

Abstract
Background
Cardiac remodeling after acute myocardial infarction (AMI) is an important process. The present study aimed to assess the protective effects of astaxanthin (ASX) on cardiac remodeling after AMI.
Methods
The study was conducted between April and September 2018. To create a rat AMI model, rats were anesthetized, and the left anterior descending coronary artery was ligated. The rats in the ASX group received 10 mg·kg−1·day−1 ASX by gavage for 28 days. On the 1st day after AMI, but before ASX administration, six rats from each group were sacrificed to evaluate changes in the heart function and peripheral blood (PB) levels of inflammatory factors. On the 7th day after AMI, eight rats from each group were sacrificed to evaluate the PB levels of inflammatory factors and the M2 macrophage count using both immunofluorescence (IF) and flow cytometry (FC). The remaining rats were observed for 28 days. Cardiac function was examined using echocardiography. The inflammatory factors, namely, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-10, were assessed using enzyme-linked immunosorbent assay. The heart weight/body weight (BW), and lung weight (LW)/BW ratios were calculated, and myocardial fibrosis in the form of collagen volume fraction was measured using Masson trichrome staining. Hematoxylin and eosin (H&E) staining was used to determine the myocardial infarct size (MIS), and TdT-mediated dUTP nick-end labeling staining was used to analyze the myocardial apoptosis index. The levels of apoptosis-related protein, type I/III collagen, transforming growth factor β1 (TGF-β1), metalloproteinase 9 (MMP9), and caspase 3 were assessed by Western blotting. Unpaired t-test, one-way analysis of variance, and non-parametric Mann-Whitney test were used to analyze the data.
Results
On day 1, cardiac function was worse in the ASX group than in the sham group (left ventricular end-systolic diameter [LVIDs]: 0.72 ± 0.08 vs. 0.22 ± 0.06 cm, t = −11.38; left ventricular end-diastolic diameter [LVIDd]: 0.89 ± 0.09 vs. 0.48 ± 0.05 cm, t = −9.42; end-systolic volume [ESV]: 0.80 [0.62, 0.94] vs. 0.04 [0.03, 0.05] mL, Z = −2.89; end-diastolic volume [EDV]: 1.39 [1.03, 1.49] vs. 0.28 [0.22, 0.32] mL, Z = −2.88; ejection fraction [EF]: 0.40 ± 0.04 vs. 0.86 ± 0.05, t = 10.00; left ventricular fractional shortening [FS] rate: 0.19 [0.18, 0.20] %FS vs. 0.51 [0.44, 0.58] %FS, Z = −2.88, all P < 0.01; n = 6). The levels of inflammatory factors significantly increased (TNF-α: 197.60 [133.89, 237.94] vs. 50.48 [47.21 57.10] pg/mL, Z = −2.88; IL-1β: 175.23 [160.74, 215.09] vs. 17.78 [16.83, 19.56] pg/mL, Z = −2.88; IL-10: 67.64 [58.90, 71.46] vs. 12.33 [11.64, 13.98] pg/mL, Z = −2.88, all P < 0.01; n = 6). On day 7, the levels of TNF-α and IL-1β were markedly lower in the ASX group than in the AMI group (TNF-α: 71.70 [68.60, 76.00] vs. 118.07 [106.92, 169.08] pg/mL, F = 42.64; IL-1β: 59.90 [50.83, 73.78] vs. 151.60 [108.4, 198.36] pg/mL, F = 44.35, all P < 0.01, n = 8). Conversely, IL-10 levels significantly increased (141.84 [118.98, 158.36] vs. 52.96 [42.68, 74.52] pg/mL, F = 126.67, P < 0.01, n = 8). The M2 macrophage count significantly increased (2891.42 ± 211.29 vs. 1583.38 ± 162.22, F = 274.35, P < 0.01 by immunofluorescence test; 0.96 ± 0.18 vs. 0.36 ± 0.05, F = 46.24, P < 0.05 by flowcytometry test). On day 28, cardiac function was better in the ASX group than in the AMI group (LVIDs: 0.50 [0.41, 0.56] vs. 0.64 [0.56, 0.74] cm, Z = −3.60; LVIDd: 0.70 [0.60, 0.76] vs. 0.80 [0.74 0.88] cm, Z = −2.96; ESV: 0.24 [0.18, 0.45] vs. 0.58 [0.44, 0.89] mL, Z = −3.62; EDV: 0.76 [0.44, 1.04] vs. 1.25 [0.82, 1.46] mL, Z = −2.54; EF: 0.60 ± 0.08 vs. 0.50 ± 0.12, F = 160.48; %FS: 0.29 [0.24, 0.31] vs. 0.20 [0.17, 0.21], Z = −4.43, all P < 0.01; n = 16). The MIS and LW/BW ratio were markedly lower in the ASX group than in the AMI group (myocardial infarct size: 32.50 ± 1.37 vs. 50.90 ± 1.73, t = 23.63, P < 0.01, n = 8; LW/BW: 1.81 ± 0.15 vs. 2.17 ± 0.37, t = 3.66, P = 0.01, n = 16). The CVF was significantly lower in the ASX group than in the AMI group: 12.88 ± 2.53 vs. 28.92 ± 3.31, t = 10.89, P < 0.01, n = 8. The expression of caspase 3, TGF-β1, MMP9, and type I/III collagen was lower in the ASX group than in the AMI group (caspase 3: 0.38 ± 0.06 vs. 0.66 ± 0.04, t = 8.28; TGF-β1: 0.37 ± 0.04 vs. 0.62 ± 0.07, t = 6.39; MMP9: 0.20 ± 0.06 vs. 0.40 ± 0.06, t = 4.62; type I collagen: 0.42 ± 0.09 vs. 0.74 ± 0.07, t = 5.73; type III collagen: 0.13 ± 0.02 vs. 0.74 ± 0.07, t = 4.32, all P < 0.01; n = 4).
Conclusions
ASX treatment after AMI may promote M2 macrophages and effectively attenuate cardiac remodeling by inhibiting inflammation and reducing myocardial fibrosis.
Keywords: Astaxanthin, Macrophages, Myocardial infarction
Introduction
Coronary artery disease (CAD) is the leading pathophysiological cause of acute myocardial infarction (AMI) and the most important cause of heart failure (HF) worldwide. The development of CAD conferred a relative HF risk of 8.1 in human beings, which is more than four-fold higher than the relative HF risk due to other major risk factors.[1]
The progression of HF is closely associated with cardiac remodeling, a process involving molecular, cellular, and interstitial events leading to clinically relevant changes in the shape, size, and mass of the heart after cardiac injury.[2] After myocardial infarction (MI), cardiac remodeling reduces the ventricular wall pressure and temporarily maintains the cardiac pump function. However, it eventually evolves into a destructive maladaptive alteration, leading to HF and even death.[3,4] Therefore, clinicians must identify effective therapeutic targets to prevent excessive myocardial fibrosis when treating ventricular remodeling after AMI.
Astaxanthin (ASX) is a xanthophyll carotenoid that contains two oxygenated groups on each ring structure, which account for the enhanced antioxidant activity.[5] ASX is a stronger peroxide radical scavenger than beta-carotene, alpha-tocopherol, alpha-carotene, lutein, and lycopene.[6] Studies have reported that ASX can reduce renal fibrosis, improve renal cell apoptosis, fight cancer, and increase the potential of stem cells.[7,8] It can also prevent vocal cord scarring in the early stages of wound healing by regulating the oxidative stress response.[9] It follows that ASX has a strong protective effect in the myocardium. The present study aimed to determine whether ASX improves cardiac remodeling after AMI.
Methods
Animals
All experimental protocols involving rats were performed between April 2018 and September 2018 and were approved by both the Animal Care and Use Committee and the Ethics Committee for Animal Research (No. 20171005) at our institution. Clean male Sprague-Dawley rats aged 6 to 8 weeks and weighing 220 to 250 g were housed in a clean room at a constant temperature (22–25°C) under a 12:12 h light-dark cycle. The rats were purchased from Hunan SJA Laboratory Animal Co. Ltd. (Hunan, China license, SCXK [Xiang] 2011-0003) and provided food and water ad libitum. The present study was performed in accordance with the recommendations of the Guide for the National Science Council of China.
Preparation of reagents and instruments
ASX was purchased from Sigma-Aldrich (Shanghai, China) and stored in the dark at −20°C. To obtain a suspension, it was dissolved in olive oil and stored in the dark at 4°C. Enzyme-linked immunosorbent assay (ELISA) kits against tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-10 were acquired from Neobioscience (Shenzhen, China). TdT-mediated dUTP nick-end labeling (TUNEL) kits and bovine serum albumin were purchased from Roche Molecular Systems Inc. (Shanghai, China). Western blotting in the present study used primary antibodies against transforming growth factor β1 (TGF-β1) (Abcam, Shanghai, China; ab92486, 1:500), glyceraldehyde-3-phosphate dehydrogenase (Abcam; ab37168, 1:10,000), type III collagen (Abcam; ab7778, 1:500), metalloproteinase 9 (MMP9) (Abcam; ab76003, 1:1000), caspase 3 (Abcam; ab49822, 1:500), and type I collagen (Affbiotech, Changzhou, China; AF7001, 1:1000). The secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit IgG (Aspen, Wuhan, China; AS1107, 1:10,000). In the immunofluorescence (IF) test, a mouse primary antibody against arginase-1 (Arg-1) (Santa Cruz Biotechnologies, Shanghai, China; sc-271430, 1:50) was used, while the secondary antibody was Cy3-labeled goat anti-mouse IgG (Aspen; AS-1111, 1:50). The primary antibodies used in flow cytometry (FC) test were as follows: anti-Arg-1 (Novus, Shanghai, China; NBP1-32731, 100 μL), fluorescein isothiocyanate (FITC) mouse anti-rat CD11b (BD, Shanghai China; Biosciences, 561684, 50 μg), and anti-CD68/SR-D1 (Novus; NB600-985APC, 100 μL). The secondary antibody used in FC test were as follows: anti-rabbit IgG (H + L), F(ab’)2 fragment (PE Conjugate) (Cell Signaling Technology, Shanghai, China; 8885s, 250 mL), Rabbit IgG Isotype Control [Unconjugated] (Novus; AB-105-C, 1 mg). Inverted Fluorescence Microscope (Olympus, IX51, Japan), image system of the inverted fluorescence microscope (Q-imaging, MicroPublisher, Canada), and IMS image analysis system (Wuhan, China) were used in the study.
Experimental design
The rats were randomly distributed to one of three groups as follows: a sham group (n = 30) in which rats received olive oil (10 mL/kg) by gavage, with no ligation of the left anterior descending (LAD) coronary artery; an AMI group (n = 30) in which rats received olive oil (10 mL/kg) by gavage for 28 days after LAD coronary artery ligation; an ASX group (n = 30) in which rats received ASX (10 mL/kg) by gavage for 28 days after LAD artery ligation. ASX was administered at a concentration of 1 mg/mL, and the rats were administered equal volumes of either olive oil or ASX for 28 consecutive days.
Data were collected at three-time points in the present study. Day 1: 24 h after LAD coronary artery ligation and before ASX administration, six rats were randomly selected from each group to have their cardiac function evaluated by echocardiography; these animals were then sacrificed and their inflammatory factors were determined using ELISA. Day 7: on the 7th day after LAD coronary artery ligation, eight rats were randomly selected from each group and sacrificed to evaluate the macrophage counts in their peripheral blood (PB) using FC, as well as in the border zone using IF. Their inflammatory factors were also determined using ELISA. Day 28: on the 28th day after LAD coronary artery ligation, all remaining rats from each group were subjected to echocardiography and sacrificed. Their relative heart and lung weight (LW), as well as their MI size, were assessed using hematoxylin and eosin (H&E) staining, and their myocardial apoptosis index (AI) was determined using TUNEL staining. The levels of collagen deposition were determined using Masson trichrome staining, and the expression of relative myocardial fibrosis-related proteins was determined using Western blotting.
Rat model of AMI
A rat model of AMI was established by permanently ligating the LAD coronary artery, as previously reported.[10,11] Rats were fasted for 24 h, given water only, before surgery. They were weighed and anesthetized by intraperitoneal injection of 40 mg/kg pentobarbital sodium (Sigma Aldrich, Shanghai, China). Their chest was opened between the third and fourth ribs and the LAD coronary artery was ligated 2 to 3 mm from its origin, between the left pulmonary artery cone and left atrium appendage, using a 6-0 silk thread. To verify the success of the model, rapid discoloration of the ischemic area from pink to gray or dark red was observed below the ligation line (anterior ventricular wall and apex), along with an immediate elevation of up to 0.2 mV in the ST segment that persisted for 30 min after ligation. For 3 consecutive days after surgery, each rat was given 800,000 U of penicillin per day to prevent infection.
The rats in the sham group underwent the same procedure, except that only the puncture line was created from the left atrium appendage to the left pulmonary artery cone, with no ligation of the LAD coronary artery after the chest was opened.
Changes in myocardial structure and detection of infarct size
H&E staining was used to determine histopathological changes in myocardial structure and infarct size. Freshly removed heart tissue was fixed using 4% paraformaldehyde for at least 24 h and then dehydrated in different concentrations of ethanol. The dehydrated sample was then embedded in paraffin wax. Sections 4 μm thick were cut and stored at room temperature. These were stained using H&E to observe pathological changes in cardiomyocytes and calculate the infarction area size under a digital microscope. Myocardial infarct size (MIS) was calculated as follows: MIS (%) = (scar arc length × 2)/(endocardial circumference + epicardial circumference) × 100%.
Inflammatory factors and macrophage detection
We detected the level of inflammatory factors twice. On day 1 after LAD coronary artery ligation and before ASX administration, we detected the cytokine levels in rat PB to determine the success of the model. On day 7 after the rat AMI model was established, we detected inflammatory factors in rat PB. We analyzed the M2 macrophage count in PB using FC and the border zone using IF staining, as reported previously.[12]
Inflammatory factor tests
ELISA was used to detect TNF-α, IL-1β, and IL-10. On the 1st and 7th day after LAD coronary artery ligation blood samples were collected, placed at room temperature for 1 h, and then centrifuged at 2000×g for 10 min at 4°C. TNF-α, IL-1β, and IL-10 were detected in the supernatant according to the ELISA kit instructions.
Flow cytometry
From each rat, 2 mL of PB was collected and the volume was brought to 10 mL using red blood cell lysis buffer; the samples were lysed for 15 min in the dark at room temperature and then centrifuged 2 to 3 times at 450×g for 5 min. The cells were incubated using Arg-1 antibody, FITC-conjugated mouse anti-rat, and CD68/SR-D1 antibody at room temperature for 20 min. The samples were then centrifuged at 450×g for 5 min, and the supernatant was discarded. Next, the remaining cells were incubated in the PE-conjugated anti-rabbit IgG (H + L) F(ab’)2 fragment secondary antibody at room temperature for 40 min. The samples were centrifuged at 450×g for 5 min, and the supernatant was discarded. The remaining cells were dissolved in 200 μL phosphate-buffered saline (PBS) for FC. Cell suspensions were stained using Arg-1 antibody, CD68/SR-D1 antibody, and FITC-conjugated mouse anti-rat CD11b. M2 macrophages were defined as CD68/CD11b/Arg-1-positive and detected using FC.
Immunofluorescence
The paraffin sections were dewaxed in xylene, dehydrated using an alcohol concentration gradient, incubated in hydrogen peroxide for 10 min, and soaked in PBS for 5 min. Subsequently, antigen retrieval was performed. The primary and secondary antibodies were added sequentially. The cardiac sections were then incubated with antibodies directed against Arg-1 at a ratio of 1:50. The fluorescent-labeled antibodies were observed using an inverted microscope, and images were captured using the MicroPublisher imaging system (Q-Imaging).
Evaluation of cardiac function, apoptosis, and fibrosis
On the 1st and 28th days after AMI, we evaluated cardiac function twice using echocardiography. We calculated the apoptotic index (AI) based on TUNEL staining and observed myocardial fibrosis using Western blotting and Masson trichrome staining on the 28th day after AMI.
Echocardiography
The rats were anesthetized using an intraperitoneal injection of 40 mg/kg of pentobarbital sodium. While breathing uniformly, the rats were placed on their backs, with their heads and limbs fixed on a flat plate. The hair was removed from the left front chest, exposing the skin. An ultrasound diagnostic system (GE, Shanghai, China; Vivid 7) and an S4 probe with a frequency of 2.5 Hz were employed. The probe was placed on the left side of the breastbone to find the left section of the shaft using two-dimensional ultrasound, guided by the left ventricular outflow tract. Next, the probe was rotated 90° to find the level of the left ventricular papillary muscle in the cross-sectional tangent plane, acquiring two-dimensional Doppler echocardiography. We recorded the following parameters using the Ultrasonic System software: left ventricular end-systolic posterior wall thickness, left ventricular end-diastolic posterior wall thickness, left ventricular end-systolic diameter (LVIDs), left ventricular end-diastolic diameter (LVIDd), end-systolic volume (ESV), end-diastolic volume (EDV). Ejection fraction (EF) and % fractional shortening (FS) were calculated by the ultrasonic system software.
Masson trichrome staining was performed to observe cardiac collagen deposition and cardiac structure changes, as well as to calculate the collagen volume fraction. Slices were sequentially dewaxed in different concentrations of xylene and ethanol solution. The nuclei were then stained using iron hematoxylin and differentiated using hydrochloric acid alcohol for 10 s. The ammonia was allowed to turn blue and the dip was then dyed using Ponceau for 7 min; it was then differentiated using phosphomolybdic acid and aniline 3 min. The blue fiber was stained using collagen fibers for 5 min, and the sections were placed in glacial acetic acid solution for 1 min. Finally, the fiber was dehydrated in different concentrations of xylene and ethanol solutions. Quantitative analysis was performed using the IMS Image Analysis System.
TUNEL staining and AI test
According to the sample size, the appropriate amounts of reagent 1 (TdT) and reagent 2 (dUTP) from the TUNEL kit were mixed at a ratio of 1:9. The tissue was covered, and the sections were placed in a wet box and incubated for 60 min in a 37°C water bath. The slides were rinsed three times in PBS for 5 min each, and the appropriate amount of 4’, 6-diamidino-2-phenylindole stain was added dropwise. The cells were sealed on a new glass slide, and anti-fluorescence quenched tablets were added. The slides were observed under a fluorescence microscope (Olympus IX51) and photographed. Apoptotic cells exhibited green fluorescence. Four visual fields of each tissue were randomly selected to calculate the AI using MicroPublisher imaging system (Q-imaging). The following equation was used for AI calculation: AI = number of apoptotic nuclei/number of total cardiac nuclei × 100%.
Western blotting analysis
Western blotting was performed to detect the expression of the target proteins in cardiac tissue. Approximately 200 to 500 mg of tissue from the infarcted border zone was cut into pieces for total protein extraction. The same amount of protein (40 μg) was loaded on sodium dodecyl sulfate-polyacrylamide gels (8%–20%) for gel electrophoresis and then transferred to 0.45 μm polyvinylidene fluoride membranes. Primary antibodies were added to the resolved proteins and incubated overnight at 4°C. The primary antibodies were then removed, and the samples were incubated with the secondary antibodies for 1 h at room temperature. The gray values of the target bands were analyzed using AlphaEase FC processing software (Alpha Innotech, California, USA).
Statistical analysis
All data are presented as the mean ± standard deviation or median (P25, P75) and analyzed using SPSS 21.0 software (Chicago, IL, USA). Graphs were plotted using GraphPad Prism 7.0 software (GraphPad Software Inc., San Diego, CA, USA). Comparisons between two groups were performed using the unpaired t-test or non-parametric Mann-Whitney test. One-way analysis of variance was used for multiple comparisons. Two-tailed differences were considered statistically significant at P values <0.05.
Results
AMI induced significant deterioration in cardiac function and increased inflammatory factors
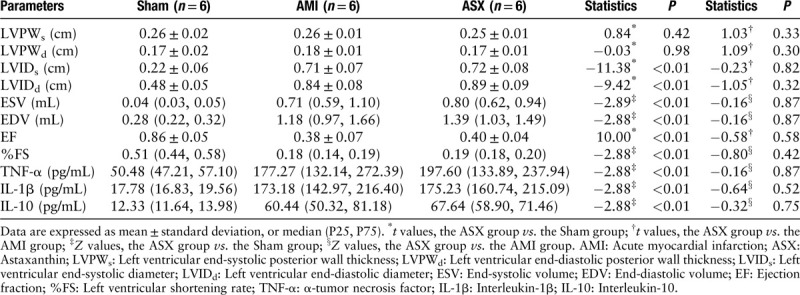
After LAD coronary artery ligation, the function of the rats’ hearts deteriorated in both AMI and ASX groups, and no significant difference was observed between the two groups. This demonstrated that we successfully constructed the AMI model to compare the ASX group with the sham group (LVIDs: 0.72 ± 0.08 vs. 0.22 ± 0.06 cm, t = −11.38; LVIDd: 0.89 ± 0.09 vs. 0.48 ± 0.05 cm, t = −9.42; ESV: 0.80 [0.62, 0.94] vs. 0.04 [0.03, 0.05] mL, Z = −2.89; EDV: 1.39 [1.03, 1.49] vs. 0.28 [0.22, 0.32] mL, Z = −2.88; EF: 0.40 ± 0.04 vs. 0.86 ± 0.05, t = 10.00; %FS: 0.19 [0.18, 0.20] vs. 0.51 [0.44, 0.58], Z = −2.88; all P < 0.01; n = 6) [Table 1]. In addition, the level of inflammatory factors was increased before ASX administration (ASX vs. Sham: TNF-α, 197.60 [133.89, 237.94] vs. 50.48 [47.21, 57.10] pg/mL, Z = −2.88; IL-1β, 175.23 [160.74, 215.09] vs. 17.78 [16.83, 19.56] pg/mL, Z = −2.88; IL-10, 67.64 [58.90, 71.46] vs. 12.33 [11.64, 13.98] pg/mL, Z = −2.88, all P < 0.01; n = 6) [Table 1].
Table 1.
Echocardiographic parameters and inflammatory factors level on day 1 after AMI before ASX administration.
ASX significantly inhibited expression of pro-inflammatory cytokines and promoted the expression of anti-inflammatory cytokines on day 7 after myocardial infarction
The TNF-α and IL-1β levels were significantly lower in the ASX group than in the AMI group on the 7th day after AMI (TNF-α: 71.70 [68.60, 76.00] vs. 118.07 [106.92, 169.08] pg/mL, F = 42.64; IL-1β: 59.90 [50.83, 73.78] vs. 151.60 [108.44, 198.36] pg/mL, F = 44.35, all P < 0.01, n = 8), while the anti-inflammatory factor IL-10 level was significantly higher in the ASX group than in the AMI group (141.84 [118.98, 158.36] vs. 52.96 [42.68, 74.52], F = 126.67, P < 0.01; n = 8) [Table 2].
Table 2.
Inflammatory factors on day 7 after AMI.

ASX treatment improved cardiac function
The cardiac echocardiographic parameters were significantly worse in the AMI group. Conversely, the ASX group showed that cardiac function deterioration reduced, and the results were statistically significant compared with the AMI group (LVIDs: 0.50 [0.41, 0.56] vs. 0.64 [0.56, 0.74] cm, Z = −3.60; LVIDd: 0.70 [0.60, 0.76] vs. 0.80 [0.74, 0.88] cm, Z = −2.96; ESV: 0.24 [0.18, 0.45] vs. 0.58 [0.44, 0.89] mL, Z = −3.62; EDV: 0.76 [0.44, 1.04] vs. 1.25 [0.82, 1.46] mL, Z = −2.54, all P < 0.05; EF: 0.60 ± 0.08 vs. 0.50 ± 0.12, F = 160.48; %FS: 0.29 [0.24, 0.31] vs. 0.20 [0.17, 0.21], Z = −4.43, all P < 0.01; n = 16) [Table 3]. The relative LW ratio was significantly lower in the ASX group than in the AMI group (LW/body weight [BW]: 1.81 ± 0.15 vs. 2.17 ± 0.37, t = 3.66, P = 0.01; n = 16) [Table 4].
Table 3.
Echocardiographic parameters on day 28 after AMI.
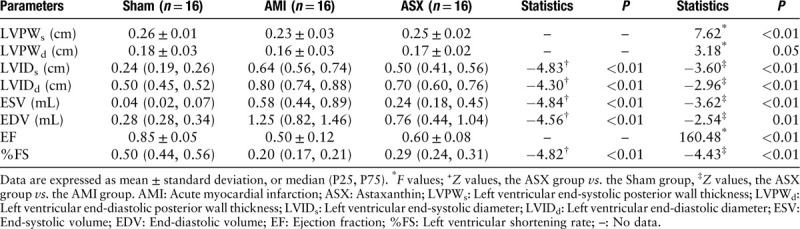
Table 4.
Relative heart/lung weight ratio on day 28 after AMI.

ASX reduced myocardial infarction size after AMI
After 28 days of AMI, the heart samples in the AMI group were visually dilated, and myocardial transmural necrosis in the infarcted area was pale and thin. However, the infarct size in the ASX group was smaller. H&E-stained samples from the sham group revealed that the cardiomyocytes were uniformly arranged, and the capsule was intact. However, the cell gaps were uniform, and the muscle fibers were completely aligned. In the AMI group, the cardiomyocytes were disorganized, and the capsule was damaged. Moreover, the number of nuclei had decreased and the muscle fibers had shrunk or even broken. ASX treatment alleviated these myocardial pathological changes after AMI. On day 28 after AMI, the count of inflammatory cells infiltrated the infarcted area and border zone was less than that in the earlier stages after AMI. Several inflammatory factors, including macrophages, were visible in the visual field [Figure 1A–1C]. For the heart cavity section, myocardial infarction led to a serious enlargement of the left ventricle in rats of the AMI group. A large number of myocardial fibers had broken down and were replaced by collagen fibers in the infarcted area. However, ASX administration ameliorated this change, especially in cases of smaller MIS, where the condition of the myocardial cells in the infarcted area also improved significantly. Although many necrotic and broken myocardiocytes were still present, the necrosis was incomplete; thus, the heart retained part of its normal structure [Figure 1E and 1F]. The infarct size was significantly smaller in the ASX group rats than in the AMI group rats (MIS [%]: 32.50 ± 1.37 vs. 50.90 ± 1.73, t = 23.63, P < 0.01; n = 8).
Figure 1.
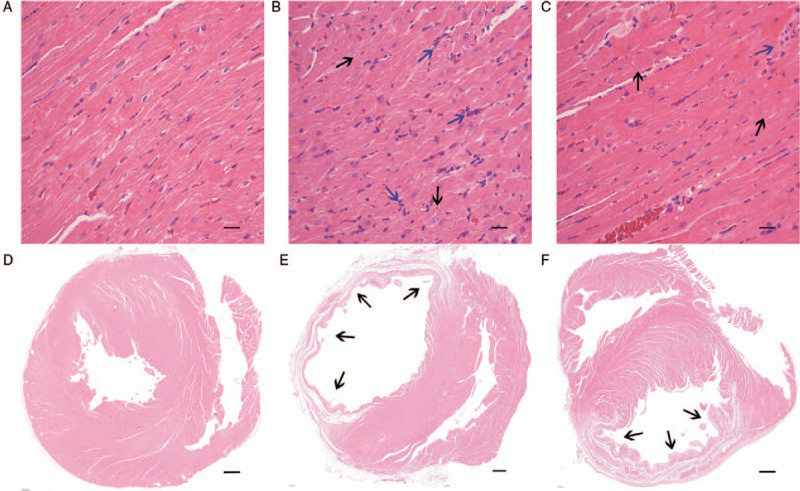
ASX significantly reduced myocardial tissue abnormalities on day 28 after AMI. (A–C) Representative H&E-stained sections of the heart tissues from the Sham, AMI, and ASX groups (n = 8 per group). (D–F) Representative H&E-stained sections of the heart cavity from the Sham, AMI, and ASX groups (n = 8 per group). The blue arrows indicate infiltrated inflammatory cells. The black arrows indicate infarcted myocardium. Scale bar = 1000 μm. AMI: Acute myocardial infarction; ASX: Astaxanthin; H&E: Hematoxylin and eosin.
ASX reduced collagen production after AMI
On day 28 after AMI, the collagen fibers were stained blue and the myocardium red. There was no obvious fibroplasia in the sham group, while in the AMI group, there was myocardial hyperplasia, and the cardiomyocytes were disorganized. In the ASX group, cardiac fibrosis was significantly less than in the AMI group, and the collagen fibers were essentially aligned [Figure 2A–2C]. The collagen volume fraction in the ASX group was considerably lower than in the AMI group (12.88 ± 2.53 vs. 28.92 ± 3.31, P < 0.01; n = 8) [Figure 2D].
Figure 2.
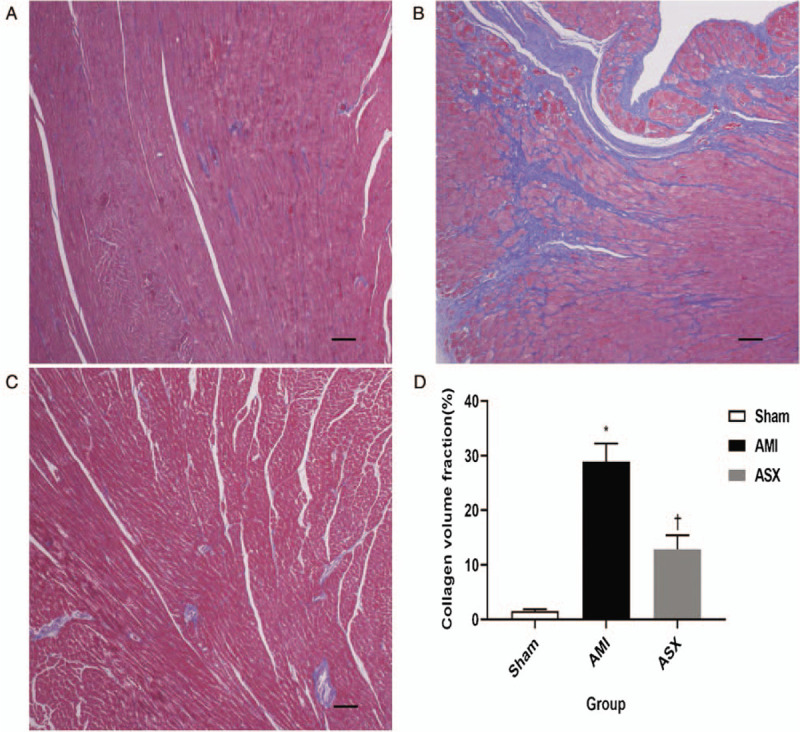
ASX reduced collagen fiber content in the border zone on day 28 after AMI. (A–C) Representative Masson trichrome-stained sections of the heart tissue from the Sham, AMI, and ASX groups (n = 8 per group) observed under a digital microscope. The blue striped structure is the proliferative collagen fiber. (D) Quantitative analysis of collagen volume fraction. ∗P < 0.01 vs. sham; +P < 0.01 vs. the AMI group. Scale bar = 100 μm. AMI: Acute myocardial infarction; ASX: Astaxanthin.
The proportion of M2 macrophages was higher in the ASX group than in the AMI group on day 7 after myocardial infarction
We used IF staining and FC to quantitatively analyze macrophages. In the IF-stained paraffin sections, macrophage nuclei were stained blue and surface markers red [Figure 3A–3C]. The integrated optical density was higher in the ASX group than in the AMI group, indicating that the infiltration of M2 macrophages into the border zone was greater in the ASX group than in the AMI group on day 7 after AMI (1583.38 ± 162.22 vs. 199.49 ± 91.09, 2891.42 ± 211.29 vs. 1583.38 ± 162.22, F = 274.35, all P < 0.01; n = 4) [Figure 3D]. Similar results were obtained in the detection of M2 macrophages in PB samples using FC. In the FC test, M2 macrophages were defined as CD11b/CD68/Arg-1-positive cells; in the flow chart, the area of the Q2-1 quadrant represents the proportion of M2 macrophages among all cells in the tested sample [Figure 4A–4C]. The proportion of M2 macrophages was higher in the ASX group, which might be why ASX improved myocardial remodeling after AMI (0.36 ± 0.05 vs. 0.26 ± 0.06, 0.96 ± 0.18 vs. 0.36 ± 0.05, F = 46.24, all P < 0.01; n = 4) [Figure 4D].
Figure 3.
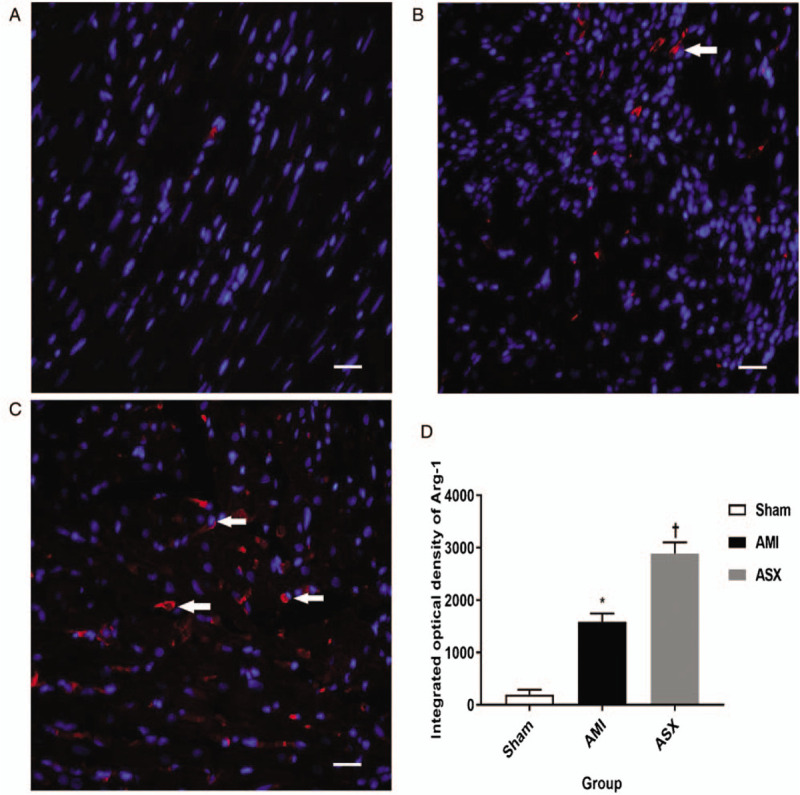
M2 macrophage count increased in the repair stages, as shown by IF staining on day 7 after AMI. (A–C) Representative IF staining sections of the heart from the Sham, AMI, and ASX groups (n = 4 per group) tissue observed under a fluorescence microscope. (D) Quantitative histogram of IF data. ∗P < 0.01 vs. sham; +P < 0.01 vs. the AMI group. Scale bar = 20 μm. AMI: Acute myocardial infarction; Arg-1: Arginase-1; ASX: Astaxanthin; IF: Immunofluorescence; IOD: Integrated optical density.
Figure 4.
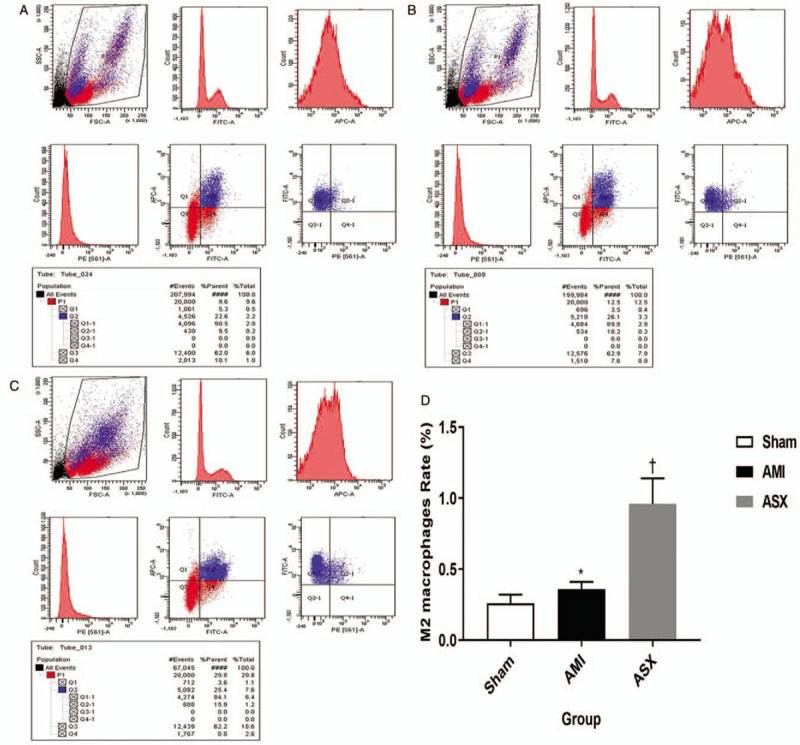
M2 macrophage count increased in the repair stages, as shown by FC on day 7 after AMI. (A–C) Representative FC results from the Sham, AMI, and ASX groups (n = 4 per group). The Q2-1 quadrant zone in the chart shows the proportion of M2 macrophages among all cells in the tested sample. (D) Quantitative histogram of the FC data. ∗P < 0.05 vs. sham; +P < 0.05 vs. the AMI group. AMI: Acute myocardial infarction; ASX: Astaxanthin; FC: Flow cytometry.
ASX inhibited apoptosis of cardiomyocytes in the border zones
The apoptotic cardiomyocytes were stained green under fluorescence microscopy after TUNEL staining. Almost no green spots were observed in the sham group, while a large number were observed in the AMI group. Apoptosis of cardiomyocytes was reduced significantly in the ASX group [Figure 5A–5C]. The AI of the cardiomyocytes was significantly higher in the AMI group than in the sham group, and ASX treatment reversed this difference (2.17 ± 0.44 vs. 0.28 ± 0.08, 1.45 ± 0.03 vs. 2.17 ± 0.44; F = 54.02, all P < 0.01; n = 4) [Figure 5D].
Figure 5.
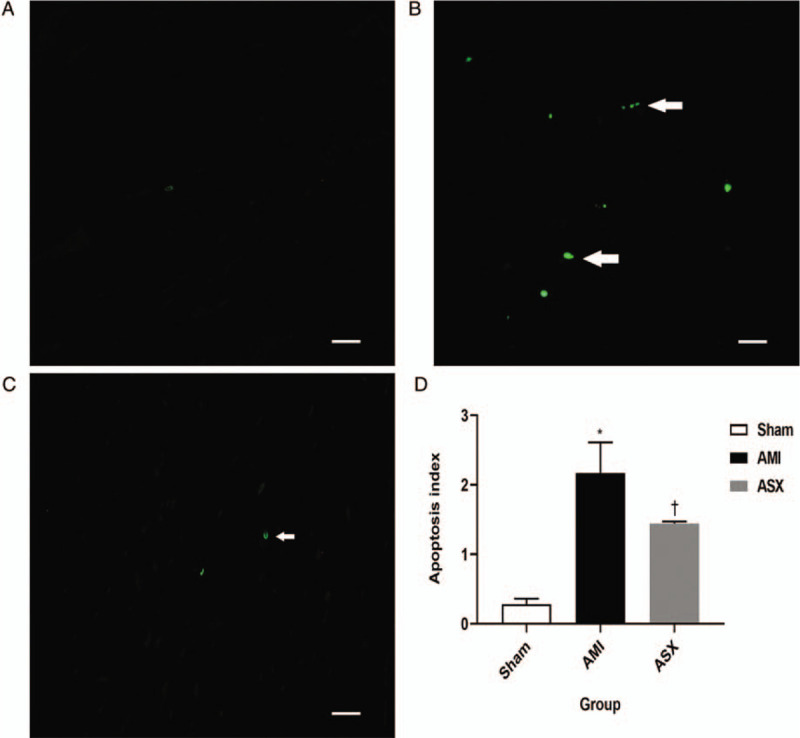
ASX reduced cardiomyocyte apoptosis on day 28 after AMI. (A–C) Representative TUNEL staining sections of the heart in the Sham, AMI, and ASX groups (n = 4 per group) observed under a fluorescence microscope. The green spot indicated by the white arrow is the apoptotic cardiomyocyte. (D) Quantitative histogram of the AI data on day 28. ∗P < 0.05 vs. sham; +P < 0.05 vs. the AMI group. Scale bar = 20 μm. AI: Apoptosis index; AMI: Acute myocardial infarction; ASX: Astaxanthin; TUNEL: Terminal dUTP Nick-end labeling.
ASX reduced the levels of myocardial fibrosis-related proteins of after AMI
We evaluated the levels of classical protein markers associated with myocardial fibrosis after AMI, including caspase 3, TGF-β1, MMP9, and collagen I/III on day 28 after AMI. In the AMI group, levels of these indicators were higher than in the sham group (caspase 3: 0.66 ± 0.04 vs. 0.10 ± 0.03, TGF-β1: 0.62 ± 0.07 vs. 0.15 ± 0.02, MMP9: 0.40 ± 0.06 vs. 0.05 ± 0.01, type I collagen: 0.74 ± 0.07 vs. 0.15 ± 0.04, type III collagen: 0.24 ± 0.05 vs. 0.05 ± 0.01, n = 4), and ASX significantly reduced expression of these proteins (caspase 3: 0.38 ± 0.06 vs. 0.66 ± 0.04, F = 170.02, TGF-β1: 0.37 ± 0.04 vs. 0.62 ± 0.07, F = 101.85, MMP9: 0.20 ± 0.06 vs. 0.40 ± 0.06, F = 50.68, collagen I: 0.42 ± 0.09 vs. 0.74 ± 0.07, F = 72.31, collagen III: 0.13 ± 0.02 vs. 0.24 ± 0.05, F = 42.91; all P < 0.01; n = 4) [Figure 6A–6E].
Figure 6.
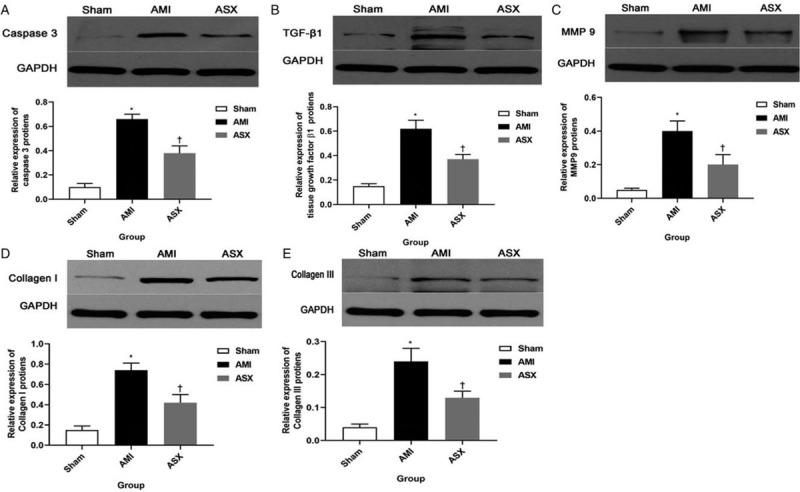
ASX reduced the expression of myocardial fibrosis-related proteins. (A) Representative Western blotting and quantitative densitometry analysis images for caspase 3. (B) Representative Western blotting and quantitative densitometry analysis images for TGF-β1. (C) Representative Western blotting and quantitative densitometry analysis images for MMP9. (D) Representative Western blotting and quantitative densitometry analysis images for collagen I. (E) Representative Western blotting and quantitative densitometry analysis images of collagen III. (n = 4). ∗P < 0.01 vs. sham; +P < 0.01 vs. the AMI group. All protein levels are normalized to GAPDH levels. AMI: Acute myocardial infarction; ASX: Astaxanthin; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; MMP9: Metalloproteinase 9; TGF-β1: Transforming growth factor β1.
Discussion
Pathological cardiac remodeling comprises structural and functional changes in the left ventricle in response to cardiovascular damage or pathogenic risk factors. It is associated with inflammation and myocardial fibrosis. Generally, there are three different phases after AMI, namely, the inflammatory infiltration phase (hours to days), subsequent proliferative phase (days to weeks), and maturation phase (several weeks).[13,14] The inflammatory response to AMI plays a crucial role in determining the infarct size and subsequent adverse cardiac remodeling. According to previous studies, the 7th day after AMI is an important time-point for the transformation from a pro-inflammatory phenotype to an anti-inflammatory phenotype.[15]
Many studies have confirmed that ASX has a strong organ-protective effect.[16,17] In respective review articles, Fassett and Coombes asserted that ASX has a strong protective effect in mouse, rat, and rabbit myocardial ischemia-reperfusion models and that it significantly reduces MIS by reducing inflammation.[18] Xue et al[19] demonstrated that ASX suppresses oxidative stress by activating the Nrf2/HO-1 pathway, thus preventing coronary microembolization-induced cardiomyocyte apoptosis and ameliorating cardiac dysfunction in rats. Garrett et al[20] pretreated Sprague-Dawley rats with different doses of ASX (25, 50, and 75 mg·kg−1·day−1 for 4 consecutive days) and subsequently developed a myocardial ischemia-reperfusion model. Both hemodynamic changes and infarction areas were significantly smaller after myocardial ischemia in the ASX-treated rats and that the change was positively correlated with the blood drug concentration. They developed a dog model of myocardial ischemia-reperfusion model and found similar results.[21] Lauver et al[22] pretreated rabbits intravenously using disodium disuccinate ASX for 4 consecutive days at a dose of 50 mg·kg−1·day−1. On the 5th day, the rabbits underwent 30 min of coronary artery occlusion, followed by a 3 h reperfusion period, leading to significantly lower complement activation and myocardial infarct size.
We permanently ligated the rat LAD coronary artery to develop an AMI model. We identified changes in cardiac function after AMI using echocardiography and relative LW ratio. Deterioration of LVIDd, EDV, EF, and %FS, as well as increased relative LW ratio, are typical manifestations of cardiac insufficiency after AMI. Our results indicated that cardiac function in rats was significantly worse after AMI and that ASX intervention improved cardiac function and reduced MIS. These findings were consistent with the histological changes evaluated using H&E, Masson trichrome, and TUNEL staining.
In previous studies, researchers have paid more attention to the protective effect of ASX on myocardial ischemic-reperfusion injury; therefore, most former investigations have pretreated or used short-term high-dose administration. The present study focused on observing myocardial fibrosis or cardiac remodeling after AMI; therefore, we used a single small dose and long post-infarct treatment time. We administered 10 mg/kg ASX to rats daily for 28 days after AMI, and the total amount of the drug was similar to that used in the above study.
Macrophages are innate immune cells that infiltrate the infarct zone early after AMI. They are involved in inflammation and repair. One recent study demonstrated that heart repair was improved when macrophages were switched from the classically activated macrophage (M1) with a pro-inflammatory phenotype into the alternatively activated macrophage (M2) with an anti-inflammatory phenotype.[23] In the proliferative stage, more M2 macrophages are recruited, while several typically pro-inflammatory factors, such as TNF-α and IL-1β, are secreted less, while IL-10, a classic anti-inflammatory factor, is secreted from the infarct zone to reduce damage and activate tissue repair. In the present study, we detected macrophages in the border zone using IF straining and in the PB using FC. M2 macrophages have many surface markers, such as CD11b/CD68, F4/80, CD206, induced nitric oxide synthase, and Arg-1. We detected the classical Arg-1 using IF staining and CD11b/CD68/Arg-1 using FC to analyze the cell surface. The main type of infiltrating macrophage in the border zone was the M2 on the 7th day after infarction. The number of M2 type macrophages in the ASX group was significantly higher than that in the AMI group. Another important result supporting macrophage polarization was the expression of inflammatory factors at different time points. M2 macrophages mainly secrete anti-inflammatory factors, such as IL-10 and IL-13.[24,25] We measured TNF-α, IL-1β, and IL-10 in the PB on day 7 after AMI using ELISA. The results were consistent with changes in macrophage polarization. The pro-inflammatory factors TNF-α and IL-1β increased significantly, and ASX treatment reversed this change. Conversely, the IL-10 concentration was higher in the ASX group than in the AMI group. The results show that ASX alleviated inflammation after AMI and promoted damage repair and that the mechanism of this protective effect may be related to ASX-induced macrophage polarization.[26,27] Peng et al[28] transplanted human umbilical cord blood mesenchymal stem cells (hUCB-MSCs) via the tail vein into a mouse model of AMI. The results demonstrated that intravenous transplantation of hUCB-MSCs could reduce the inflammatory response by stimulating the conversion of intracardiac and extracardiac macrophage subtype from M1 to M2. Besides, they could improve cardiac function and protect infarcted myocardium. Heinen et al[29] demonstrated that treatment using insulin-like growth factor 1 (IGF1) for 3 days after AMI improved cardiac function after 1 and 4 weeks. IGF1 induced an M2-like anti-inflammatory phenotype in bone marrow-derived macrophages and enhanced the number of anti-inflammatory macrophages in heart tissue on day 3 after AMI in vivo.
Myocardial fibrosis after AMI is a feature of adverse left ventricle remodeling. Using Western blotting, we detected type I and III collagen fibers, MMP9, TGF-β1, and caspase 3 in the border zone of rats on day 28 after AMI. The results showed that ASX treatment reduced type I/III collagen expression, which was consistent with the results of Masson staining. The expression of related proteins MMP9 and TGF-β1 were lower than that in the AMI group. The expression of the apoptosis-related protein caspase 3 was also inhibited in the ASX group.
To summarize, we preliminarily demonstrated that ASX has a protective effect on myocardial fibrosis after AMI. This effect may be related to macrophage polarization, but more research is needed to explore the signaling pathway and to provide a new target for clinical treatment of myocardial remodeling after AMI.
There were some limitations in the present study. Firstly, we observed the protective effect of ASX on myocardial fibrosis, but further study is needed to elucidate the signaling pathway. Moreover, we did not use M2 macrophage inhibition to fully verify the correlation between ASX intervention and M2 quantity change, so the evidence that ASX promotes M2 polarization is weak.
In conclusion, ASX treatment after AMI induced by ligating the LAD artery in rats can attenuate cardiac fibrosis and promotes M2 macrophages polarization. More studies are required to determine the exact mechanism.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81772044).
Conflicts of interest
None.
Footnotes
How to cite this article: Pan X, Zhang K, Shen C, Wang X, Wang L, Huang YY. Astaxanthin promotes M2 macrophages and attenuates cardiac remodeling after myocardial infarction by suppression inflammation in rats. Chin Med J 2020;133:1786–1797. doi: 10.1097/CM9.0000000000000814
Xia Pan and Kai Zhang contributed equally to this work.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Executive summary: heart disease and stroke statistics–2016 update: a report from the American Heart Association. Circulation 2016; 133:447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 2000; 35:569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 3.Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010; 122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol 2015; 89:1401–1438. doi: 10.1007/s00204-015-1477-x. [DOI] [PubMed] [Google Scholar]
- 5.Guerin M, Huntley ME, Olaizola M. Haematococcus astaxanthin: applications for human health and nutrition. Trends Biotechnol 2003; 21:210–216. doi: 10.1016/S0167-7799(03)00078-7. [DOI] [PubMed] [Google Scholar]
- 6.Nagendraprabhu P, Sudhandiran G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFkB and COX-2. Invest New Drugs 2011; 29:207–224. doi: 10.1007/s10637-009-9342-5. [DOI] [PubMed] [Google Scholar]
- 7.Liu G, Shi Y, Peng X, Liu H, Peng Y, He L. Astaxanthin attenuates adriamycin-induced focal segmental glomerulosclerosis. Pharmacology 2015; 95:193–200. doi: 10.1159/000381314. [DOI] [PubMed] [Google Scholar]
- 8.Kim JH, Nam SW, Kim BW, Choi W, Lee JH, Kim WJ, et al. Astaxanthin improves stem cell potency via an increase in the proliferation of neural progenitor cells. Int J Mol Sci 2010; 11:5109–5119. doi: 10.3390/ijms11125109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizuta M, Hirano S, Hiwatashi N, Tateya I, Kanemaru S, Nakamura T, et al. Effect of astaxanthin on vocal fold wound healing. Laryngoscope 2014; 124:E1–E7. doi: 10.1002/lary.24197. [DOI] [PubMed] [Google Scholar]
- 10.Tran BH, Huang C, Zhang Q, Liu X, Lin S, Liu H, et al. Cardioprotective effects and pharmacokinetic properties of a controlled release formulation of a novel hydrogen sulfide donor in rats with acute myocardial infarction. Biosci Rep 2015; 35:e216.doi: 10.1042/BSR20140185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muthuramu I, Lox M, Jacobs F, De Geest B. Permanent ligation of the left anterior descending coronary artery in mice: a model of post-myocardial infarction remodelling and heart failure. J Vis Exp 2014; 94:e52206.doi: 10.3791/52206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu W, Zhang X, Zhao M, Zhang X, Chi J, Liu Y, et al. Activation in M1 but not M2 macrophages contributes to cardiac remodeling after myocardial infarction in rats: a critical role of the calcium sensing receptor/NRLP3 inflammasome. Cell Physiol Biochem 2015; 35:2483–2500. doi: 10.1159/000374048. [DOI] [PubMed] [Google Scholar]
- 13.Gombozhapova A, Rogovskaya Y, Shurupov V, Rebenkova M, Kzhyshkowska J, Popov SV, et al. Macrophage activation and polarization in post-infarction cardiac remodeling. J Biomed Sci 2017; 24:13.doi: 10.1186/s12929-017-0322-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C, et al. Mechanisms contributing to cardiac remodelling. Clin Sci (Lond) 2017; 131:2319–2345. doi: 10.1042/CS20171167. [DOI] [PubMed] [Google Scholar]
- 15.Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 2018; 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Agamy SE, Abdel-Aziz AK, Wahdan S, Esmat A, Azab SS. Astaxanthin ameliorates doxorubicin-induced cognitive impairment (chemobrain) in experimental rat model: impact on oxidative, inflammatory, and apoptotic machineries. Mol Neurobiol 2018; 55:5727–5740. doi: 10.1007/s12035-017-0797-7. [DOI] [PubMed] [Google Scholar]
- 17.Okazaki Y, Okada S, Toyokuni S. Astaxanthin ameliorates ferric nitrilotriacetate-induced renal oxidative injury in rats. J Clin Biochem Nutr 2017; 61:18–24. doi: 10.3164/jcbn.16-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fassett RG, Coombes JS. Astaxanthin: a potential therapeutic agent in cardiovascular disease. Mar Drugs 2011; 9:447–465. doi: 10.3390/md9030447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue Y, Sun C, Hao Q, Cheng J. Astaxanthin ameliorates cardiomyocyte apoptosis after coronary microembolization by inhibiting oxidative stress via Nrf2/HO-1 pathway in rats. Naunyn Schmiedebergs Arch Pharmacol 2019; 392:341–348. doi: 10.1007/s00210-018-1595-0. [DOI] [PubMed] [Google Scholar]
- 20.Gross GJ, Lockwood SF. Cardioprotection and myocardial salvage by a disodium disuccinate astaxanthin derivative (Cardax). Life Sci 2004; 75:215–224. doi: 10.1016/j.lfs.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Gross GJ, Lockwood SF. Acute and chronic administration of disodium disuccinate astaxanthin (Cardax) produces marked cardioprotection in dog hearts. Mol Cell Biochem 2005; 272:221–227. doi: 10.1007/s11010-005-7555-2. [DOI] [PubMed] [Google Scholar]
- 22.Lauver DA, Lockwood SF, Lucchesi BR. Disodium disuccinate astaxanthin (Cardax) attenuates complement activation and reduces myocardial injury following ischemia/reperfusion. J Pharmacol Exp Ther 2005; 314:686–692. doi: 10.1124/jpet.105.087114. [DOI] [PubMed] [Google Scholar]
- 23.Hasan AS, Luo L, Yan C, Zhang TX, Urata Y, Goto S, et al. Cardiosphere-derived cells facilitate heart repair by modulating M1/M2 macrophage polarization and neutrophil recruitment. PLoS One 2016; 11:e165255.doi: 10.1371/journal.pone.0165255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinberger T, Schulz C. Myocardial infarction: a critical role of macrophages in cardiac remodeling. Front Physiol 2015; 6:107.doi: 10.3389/fphys.2015.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korf-Klingebiel M, Reboll MR, Klede S, Brod T, Pich A, Polten F, et al. Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat Med 2015; 21:140–149. doi: 10.1038/nm.3778. [DOI] [PubMed] [Google Scholar]
- 26.Diao W, Chen W, Cao W, Yuan H, Ji H, Wang T, et al. Astaxanthin protects against renal fibrosis through inhibiting myofibroblast activation and promoting CD8(+) T cell recruitment. Biochim Biophys Acta Gen Subj 2019; 1863:1360–1370. doi: 10.1016/j.bbagen.2019.05.020. [DOI] [PubMed] [Google Scholar]
- 27.Farruggia C, Kim MB, Bae M, Lee Y, Pham TX, Yang Y, et al. Astaxanthin exerts anti-inflammatory and antioxidant effects in macrophages in NRF2-dependent and independent manners. J Nutr Biochem 2018; 62:202–209. doi: 10.1016/j.jnutbio.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Peng Y, Chen B, Zhao J, Peng Z, Xu W, Yu G. Effect of intravenous transplantation of hUCB-MSCs on M1/M2 subtype conversion in monocyte/macrophages of AMI mice. Biomed Pharmacother 2019; 111:624–630. doi: 10.1016/j.biopha.2018.12.095. [DOI] [PubMed] [Google Scholar]
- 29.Heinen A, Nederlof R, Panjwani P, Spychala A, Tschaidse T, Reffelt H, et al. IGF1 treatment improves cardiac remodeling after infarction by targeting myeloid cells. Mol Ther 2019; 27:46–58. doi: 10.1016/j.ymthe.2018.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]