

Abstract
M. avium-infected macrophages undergo apoptosis, but M. tuberculosis suppresses apoptosis and triggers necrosis
Although distinct, Mycobacterium tuberculosis and Mycobacterium avium both infect and survive in host macrophages. M. tuberculosis is more virulent, specifically adapted to humans, and grows poorly outside of the host. In contrast, M. avium can survive in the environment and also can infect several different host species in addition to humans, including birds, pigs, dogs, and monkeys.
M. tuberculosis is primarily a pathogen of the respiratory tract, whereas M. avium, which can infect the lungs, can also gain access to hosts by crossing the intestinal barrier. Although M. avium infections in healthy individuals are rarely disseminated, this bacterium can persist within macrophages in mesenteric lymph nodes. Both types of bacteria survive within host mononuclear phagocytes, inhibiting vacuole acidification and phagosome-lysosome fusion. However, while the reactive nitrogen intermediates (RNIs) in activated macrophages kill M. tuberculosis, M. avium is resistant to RNIs.
Following infection, virulent mycobacteria replicate within macrophages of susceptible hosts and subsequently infect surrounding macrophages. To spread the infection, mycobacteria escape one macrophage, probably once it becomes dysfunctional, to infect another. In the case of M. avium, infected macrophages undergo apoptosis, which triggers the escape of the bacterium, enabling it to spread to other cells. Meanwhile, M. tuberculosis inhibits apoptosis and exits cells by inducing necrosis.
Apoptosis Amounts to an Innate Immune Response against M. tuberculosis
A number of pathogens interfere with host cell apoptosis, either promoting it or suppressing it. For example, some pathogens, such as Shigella, Salmonella, and Legionella, activate pro-caspase-1, which is a principal agent of the apoptosis cascade. However, other pathogens, including Mycobacterium tuberculosis, inhibit apoptosis.
Apoptosis is a form of innate immunity against some pathogens. Both extrinsic factors, such as cytokines induced by phagocytized bacteria, or intrinsic stimuli, such as host proteins that, once activated, increase the permeability of the mitochondrial membrane, can trigger apoptosis. In some cases, intracellular pathogens disable cells, such as mononuclear phagocytes, as a means of establishing an infection. However, the disabling of host cells sometimes backfires when those cells no longer provide nutrients or other environmental conditions that the pathogen needs to replicate and survive.
Recent studies demonstrate that macrophages and lymphocytes of patients with tuberculosis undergo apoptosis. Moreover, experiments in vitro establish that macrophages infected with viable virulent strains of M. tuberculosis undergo apoptosis with less frequency than macrophages infected with attenuated strains of M. tuberculosis. Strain virulence in mice correlates directly to the extent to which apoptosis is inhibited. Thus, virulent strains of M. tuberculosis apparently suppress apoptosis in host cells. Conversely, macrophage apoptosis appears to provide host cells a means for killing other types of intracellular microorganisms, with host cells likely initiating apoptosis following infection. Secreted bacterial proteins likely interact with proteins in the host-cell cytoplasm, members of the Nod-leucine-rich repeats (LRR-CARD) family, to trigger this process in macrophages.
M. tuberculosis and Macrophage Apoptosis
Hardy Kornfeld of the University of Massachusetts Medical School in Worcester once suggested that M. tuberculosis inhibits macrophage apoptosis by decreasing the number of tumor necrosis factor-receptor-1 (TNF-R1) molecules on the phagocytic cell surface and lowering its secretion of interleukin-10. After M. tuberculosis infects macrophages, TNF-α secretion is suppressed, with the speed and intensity of suppression being strain-dependent. Some other M. tuberculosis strains are thought to induce TNF-α production. In addition, the response of M. tuberculosis-infected macrophage to TNF-α stimulation is impaired.
We propose that TNF-α is secreted upon bacterial uptake, and its synthesis and secretion then decrease progressively, while TNFR1 is down-regulated. However, at least in infected macrophage monolayers, M. tuberculosis does not fully inhibit apoptosis, which starts by day 3 and peaks at day 4 or 5. By day 5, secretion of TNF-α is probably down-regulated in some, but not all, macrophage cells.
Both M. avium and M. tuberculosis can down-regulate interleukin-12 (IL-12), a cytokine that macrophages produce, according to reports from ours as well other labs. However, when M. tuberculosis infects macrophages, it suppresses apoptosis triggered by other extracellular stimuli, such as recombinant TNF-α. This finding suggests that the TNFR1 mechanism, as well as the decreased secretion of TNF-α, do not completely explain bacterium-associated suppression.
Live and thriving bacteria are better than dead bacteria at blocking apoptosis. However, treating M. tuberculosis-infected macrophages with rifampin at levels that inhibit protein synthesis without affecting bacterial viability significantly increases apoptosis, suggesting that newly synthesized M. tuberculosis proteins are required to inhibit extrinsic apoptosis. In sum, M. tuberculosis requires ongoing protein synthesis to inhibit macrophage apoptosis in vitro and in vivo.
M. avium Does Not Suppress Macrophage Apoptosis
Unlike M. tuberculosis, M. avium apparently does not suppress macrophage apoptosis. Indeed, we and others showed that, when infected with M. avium, both human and mouse macrophages undergo apoptosis.
Many but not all of the infecting M. avium organisms appear to be killed following macrophage apoptosis, and thus some M. avium bacteria survive to infect other macrophages, thereby disseminating. In fact, M. avium can exit the phagocyte vacuole, and subsequently the cell, after apoptosis. Other bacteria, such as Listeria monocytogenes, also escape the vacuole, but in this case by a listeriolysin-dependent mechanism. Although Mycobacterium marinum similarly acidifies the phagocytic vacuole, its escape is not known to be associated with secretion of a lytic enzyme.
Using video microscopy, we found that some M. avium cells leave the phagocytic cell, while others remain within the apoptotic body and soon are ingested by another macrophage. Whether the failure of some bacteria to escape apoptotic cells is due to the lack of an efficient propulsion mechanism or a failure to come into contact with the cytoplasmic membrane is being investigated. Although M. tuberculosis and M. avium were considered to behave fairly similarly when occupying macrophages, evidence of their differences is accumulating.
Genes Associated with the Spreading of Mycobacteria among Host Cells
M. tuberculosis and M. avium spread from one host cell to another by different mechanisms. M. avium escape from macrophages following apoptosis, whereas M. tuberculosis partially inhibits macrophage apoptosis.
We recently identified an operon of M. tuberculosis that can inhibit apoptosis. The operon is composed of seven genes: four of them resemble a pilus operon, the fifth gene encodes a transmembrane protein, and the other two genes encode secreted proteins. When a construct carrying the two genes encoding those two secreted M. tuberculosis proteins transfects macrophages, tumor necrosis factor-α (TNF-α) and staurosporine-mediated apoptosis are inhibited. The mechanism(s) of action of the two secreted proteins is under investigation.
Based on the homology between four genes in this M. tuberculosis operon and type IV pili genes, we hypothesized that the M. tuberculosis genes might encode a secretion apparatus for the apoptosis-inhibiting proteins. In attempting to visualize such an apparatus, freeze-fraction transmission electron microscopy showed that both the bacterial and vacuole membranes have pores. The M. tuberculosis pores show two different morphologies; one type is similar to porins that are found in Mycobacterium smegmatis, while the other pores contain a protruding edge.
Other M. tuberculosis genes appear to be involved in inhibiting macrophage apoptosis. For instance, several encode enzymes that appear to be involved in synthesizing and degrading lipids. Another encodes mmpL8, a membrane protein that is involved in transporting fatty acids. All of these genes are associated with inactivating the extrinsic pathway of apoptosis. In fact, M. tuberculosis infections of macrophages do not inhibit the intrinsic pathway, raising the possibility of a link between this pathway and escape of M. tuberculosis from the host cell, which occurs secondary to necrosis. Many studies indicate that the pathogenesis-related region RD-1 and, more specifically, the genes encoding proteins ESAT-6 and CPF-10, are directly involved in macrophage cytotoxicity and spread of this bacterial pathogen. The mechanisms remain unknown.
DNA array studies of macrophages infected with M. tuberculosis mutants that do not inhibit apoptosis show that the wild-type bacterium inhibits expression of TNF-α and induces expression of TNF-RI. When macrophages are infected with one of these mutants, there is a 25-fold increase in expression of the pro-apoptotic gene bim-L. Apparently, one of the secreted proteins blocks expression of bim-L.
We found four M. avium mutants that do not escape macrophages upon apoptosis. Although these mutants escape the phagosome, they cannot exit the cell. We do not know whether the vacuole membrane disintegrates upon apoptosis or whether the virulent bacteria respond to changes in the vacuoles by triggering the disintegration of the vacuole membrane.
M. avium does not seem to accumulate actin at its poles. In Mycobacterium marinum, however, polar actin appears responsible for propagating these bacteria in macrophage cytoplasm, according to Luisa Stamm of the University of California, San Francisco and her collaborators. Assuming M. avium lacks actin propulsion implies that these bacteria encounter host-cell cytoplasmic membranes with low efficiency. However, once the host cell dies, the volume of the cytoplasm decreases, thus bringing these bacteria into closer contact with the cell membrane. We have identified several genes that apparently enable M. avium to escape the cell membrane. These phospholipases or other enzymes with lytic activity might need the cytoplasmic environment to be activated before M. avium can be released.
FIGURE 1.
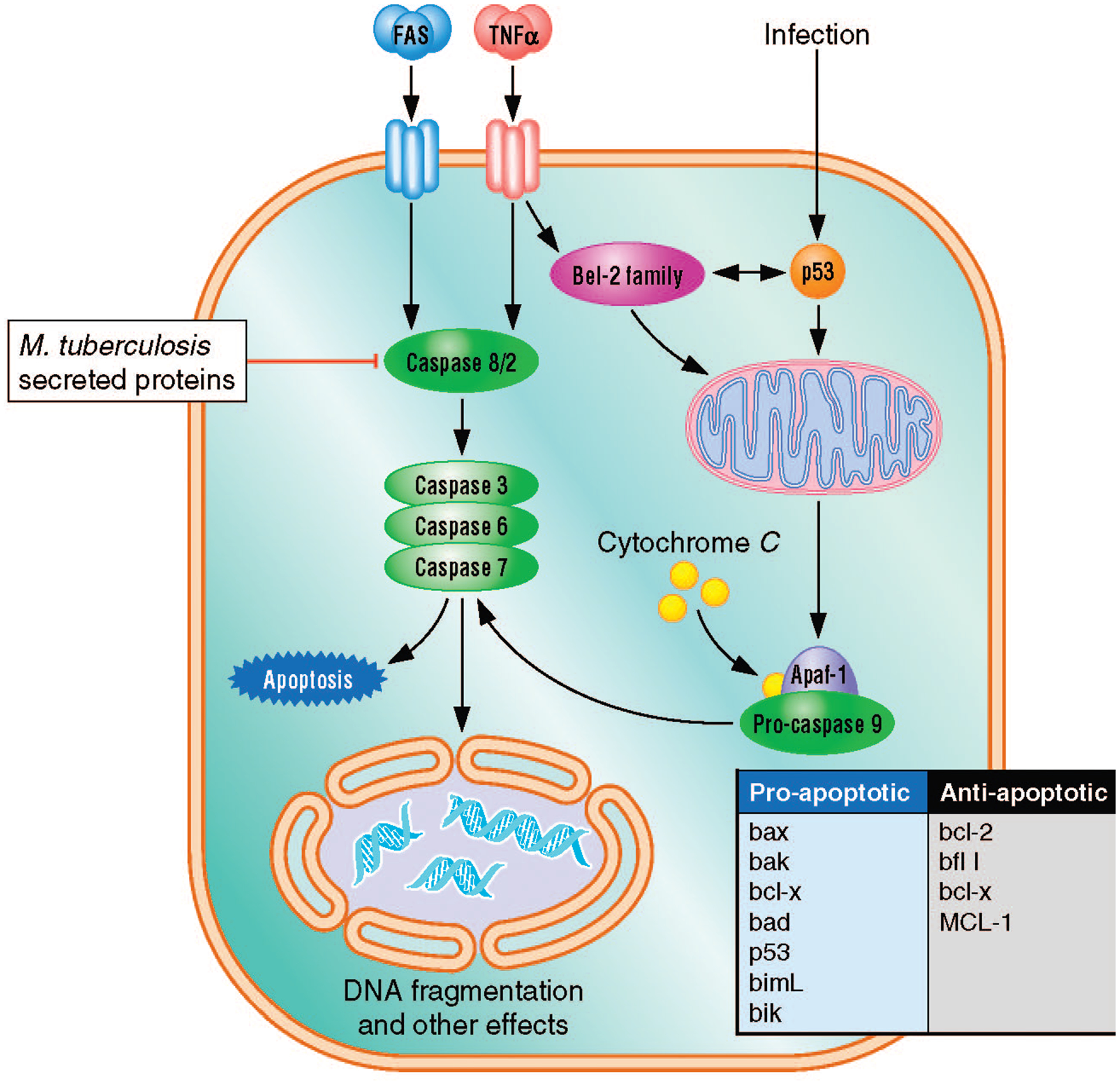
Main pathways of apoptosis. Infecting macrophage cells with a virulent strain of M. tuberculosis inhibits apoptosis by the extrinsic pathway. When M. tuberculosis-infected macrophages are exposed to TNF-α, caspase-8 cannot be activated. The existence of additional targets in the pathway is possible.
FIGURE 2.
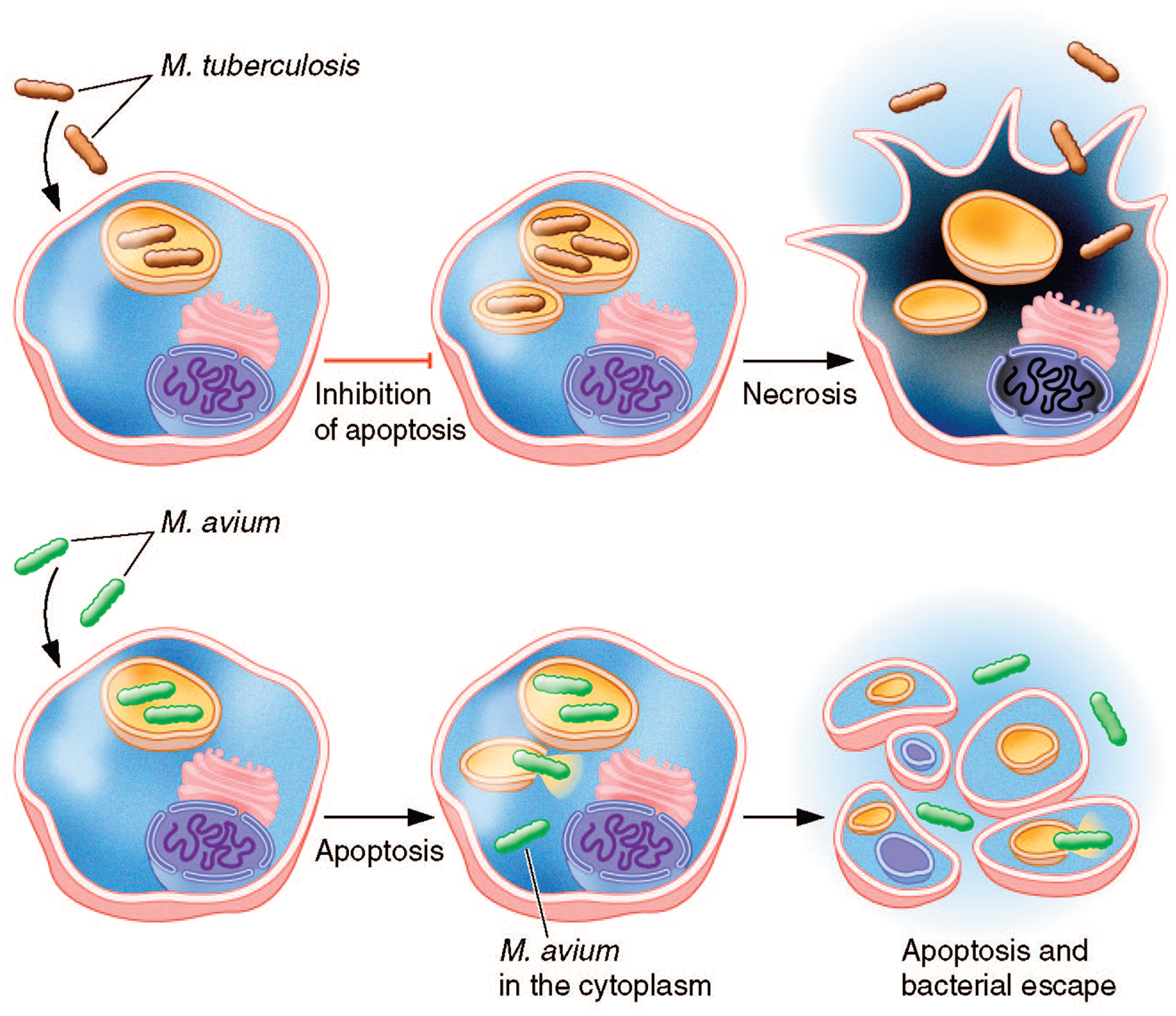
M. tuberculosis and M. avium interact differently with macrophages. For example, M. avium-infected macrophages undergo apoptosis, which triggers the escape of M. avium from the cell, whereas M. tuberculosis-infected macrophages suppress apoptosis, and the bacterium ultimately escapes the macrophages by inducing necrosis.
Summary.
Between Mycobacterium tuberculosis and Mycobacterium avium, the former is more virulent, specifically adapted to humans, and grows poorly on its own, whereas M. avium survives in the environment and readily infects several host species.
M. avium-infected macrophages undergo apoptosis, whereas M. tuberculosis suppresses apoptosis but allows necrosis.
A M. tuberculosis operon that inhibits apoptosis contains seven genes, including four that resemble a pilus operon, a fifth that encodes a transmembrane protein, and two genes that encode secreted proteins; still other genes appear to be involved in inhibiting this process.
Several genes, encoding phospholipases or other enzymes with lytic activity, apparently enable M. avium to escape the cell membrane.
Contributor Information
Luiz E. Bermudez, Department of Biomedical Sciences, College of Veterinary Medicine, and the Department of Microbiology, College of Science, Oregon State University, Corvallis..
Lia Danelishvili, Department of Biomedical Sciences, Oregon State University, Corvallis..
Julie Early, Department of Biomedical Sciences, College of Veterinary Medicine, and the Molecular and Cell Biology Program, Oregon State University, Corvallis..
SUGGESTED READING
- Bermudez LE, Parker A, and Goodman JR. 1997. Growth within macrophages increases the efficiency of Mycobacterium avium in invading other macrophages by a complement receptor-independent pathway. Infect. Immun 65:1916–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez LE, Petrofsky M , and Sangari F. 2004. Intracellular phenotype of Mycobacterium avium enters macrophages primarily by a macropinocytosis-like mechanism and survives in a compartment that differs from that with extracellular phenotype. Cell. Biol. Int 28:411–419. [DOI] [PubMed] [Google Scholar]
- Danelishvili L, McGarvey J, Li YJ , and Bermudez LE. 2003. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell. Microbiol 5:649–660. [DOI] [PubMed] [Google Scholar]
- Dobos KM, Spotts EA, Quinn FD , and King CH. 2000. Necrosis of lung epithelial cells during infection with Mycobacterium tuberculosis is preceded by cell permeation. Infect. Immun 68:6300–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ , and Kornfeld H. 1997. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect. Immun 65:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy A, Laochumroonvorapong P , and Kaplan G. 1994. Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guerin. J. Exp. Med 180:1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Tamayo MH, Gonzalez-Juarrero M, Orme IM , and Ordway DJ. 2006. Virulent clinical isolates of Mycobacterium tuberculosis grow rapidly and induce cellular necrosis but minimal apoptosis in murine macrophages. J. Leukocyte Biol 79:80–86. [DOI] [PubMed] [Google Scholar]
- Rojas M, Olivier M, Gros P, Barrera LF , and Garcia LF. 1999. TNF-alpha and IL-10 modulate the induction of apoptosis by virulent Mycobacterium tuberculosis in murine macrophages. J. Immunol 162:6122–6131. [PubMed] [Google Scholar]
- Sly LM, Hingley-Wilson SM, Reiner NE , and McMaster WR. 2003. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J. Immunol 170:430–437. [DOI] [PubMed] [Google Scholar]
- Stamm LM, Morisaki JH, Gao LY, Jeng RL, McDonald KL, Roth R, Takeshita S, Heuser J, Welch MD , and Brown EJ. 2003. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J. Exp. Med 198:1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]