusion was identified both in the diagnostic B-ALL and in the relapsed AML with identical breakpoints. In addition, the diagnostic and relapse specimens both showed identical 12p13 deletions. This effectively eliminates the possibility that the relapsed disease represents a therapy-related AML.
Besides mutations leading to loss of CD19 expression, a myeloid lineage switch has also been observed as a relapse mechanism post-CD19-directed immunotherapy, and its probability may be dependent on the genetic subtype and key oncogenic driver in leukemia cells. The lineage switch under CD19 CAR-T selective pressure may occur through reprogramming and dedifferentiation of previously committed B-lymphoid blasts, or more likely through myeloid differentiation of a noncommitted leukemic stem cell.2,3 Lineage switch is a rare event in ALL, but has been well described in KMT2A-rearranged cases after intensive chemotherapy, and recently also after treatment with CD19-targeting agents. The propensity for a lineage switch in KMT2A-rearranged leukemias has been ascribed to their differentiation arrest at the early pro-B cell stage.2 Based on some shared features with KMT2A-rearranged ALLs (including lack of CD10 expression, co-expression of myeloid markers, enrichment for hematopoietic stem cell expression signatures,11 and high incidence in mixed phenotype leukemias), we hypothesize that ALLs with the TCF3-ZNF384 fusion may also have increased probability of relapse with lineage switch post-anti-CD19 immunotherapy
Patients who relapse with CD19 negative disease after CAR-T treatment have a very poor prognosis; understanding risk factors for such relapse and developing novel prevention and treatment strategies are thus of the highest importance. Additional data need to be collected to determine if ZNF384 fusions represent a risk factor for lineage switch following CD19-directed therapy.
ACKNOWLEDGMENTS
M.A.P. received a support by U10 HL069254 and a Johnny Cristopher Children’s Charitable Foundation St. Baldrick’s Consortium Grant. A.S.W. received a support in part by NCI P30CA014089. We gratefully acknowledge the patient and family and the clinical care and research teams.
Abbreviations:
- AML
acute myeloid leukemia
- B-ALL
B-cell acute lymphoblastic leukemia
- BM
bone marrow
- CAR-T
chimeric antigen receptor T cell
- CMA
chromosomal microarray
- HSCT
hematopoietic stem cell transplant
- MRD
minimal residual disease
- NGS
next-generation sequencing
Footnotes
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
REFERENCES
- 1.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J. 2016;14:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacoby E, Nguyen SM, Fountaine TJ, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;27:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagel I, Bartels M, Duell J, et al. Hematopoietic stem cell involvement in BCR-ABL1-positive ALL as potential mechanism of resistance to blinatumomab therapy. Blood. 2017; 130:2027–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gocho Y, Kiyokawa N, Ichikawa H, et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2015;29(12):2445–2448. [DOI] [PubMed] [Google Scholar]
- 7.Liu YF, Wang BY, Zhang WN, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian M, Zhang H, Kham SK, et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 2017;27:185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasuda T, Tsuzuki S, Kawazu M, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569–574. [DOI] [PubMed] [Google Scholar]
- 10.Alexander TB, Gu Z, Choi JK, et al. Genomic landscape of pediatric mixed phenotype acute leukemia. Blood. 2016;128:454. [Google Scholar]
- 11.Hirabayashi S, Ohki K, Nakabayashi K, et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102:118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armstrong SA, Mabon ME, Silverman LB, et al. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004;103:3544–3546. [DOI] [PubMed] [Google Scholar]