

Abstract
Circulating tumor cells (CTCs) are extremely rare in the blood, yet they account for metastasis. Notably, it was reported that CTC clusters (CTCCs) can be 50–100 times more metastatic than single CTCs, making them particularly salient as a liquid biopsy target. Yet they can split apart and are even rarer, complicating their recovery. Isolation by filtration risks loss when clusters squeeze through filter pores over time, and release of captured clusters can be difficult. Deterministic lateral displacement is continuous but requires channels not much larger than clusters, leading to clogging. Spiral inertial focusing requires large blood dilution factors (or lysis). Here, we report a microfluidic chip that continuously isolates untouched CTC clusters from large volumes of minimally (or undiluted) whole blood. An array of 100-μm-wide channels first concentrates clusters in the blood, and then a similar array transfers them into a small volume of buffer. The microscope-slide-sized PDMS device isolates individually-spiked CTC clusters from >30 mL/hour of whole blood with 80% efficiency into enumeration (fluorescence imaging), and on-chip yield approaches 100% (high speed video). Median blood cell removal (in base-10 logs) is 4.2 for leukocytes, 5.5 for red blood cells, and 4.9 for platelets, leaving less than 0.01% of leukocytes alongside CTC clusters in the product. We also demonstrate that cluster configurations are preserved. Gentle, high throughput concentration and separation of circulating tumor cell clusters from large blood volumes will enable cluster-specific diagnostics and speed the generation of patient-specific CTC cluster lines.
Graphical Abstract
Rare CTC clusters can be purified intact from large blood volumes with a continuous three-stage non-equilibrium inertial separation array (NISA).
Introduction
Circulating tumor cells (CTCs) seed metastasis by traveling through the peripheral blood of cancer patients 1, making them key actors in cancer progression and an important liquid biopsy target for personalized oncology. Circulating tumor cell clusters (CTCCs) have been inferred as fifty-fold more metastatic 2 than CTCs, and they have distinct epigenetic markers 3. Because CTC clusters enter the bloodstream with their nearest neighbors from the tumor, they are hypothesized to be easier to culture for drug testing 4. However, they can be weakly bound to each other and are even rarer than single CTCs 5,6. Isolating CTCCs from large blood volumes will unlock a unique reservoir of biomarkers linked to a most metastatic population of tumor cells 7, and it will enable frequent, minimally-invasive sampling of the CTCC genome, transcriptome, and proteome, and in vitro drug testing on patient-specific cell lines.
However, CTCC isolation is a major technical challenge. Even from metastatic patients, there may be just 1–2 clusters in a tube of blood (7.5 mL nominal volume), so it will be necessary to sift through multiple vacutainers to achieve a reliable CTCC-based assay. In four tubes of blood, there are about 150 billion erythrocytes (RBCs), 9 billion platelets, and 180 million leukocytes (WBCs). These must be removed to a high degree without losing any CTCCs. Moreover, separation should be rapid to avoid drift or degradation of CTCC-derived molecular signatures, yet gentle to avoid breaking clusters and uncoupling their linked bioinformatic signatures. These constraints directly oppose each other.
Three primary technologies are used to isolate CTCCs. First, CTC clusters can be captured by positive selection, (e.g. by EpCAM antibody binding 8–10). However, not all CTCs express membrane-bound EpCAM, for example after the epithelial-mesenchymal transition 11,12 or in non-epithelial melanoma. Second, WBCs can be removed from CTCCs by targeting CD45. This negative selection strategy is employed by our CTC-iChip 13, where bead-labeled leukocytes are removed by magnetic activated cell sorting (MACS), after RBC size-sorting upstream. However, CTC clusters can be lost in negative selection if they associate with WBCs 14. A third approach is simplest, separating CTCCs from blood by their larger size or different shape. Single CTCs often overlap with WBCs in size 13,15, yet even two-cell clusters are sufficiently larger than WBCs to allow a clean separation, and when sorted by their long-axis16, this difference can be maximized.
Size-based filtration is a simple way to isolate CTCCs 17,18, yet clusters can squeeze through surprisingly tiny pores if given enough time 19. To eliminate this loss, a microfluidic see-saw can trap CTCCs at low shear stress, but release is complicated by nonspecific adhesion to the substrate 20. More recently, deterministic lateral displacement (DLD) was used to isolate CTCCs 16. DLDs require channels near in size to sorted cells to ensure bumping across streamlines and thus struggle to process large blood volumes without clogging 21 (platelet-targeted additives can aid processing 22).
To minimize clogging, channels can be enlarged and flow rates increased to reduce residence time of blood in the chip. Fluid forces can also be leveraged to shift CTCCs and blood cells away from channel walls, further reducing clogging and facilitating separation in larger channels. Inertial focusing 23 and acoustic focusing 24 are particularly useful. For example, inertial lift has been used to trap large cells in micro-vortex traps, processing blood at 22.5 mL/hr 25, but wall shear stress is too high in narrow intervening channels to keep CTCCs intact (up to 1000 Pa, 200× peak arterial). Inertial focusing in spirals has lower wall shear stress and can process blood at about 3 mL/hr with dilution 26. Yet spirals require Dean flow 27 to drag cells laterally, resulting in mixing and carryover to the product of unfocused material (e.g. RBC fragments, platelets, plasma). Carryover can be reduced by lysing the RBCs 28. However, this precludes apheresis, and adds stress to clusters. Acoustics can also process blood at low shear 29, yet purity is lower in comparison.
Recently, we created a microfluidic method for size-sorting, a non-equilibrium inertial separation array (NISA) 30, combining inertial focusing with repetitive flow-shifting. In contrast to DLD, where cells cross streamlines when nudged by a post, NISA devices use inertial lift to push cells away from channel walls in a size-dependent manner. Instead of small DLD posts and narrow channels, NISA devices incorporate longer rectangular islands, between which inertial focusing occurs in relatively wider channels (see Fig. 1A). At the end of each channel segment between islands, about 3–4% of the flow is siphoned through a narrow gap between one island and the next. Because larger cells remain in the channels and smaller cells are siphoned, this progressively moves large cells to an upper product lane, leaving small cells to exit below. By injecting 15–20% sample (1:1 diluted blood), we were able to isolate leukocytes from 400 mL blood in 3 hours and remove >99.99% of RBCs, sorting 300 million cells a second in 104 parallel arrays 30. Yet wall shear stress was ~100 Pa, and channels were only 50 μm wide, not optimized for gentle isolation of CTC clusters.
Figure 1:

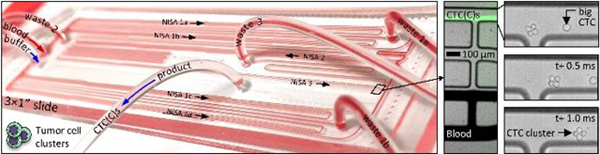
Size-based CTC and CTC cluster (CTCC) sorter built from enlarged non-equilibrium inertial separation arrays (NISA-XL). A In a first and second stage of NISA, with 100% sample injection, CTCs and CTC clusters (CTCCs) are concentrated in blood. Next, a third stage of NISA transfers CTC(C)s into a co-flowing buffer. B Photo is of fully-integrated slide-sized PDMS chip as it processes blood. The green streaks in CD are fluorescent spiked CTCs in blood or buffer (CDE locations are marked in B). E Image sequence shows one 4-cell CTC cluster and a single CTC passing the final product-waste split in stage 3. Scale bars in CDE are 100 μm.
Here, we adapt NISA to rapid, gentle, size-sorting of CTCCs from blood. By increasing separation channel width two-fold, height three-fold, and adjusting array parameters (island length, flow shift percentage), the size cutoff rises to between the largest leukocytes and smallest CTC clusters (or large single CTCs), ~15 μm. In this manner, wall shear stress falls 7× to <15 Pa in six-fold larger cross-section channels, enabling larger clusters to pass. To speed blood processing, we also created a device architecture where CTC(C)s are first concentrated by about ten times in the blood without a coflow. Then, CTC(C)s are separated into clean buffer by a final stage of NISA (15% injection of sample alongside buffer). After tuning flow rate for the desired size cutoff and establishing high yield isolation of intact CTCCs by high speed video, performance is reported for runs of 15 mL whole blood that take ~30 min each. Depletion levels are high at 4.2 for WBCs, 5.5 for RBCs, and 4.9 for platelets (in base-10 logs). In addition, five 2-cell and three 3-cell CTCCs were manually spiked into multiple 15-mL blood aliquots and detected in product wells.
Experimental
NISA-XL design.
To adapt the NISA technology for gentle isolation of large single CTCs and ultra-rare/potentially-fragile CTC clusters, we had to redesign the existing NISA devices that were built for leukocyte separation 30. First, the size cutoff had to rise from a level between RBCs and WBCs (~7μm) to above the vast majority of WBCs/smaller CTCs yet remain below that of large CTCs and two-cell clusters (~18–20μm). To settle on a NISA design, candidate devices were first generated to the appropriate length scale with the help of dynamic similarity to prior NISA chips. These designs were tested as single arrays where spiked CTC/cluster yield and blood cell depletion were the key performance metrics. Devices also needed to operate at high sample flow rate while incurring minimal wall shear stress and fit on a standard microscope slide (≤7.5 cm long) to fit fabrication constraints. Ultimately, the chosen NISA design enlarged the channels from 50 to 100 μm wide and extended to 156 μm tall while increasing channel length from 200 to 450 μm (Fig. 2A, at left). As depicted in Fig. 2A, size sorting occurs at the break between islands at the end of each channel. Here, fluid siphoning percentage was set to 3.6% of the main channel flow at each break between islands to achieve size sorting in the desired range. Particles that migrated beyond the red streamline remain on the +y side of the island, while smaller particles remain closer to the −y wall and are siphoned back to the next lane in the −y direction. The computational fluid dynamics model of one NISA-XL device (Fig. 2G) was performed with ANSYS 13 Fluent (mesh: 3.9 million prismatic elements; midplane symmetry used to cut model in half). At 200 uL/min per array (0.8 mL/min total), peak wall shear stress is <15 Pa (Fig. 2G) for about 4 seconds, close to the peak shear stresses experienced in circulation (up to about 5 Pa or much higher in stenotic arteries 31), although cancer cells will have different sensitivities to shear stress 32.
Figure 2:

Sorting mechanism, size cutoff vs. flow rate, and wall shear stress. A After inertial migration of cells away from channel side walls, CTC(C)s are separated from smaller particles at each flow siphoning gap (at bottom, a 16 μm cell passes to product side). B The cell diameter that results in 90% yield of CTCs into product is plotted against volumetric flow rate per NISA-XL device (25% of total flow rate; cells in Ficoll). C Input Mel-182–1 cell size distribution. Each data point in B is found by size-binning product/waste distributions in DE to estimate yield (plotted in F for blue diamond point in B). Gray region in B defines 95% confidence interval of fit. G A computational fluid model (Fluent) shows that peak wall shear stress is below 15 Pa at 200 μL/min (2 mPa-s viscosity).
Architecture.
Larger arrays complicate parallelization, taking ~4× space on-chip, and sample throughput was limited by low injection of 15–18% in our prior leukocyte sorting device 30. Therefore, five upstream NISA devices (in two stages) were inserted without a buffer co-flow (Fig. 1A, stages 1–2; Suppl. Fig. 1, first two boxes). Together, they concentrate large CTCs/clusters within the sample by ~10× after two NISA stages with 100% sample injection (4 in stage 1, 1 in stage 2). This also removes about 90% of the blood. Finally, one co-flow NISA stage separates the CTC/clusters as they transit to the product outflow tubing (Fig. 1A, stage 3; Suppl. Fig. 1, 3rd box). In the 100% injection section of the chip, sample with blood cells and smaller CTCs (green streaks) leaves in wastes 1–2 (Fig. 1C, lower three lanes), while large CTCs/clusters exit in the product lane of stages 1–2 (Fig. 1C, upper lane). Large cells/clusters can be seen exiting a third stage device in Fig. 1D (upper lane) alongside waste 3 (remaining sample plus co-flow buffer; lower three lanes). Large spiked cells/clusters are visible in Fig. 1CD as fluorescent streaks by virtue of the large number spiked into blood. Also note the 4-cell cluster in Fig. 1E as it exits the final product in stage 3 (high speed video; Phantom v4.2M, Vision Research). Notably, by increasing sample injection from 15% to 100% in stages 1–2, 15 mL of whole blood can be processed in 30 minutes from six NISA devices on a single standard slide-sized device (Fig. 1B) with relatively low shear stress, without even a filter to remove debris.
Fluidic control.
Before introducing sample to the chip, priming was performed through the sample inlet with 50% ethanol in deionized water, individually addressing each port in order: waste 1, waste 2, buffer inlet, waste 3, product. The channels and tubing were subsequently flushed with buffer (1% w/v F127 in PBS) via the buffer inlet in reverse order, finishing with the sample line. At this point sample processing could begin. During the run, sample and buffer were pushed through the device in one of two ways: 1) syringe pumps for sample and buffer to ensure known input flow rates, or 2) a single pressure source connected to the sample and buffer inlet reservoirs (60 mL plastic syringes). Use of pressure control allowed rocking of the blood sample to avoid settling during the run. In all cases, flow rates internal to the chip were controlled passively by built-in fluidic resistors as we did for the CTC-iChip 13 (see Suppl. Fig. 1). Blood sample viscosity is variable even with the normalization of hematocrit (HCT) to 20%; however, volumetric splits exhibited good reliability during pressure-driven experiments when compared to the design split targets (Suppl. Fig. 2). These included the flow fraction entering the product of each NISA stage as well as the injection fraction of sample to stage 3. Processed blood volume averaged 15.3 mL, blood flow rate was 30.8 mL/hr, and product volume was 1.64 mL per nominal tube of blood (7.5 mL). These data are reported for 30 runs in Suppl. Fig. 3.
Microfabrication.
Microfluidic devices were created with PDMS soft lithography techniques 33. Su8–100 was coated onto silicon wafers by spin-coating at 2000 rpm for 30 seconds, to a thickness of about 156 μm. UV photolithography (365 nm) was then used to create a single layer of microchannels on coated silicon wafers. PDMS (Sylgard 184) was then poured onto the resulting channel molds at a ratio of 9:1 base to cross-linker. After cutting cured PDMS from the mold and dicing devices, biopsy punches were used to create holes for press-fit tubing to connect sample inlet, buffer inlet, waste 1 (two), waste 2, waste 3, and product. Channels were enclosed by permanently bonding each 3×1 inch2 device to a glass microscope slide in an oxygen plasma oven. Devices were then baked immediately at 70°C for 10 minutes and later at 150°C for 3 hours with gradual ramps. This last bake helps to increase the elastic modulus of the PDMS, such that microchannel dimensions are not enlarged too much in upstream NISA modules.
Sample acquisition.
Blood samples were collected from healthy volunteers following experimental protocols reviewed and approved by the Massachusetts General Hospital IRB, where protocol numbers 2009-P-000295 and 2015-P-000656 were used to obtain informed consent from internal donors and donors at the MGH Blood Bank respectively. Some healthy samples were also ordered from Research Blood Components, LLC (Brighton, MA). Blood samples were collected from patients following experimental protocols reviewed and approved by the Dana-Farber Cancer Institute IRB, where protocol number 05–300 was used to obtain informed consent (expires on 10/21/2020). All samples were obtained in accordance with the applicable federal guidelines and regulations.
Results and discussion
Cell size cutoff.
To verify the size cutoff of the three-stage NISA device, a set of model experiments were run using two CTC lines (MGH-Mel-182–1 and MGH-Pem-22). These cell lines were isolated from blood samples of two patients bearing metastatic melanoma who were treated at Massachusetts General Hospital. These cells were chosen because their size distributions were centered around the intended size-cutoff for sorting (see Fig. 2C for MGH-Mel-182–1). MGH-Mel-182–1 suspensions had a median diameter of 16.3 μm, and MGH-Pem-22 suspensions had a median diameter of 18.8 μm (Z2 Coulter Counter Analyzer, Beckman Coulter). Instead of blood, 7% Ficoll PM 70 was used as the sample matrix. This simulated the viscosity of 1:1 diluted whole blood without obscuring spiked CTCs in the waste, enabling yields to be measured. The results are summarized in Fig. 2B, where the cell size for 90% yield is plotted against per-array flow rate (syringe pump control). To get each data point, cell diameter distributions were first measured in product (Fig. 2D) and pooled waste (Fig.2E), again by Coulter counter. Next, yield of cells was quantified as a function of diameter within 1-μm wide bins (Fig. 2F), and the cell diameter with 90% yield was estimated by curve fit. The overall fit in Fig. 2B is linear with flow rate (semi-log x-axis results in apparent non-linear trend) and exhibits the expected drop-off with flow rate (inertial focusing increases at higher Reynolds number). Note however that a population of smaller debris was found in the output fractions which was not present in the input cell cultures. This seems to have been a reaction of cells to extended storage in Ficoll while waiting to be processed by the Coulter Counter.
To minimize shear stress while still keeping high cluster yield, a per-array flow rate of 200 μL/min was chosen for remaining experiments, requiring a 10 psi input pressure to sample and buffer. With sample viscosity of 2 mPa-s, this flow rate results in a particle Reynolds number of about 0.1 for cells of diameter (a) 18.5 μm (the 90% cutoff); therefore, inertial focusing 34 is actively pushing cells near the size cutoff across streamlines. In this calculation, particle Reynolds number (Rep) is defined as Re (a/Dh)2 where Re is the channel Reynolds number (ρVDh/μ), ρ and μ are mass density and dynamic viscosity of the fluid respectively, V is mean flow velocity, and Dh is the hydraulic diameter of the focusing channel depicted at left in Fig. 2A.
Although the size cutoff could be reduced further at higher flow rates, the use of PDMS as a device material causes channels to inflate from the increased pressure. This increases channel width and requires cells to migrate even farther from the wall to remain in the product lane of a given NISA module. Therefore, to examine NISA performance at much higher flow rates, whole blood was injected into a single isolated NISA device (100% injection). The results in Suppl. Fig. 4 confirm that CTC(C) sorting (MGH-BRx-142 cells) is possible even at 2 mL/min per array of whole undiluted blood (47% HCT), and that a single array can process 20 mL of whole blood (35% HCT) four times at 4 mL/min. Hence, a full device could process as much as 320 mL of whole blood in 20 minutes. This extreme throughput is particularly notable given that the NISA devices have no filter to remove debris. Rather, they act as a continuous flow size-sorter that almost never clogs. Nevertheless, a multi-stage device operating this fast would require a rigid material like plastic to avoid inflation, and shear stress would be too high for intact cluster isolation. A better approach would be to run blood slowly at first (i.e. at 200 μL/min per array) to remove larger CTCs and clusters gently, then increase the flow rate to as high as 2 mL/min to isolate smaller single CTCs from the waste fractions.
Blood cell removal.
Just as important as size sorting large CTCs/clusters is the removal of blood cells (and smaller CTCs). To measure this depletion performance, a set of experiments were performed using the intended sample matrix (blood). Specifically, 17 chips were run, processing 15 mL whole blood each through the integrated 3-stage NISA device (Fig. 1B). To standardize the viscosity and more precisely measure depletion at a controlled flow rate, blood was diluted to 20% hematocrit by a buffer (1% w/v F127 in PBS). This is about 1:1 dilution on average. After processing through the chip, platelets (PLTs), RBCs, and WBCs were counted manually in the product using hemacytometers and Nageotte chambers depending on the cell concentrations. Vybrant DyeCycle green was used to label WBCs, while Calcein AM allowed platelets to be identified. Blood cell removal factor is defined for each cell type as total cells injected to the chip divided by total cells found in product, and it is plotted in Fig. 3 alongside the base-10 log depletion equivalent (given as a summary statistic). Briefly, platelets were removed to a level of 12 ppm (4.92 log10), red blood cells to 3 ppm (5.53 log10), and WBCs to 66 ppm (4.18 log10), where 1000 ppm indicates 99.9% removal (3-log depletion).
Figure 3:

Depletion of blood cells, including platelets (PLTs), red blood cells (RBCs), and white blood cells (WBCs). Whole blood samples (15.2 mL average blood volume; N=17) were mixed with buffer to a 20% hematocrit to standardize. Samples were processed at 10 psi input pressure with integrated device of Fig. 1B.
Though this is also a higher level of cell depletion in each case than reported for the CTC-iChip (Fachin et al., 2017), where depletion of platelets, RBCs, and WBCs is 4, 5, and 4 respectively (based on internal data), it still leaves about 3600 platelets, 15 thousand RBCs, and 400 WBCs in the product per mL of blood processed (based on a standard blood sample with 300 million platelets, 5 billion RBCs, and 6 million WBCs per mL). It would be possible to improve these depletion levels by re-running the product through additional NISA stages. Platelet removal is complicated by their association with WBCs (satellites) and self-aggregation in some donors, so platelet removal may remain below that of RBCs despite their smaller size individually.
Gentle isolation of CTC clusters.
The preferred CTC cluster isolation technology should not break up cell clusters that exist in the bloodstream. In general then, clusters should not be subjected to fluid stresses in excess of what may occur in the body, though it is worth noting that transit through a chip is much shorter than residence in circulation. We have also found that flow acceleration is particularly suited to break cell clusters (data not shown), so the NISA-XL design avoids any rapid constrictions while also keeping shear stresses below 15 Pa. To determine if CTC cluster isolation was gentle to potentially fragile CTC clusters, we observed spiked CTC clusters at all key decision points within the integrated 3-stage device. Specifically, we observed the inlet to stage 1 (just after the sample inlet port), the outlet of stage 1 (all four NISA modules in parallel), the outlet of stage 2 (one NISA module), and the outlet of stage 3 (one NISA module). To ensure that clusters were clearly visible within the sample and waste streams, Ficoll PM 70 (7% w/v in 1% F127 in PBS) was again used as a blood sample viscosity mimic. Besides keeping shear stresses comparable to that for 20% HCT blood, this also ensured volumetric splits remained within desired ranges. High speed videos at each location were manually examined to count individual spiked CTCs and clusters of varying cell number, enabling a verification that CTCCs conserved their integrity throughout the chip.
This experiment was performed for two separate cell types: MDA-MB-231 (ATCC), a standard triple negative breast cancer cell line, and MGH-BRx-142, a cultured line of CTCs directly derived from the blood of a patient with hormone receptor positive breast cancer 35. We chose MGH-BRx-142 cells in part because their clusters are fragile (as will be apparent from Fig. 5), often breaking up during insufficiently careful pipetting. MDA-MB-231 cells were chosen in part because they are a commonly used cancer cell line for spike cell experiments, allowing outside comparison. In each case, cultured cells were injected to the device unaltered, and no special handling was performed to artificially cluster the cells. For MGH-BRx-142 input suspensions, single cells represented 65% of the total with the remaining cells residing in clusters of 2.8 cells each on average. For MDA-MB-231 input suspensions, single cells represented 64% of the total with the remaining cells residing in clusters of 2.9 cells each on average.
Figure 5:

Rare cell isolation of spiked CTC clusters. To demonstrate gentle isolation of rare cancer cell clusters, small numbers of clusters (five doublets and/or three triplets, in alternate colored fluorescent pre-labels) were manually spiked into each of nine runs on the integrated three-stage device of Fig. 1B (15.7 mL blood per run on average). After scanning the whole product, overall end-to-end yields are reported at left for MGH BRx-142 and LnCAP cells. Individual input-output counts are given at right, where empty circles represent missing cells in the product, and gray circles indicate single cells shed from spiked CTC clusters. 100% yield at top.
Observed cluster distributions are broken out in Fig. 4. Note that the large clusters of 5+ cells are lumped together to improve sampling error in these less frequent event classes. Cluster cells were tallied per cell, so that one 5-cell cluster plus one 10-cell cluster counted as three 5-cell clusters when plotted. No CTC clusters were observed to enter the waste path in stages 1, 2, or 3 for either spike cell line, and output cluster size distributions closely matched the inputs. Specifically, 146 MGH-BRx-142 clusters were observed entering the stage 3 product (2.7 cells/cluster on average), and 427 MDA-MB-231 clusters were observed entering the stage 3 product (2.9 cells/cluster on average). Example CTC cluster transits into stage 3 product are shown at right in Fig. 4, highlighting rotation and orientation of CTCCs. Note that magnitudes in Fig. 4 reflect frequencies of each population relative to the others (within a cell line), yet the actual magnitude is arbitrary, being proportional to input spike number. MGH-BRx-142 cells 35 are relatively large (median diameter of 19.3 μm), and their yield of single CTCs from input to stage 3 product was 97.6%. MDA-MB-231 cells exhibited a yield of 76.4% to product at 200 μL/min per array. Moreover, we observed successful isolation of one hundred seventeen MDA-MB-231 doublets in a row at the stage 3 product, albeit in a blood viscosity mimic to enable observation. Our first cluster chip 20 yielded 70% of three-cell clusters and 41% of two-cell clusters with the same cell line.
Figure 4:

CTC clusters remain intact during sorting. Frequency of observed CTCs is reported as a function of high-speed video recordings from inlet to outlet of integrated chip. Preservation of larger clusters suggests gentle isolation in a sample of viscosity-matched buffer (7% Ficoll PM 70 in PBS, 1% F127). Standard deviations reflect sampling error from manual counts. No clusters were observed entering wastes 1–3 (N of 573). Single-cell yields were 76.4% (MDA-MB-231) and 97.6% (MGH-BRx-142). Input pressure was 10 psi in all cases. Examples in stage 3 product lane are at right (flow: left to right). Multi-exposures were created by projecting minimum intensity of transit videos (frame rate noted) onto one output frame (ImageJ), highlighting CTC cluster rotation.
Rare CTC cluster isolation from blood.
Switching to whole blood, we next tested the end to end yield of small numbers of manually-spiked CTC clusters. Specifically, either five 2-cell clusters or three 3-cell clusters were spiked into 15 mL of whole blood, mixed with buffer (1% w/v F127 in PBS) to 20% hematocrit, and processed through the chip using pressure control (10 psi). In contrast with all other spike cell experiments, where cell suspensions were mixed into Ficoll or blood samples, direct cluster spiking was performed here by micro manipulating individual fluorescent pre-labeled clusters and transferring them into the blood. Post isolation by the NISA chip, clusters were identified in the whole product volume by panning a microscope objective throughout a well plate reservoir. The results of 16 cluster-spike experiments are reported in Fig. 5 across two cell lines. Note that though cluster yield within the integrated 3-stage chip is expected to be very high (as in Fig. 4 caption), these experiments incurred variable transfer losses in manually ejecting spike clusters from the glass cell-picking capillary. This is because a gentle flow was required to avoid breaking clusters even at the point of spiking into blood, yet that risked being insufficient to dislodge partially-adhered clusters. Nevertheless, around 77% of MGH-BRx-142 clusters and 84% of LnCAP clusters were found intact in the product, demonstrating the ability to find a handful of CTC clusters in an admix of about 75 billion RBCs, 90 million WBCs, and 4.5 billion platelets. In these experiments, MGH-BRx-142 cells were again chosen in part due to their observed fragility as clusters, and LnCAP cells (ATCC; prostate cancer cell line) were chosen as a common cell line to serve as a point of comparison.
Spiked cell growth post isolation.
Besides isolation of intact clusters, we also tested whether or not isolation would affect growth rate of isolated spike CTCs and clusters. So we spiked MGH-BRx-142 cells, a CTC line derived from breast cancer patient blood at MGH, into three 15 mL blood aliquots and cultured the product fractions for six days. When compared to day-matched controls of equal input spike number, isolated spike CTCs and clusters grew by about 9.4 fold during the course of six days in vitro, not significantly different than for matched controls (Fig. 6). This cell line was chosen in part because it is relatively large (19.3 μm median by Coulter counter), nearly removing isolation yield from the comparison, leaving just growth rate as a driver of final cell numbers.
Figure 6:

Growth rates of spiked MGH BRx-142 cells after isolation with integrated chip match the controls. Images are for companion cell culture of unprocessed (control) or spiked CTC isolates (in 15 mL blood) for up to 6 days in vitro.
CTC cluster types.
The understanding of CTC clusters is rapidly evolving 36, yet it is important to consider how the NISA-XL device is positioned to capture each subtype. Three main characteristics that could affect size-sorting are: 1) the number of cells in the cluster (two to 50 is typical 36), 2) its overall size (most are 20 to 130 μm 37, but some may be >300 μm 38 ), and 3) the strength of intercellular adhesions. CTC doublets composed of small individual cells are closest in size to WBCs (though doublets including one CTC and one WBC 14 might be even smaller). Interestingly, we find that in buffer (or Ficoll), cell doublets almost always tumble end over end as seen in Fig. 4. This side-effect of inertial focusing helps increase their effective size by pushing their center of mass further from the channel wall than if they were to spin along their long axis. It is unclear if this occurs in blood where clusters cannot be clearly seen, but Fig. 5 demonstrates the ability to isolate spiked CTC doublets. As CTC cluster size increases, size sorting is easier but actual forces applied to the cluster gradually increase as the CTCC occludes more of the channel. Therefore, it was key to increase channel width to 100 μm and reduce shear stress at the wall to near the physiological level.
Regarding isolation of the largest CTC clusters (or CTMs 36), we can extrapolate from the results of others. Recently, inertial migration was applied in a straight microchannel to separate CTC clusters from buffy coat or blood 38,39. In this work, sample is injected at about 50 μL/min (1:2 to buffer) from each side of a 150 μm wide, 50 μm high, microchannel. After 2 cm, cells over 14 μm migrate into the central buffer flow, leaving most blood cells behind. Despite that sample passes through a twice-narrower sorting channel, CTMs of apparent size up to 340 μm were found in samples of head and neck cancer patient blood 38. This suggests the NISA-XL device can capture even larger clusters, and by reducing wall shear stress over ten-fold, weaker bound clusters. Interestingly, these large CTC clusters contained WBCs, as observed bound to CTC(C)s in other studies 14,17,40. In addition, CTCs were sometimes observed with platelet satellites 40, and platelets have been linked to protection of CTCs against immune targeting by natural killer cells 41. Size-sorting does not distinguish among these differences, so each cluster type should be found by the NISA-XL device.
Conclusions
In this study, we developed a high throughput microfluidic chip that can isolate intact circulating tumor cell clusters (and large single CTCs) from large blood volumes with low shear and high purity. The device has a high yield of clusters, extremely low carryover of blood cells, and it requires minimal to no dilution of blood, making it possible to construct CTC cluster apheresis. We have measured the size cutoff for cell sorting as a function of per-array flow rate, demonstrated the ability to find a handful of spiked clusters from 15-mL blood samples, and seen minimal cluster breakup on-chip by high speed imaging. We also found that cell growth rate is preserved post-isolation. To the best of our knowledge, even the reduced speeds used in the bulk of experiments represent the fastest technology for CTC cluster isolation with >2 log enrichment from blood cells that does not require trapping cells on-chip, large dilution factor of the blood, or upstream debulking methods to generate buffy coat. Specifically, our enrichment levels exceed 4 base-ten logs for WBCs, RBCs and PLTs. A single NISA module can also process very large blood volumes of 80 mL at 4 mL/min, even without an inlet filter. By allowing CTC clusters to be pulled out of very large blood volumes, the NISA-XL chip could speed generation of patient CTC lines for in vitro personalized drug testing 4. Moreover, product purity can be further improved by adding more stages of 100% injection NISA and co-flow buffer, or re-running the product through a second time. However, there will be a limit to this purification in that an unknown background of rare non-cancerous clusters may be present. This represents a potentially important population of cells to investigate on its own, particularly in the field of immunotherapy, where clusters of immune cells and cancer cells are created by therapeutic bifunctional antibodies 42. Returning to liquid biopsy of cancer, a final point to highlight is that single CTCs exiting in the NISA chip “waste” could be isolated using negative selection 13 as shown in Suppl. Fig. 5 for melanoma. By splitting CTC clusters from single CTCs, it would enable one to compare the molecular signatures of CTC clusters to single CTCs, raising the value of each biomarker.
Supplementary Material
Acknowledgments
First and foremost, we would like to acknowledge the patients whose sacrifices during very trying periods of their lives have enabled this work. This work was supported in part by the National Institute of Biomedical Imaging and Bioengineering (P41EB002503 and U01EB012493), by the National Cancer Institute (U01CA214297, 2RO1CA129933, and R01CA226871), by the American Cancer Society (132030-RSG-18-108-01-TBG), and by the Howard Hughes Medical Institute. The authors would also like to thank Risa Burr and Christina Constantino for useful discussions, Michelle Jewett and Neelima Magnus for providing some of the spike cells, Brian Chirn, Rebecca Fisher, Ben Nicholson, Haley Pleskow, Brittany Reeves, Ben Wesley, and Devon Wiley for performing some of the isolation runs, and Octavio Hurtado for microfabrication assistance.
Footnotes
Conflicts of interest
Massachusetts General Hospital has been granted patent protection for the NISA technology and for the underlying inertial focusing technology.
Notes and references
- 1.Mohme M, Riethdorf S. and Pantel K, Nat. Rev. Clin. Oncol, 2017, 14, 155–167. [DOI] [PubMed] [Google Scholar]
- 2.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA and Maheswaran S, Cell, 2014, 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, Krol I, Scheidmann MC, Beisel C, Stirnimann CU, Kurzeder C, Heinzelmann-Schwarz V, Rochlitz C, Weber WP and Aceto N, Cell, 2019, 176, 98–112.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, Stojanov P, Brachtel E, Sgroi D, Kapur R, Shioda T, Ting DT, Ramaswamy S, Getz G, Iafrate AJ, Benes C, Toner M, Maheswaran S. and Haber DA, Science (80-. ), 2014, 345, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stott SL, Hsu C, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, V Sequist L, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA and Toner M, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 18392–18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hou JM, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, Priest LJC, Greystoke A, Zhou C, Morris K, Ward T, Blackhall FH and Dive C, J. Clin. Oncol, 2012, 30, 525–532. [DOI] [PubMed] [Google Scholar]
- 7.Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JS and Ewald AJ, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, E854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kagan M, Howard D, Bendele T, Mayes J, Silvia J, Repollet M, Doyle J, Allard J, Tu N, Bui T, Russell T, Rao C, Hermann M, Rutner H. and Terstappen LWMM, J. Clin. Ligand Assay, 2002, 25, 104–110. [Google Scholar]
- 9.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA and Toner M, Nature, 2007, 450, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gleghorn JP, Pratt ED, Denning D, Liu H, Bander NH, Tagawa ST, Nanus DM, Giannakakou PA and Kirby BJ, Lab Chip, 2010, 10, 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiery JP, Nat. Rev. Cancer, 2002, 2, 442–454. [DOI] [PubMed] [Google Scholar]
- 12.Hyun Kyung-A, Goo KB, Han H, Sohn J, Choi W, Il Kim S, Il Jung H and Kim YS, Oncotarget, 2016, 7, 24677–24687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fachin F, Spuhler P, Martel-Foley JM, Edd JF, Barber TA, Walsh J, Karabacak M, Pai V, Yu M, Smith K, Hwang H, Yang J, Shah S, Yarmush R, Sequist LV, Stott SL, Maheswaran S, Haber DA, Kapur R. and Toner M, Sci. Rep, 2017, 7, 10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, Scheidmann MC, Donato C, Scherrer R, Singer J, Beisel C, Kurzeder C, Heinzelmann-Schwarz V, Rochlitz C, Weber WP, Beerenwinkel N. and Aceto N, Nature, 2019, 566, 553–557. [DOI] [PubMed] [Google Scholar]
- 15.Chen JF, Ho H, Lichterman J, Lu YT, Zhang Y, Garcia MA, Chen SF, Liang AJ, Hodara E, Zhau HE, Hou S, Ahmed RS, Luthringer DJ, Huang J, Li KC, Chung LWK, Ke Z, Tseng HR and Posadas EM, Cancer, 2015, 121, 3240–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Au SH, Edd J, Stoddard AE, Wong KHK, Fachin F, Maheswaran S, Haber DA, Stott SL, Kapur R. and Toner M, Sci. Rep, 2017, 7, 2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams DL, Martin SS, Alpaugh RK, Charpentier M, Tsai S, Bergan RC, Ogden IM, Catalona W, Chumsri S, Tang C-M and Cristofanilli M, Proc. Natl. Acad. Sci, 2014, 111, 3514–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller MC, Robinson PS, Wagner C. and O’Shannessy DJ, Cytom. Part A, 2018, 93, 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Au SH, Storey BD, Moore JC, Tang Q, Chen YL, Javaid S, Sarioglu AF, Sullivan R, Madden MW, O’Keefe R, Haber DA, Maheswaran S, Langenau DM, Stott SL and Toner M, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 4947–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT, Luo X, Bardia A, Wittner BS, Ramaswamy S, Shioda T, Ting DT, Stott SL, Kapur R, Maheswaran S, Haber DA and Toner M, Nat. Methods, 2015, 12, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loutherback K, D’Silva J, Liu L, Wu A, Austin RH and Sturm JC, AIP Adv., 2012, 2, 042107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong KHK, Edd JF, Tessier SN, Moyo WD, Mutlu BR, Bookstaver LD, Miller KL, Herrara S, Stott SL and Toner M, Lab Chip, 2018, 18, 2146–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Carlo D, Irimia D, Tompkins RG and Toner M, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 18892–18897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petersson F, Åberg L, Swärd-Nilsson AM and Laurell T, Anal. Chem, 2007, 79, 5117–5123. [DOI] [PubMed] [Google Scholar]
- 25.Sollier E, Go DE, Che J, Gossett DR, O’Byrne S, Weaver WM, Kummer N, Rettig M, Goldman J, Nickols N, McCloskey S, Kulkarni RP and Di Carlo D, Lab Chip, 2014, 14, 63–77. [DOI] [PubMed] [Google Scholar]
- 26.Hou HW, Warkiani ME, Khoo BL, Li ZR, Soo RA, Tan DSW, Lim WT, Han J, Bhagat AAS and Lim CT, Sci. Rep, 2013, 3, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dean WR and Hurst JM, Mathematika, 1959, 6, 77–85. [Google Scholar]
- 28.Warkiani ME, Khoo BL, Wu L, Tay AKP, Bhagat AAS, Han J. and Lim CT, Nat. Protoc, 2016, 11, 134–148. [DOI] [PubMed] [Google Scholar]
- 29.Dow P, Kotz K, Gruszka S, Holder J. and Fiering J, Lab Chip, 2018, 18, 923–932. [DOI] [PubMed] [Google Scholar]
- 30.Mutlu BR, Smith KC, Edd JF, Nadar P, Dlamini M, Kapur R. and Toner M, Sci. Rep, 2017, 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strony J, Beaudoin A, Brands D. and Adelman B, Am. J. Physiol. - Hear. Circ. Physiol, 1993, 265, H1787–H1796. [DOI] [PubMed] [Google Scholar]
- 32.Barnes JM, Nauseef JT and Henry MD, PLoS One, 2012, 7, e50973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duffy DC, McDonald JC, Schueller OJA and Whitesides GM, Anal. Chem, 1998, 70, 4974–4984. [DOI] [PubMed] [Google Scholar]
- 34.Di Carlo D, Lab Chip, 2009, 9, 3038–3046. [DOI] [PubMed] [Google Scholar]
- 35.Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, Yu M, Sundaresan TK, Licausi JA, Desai R, O’Keefe RM, Ebright RY, Boukhali M, Sil S, Onozato ML, Iafrate AJ, Kapur R, Sgroi D, Ting DT, Toner M, Ramaswamy S, Haas W, Maheswaran S. and Haber DA, Nature, 2016, 537, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Umer M, Vaidyanathan R, Nguyen N-T and Shiddiky MJA, Biotechnol. Adv, 2018, 36, 1367–1389. [DOI] [PubMed] [Google Scholar]
- 37.Krebs MG, Hou J-M, Sloane R, Lancashire L, Priest L, Nonaka D, Ward TH, Backen A, Clack G, Hughes A, Ranson M, Blackhall FH and Dive C, J. Thorac. Oncol, 2012, 7, 306–315. [DOI] [PubMed] [Google Scholar]
- 38.Kulasinghe A, Zhou J, Kenny L, Papautsky I. and Punyadeera C, Cancers (Basel)., 2019, 11, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou J, Kulasinghe A, Bogseth A, O’Byrne K, Punyadeera C. and Papautsky I, Microsystems Nanoeng., 2019, 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang X, Wong KHK, Khankhel AH, Zeinali M, Reategui E, Phillips MJ, Luo X, Aceto N, Fachin F, Hoang AN, Kim W, Jensen AE, Sequist LV, Maheswaran S, Haber DA, Stott SL and Toner M, Lab Chip, 2017, 17, 3498–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Jirousková M. and Degen JL, Blood, 2005, 105, 178–185. [DOI] [PubMed] [Google Scholar]
- 42.Dahlén E, Veitonmäki N. and Norlén P, Ther. Adv. Vaccines Immunother, 2018, 6, 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.