

Abstract
Efficient drug accumulation in tumor cells is essential for cancer therapy. Herein, we developed a dimeric prodrug self-delivery nanoparticles (NPs) with enhanced drug loading and bioreduction-responsiveness for triple negative breast cancer (TNBC) therapy. Specially designed camptothecin dimeric prodrug (CPTD) containing a disulfide bond was constructed to realize intracellular redox potential controlled drug release. Direct conjugation of hydrophobic CPTD to poly(ethylene glycol) PEG5000, a prodrug-based amphiphilic CPTD-PEG5000 copolymer was synthesized, which could encapsulate parental CPTD prodrug spontaneously and form ultra-stable NPs due to the highly analogous structure. Such dimeric prodrug self-delivery nanoparticles showed ultra-high stability with CMC as low as 0.75 μg/ml and remained intact during endocytosis. In addition, neurotensin (NT), a 13 amino acid ligand, was further modified on the nanoparticles for triple negative breast cancer (TNBC) targeting. Optimized NT-CPTD NPs showed improved pharmacokinetics profile and increased drug accumulation in TNBC lesions than free CPT, which largely reduced the systemic toxicity and presented an improved anti-cancer efficacy in vivo. In summary, with advantages of extremely high drug loading capacity, tumor microenvironmental redox responsiveness and targeted TNBC accumulation, NT-CPTD NPs showed its potential for effective triple negative breast cancer therapy.
Keywords: Triple negative breast cancer, Neurotensin, Camptothecin, Redox responsiveness, Prodrug
TOC graphic
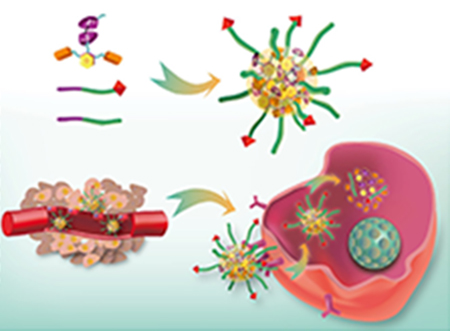
1. INTRODUCTION
Triple negative breast cancer (TNBC) is a subtype of breast cancer lacking expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2), which possesses around 20 % of all breast cancer diagnoses and is notoriously aggressive contributing to high number of metastatic lesions and poor prognosis 1. Current treatment for TNBC relies primarily on surgery, radiation and non-specific chemotherapies. However, most antitumor drugs have great limitations in clinical use, such as poor water solubility, fast clearance, limited tumor selectivity and severe side effects. In the past decades, various biocompatible nanosystems such as polymeric prodrugs2, liposomes3, polymeric micelles4, polymeric nanoparticles5 and inorganic nanoparticles6 have been developed to greatly enhance anticancer drugs’ water solubility and stability, to modulate their pharmacokinetic and pharmacodynamics profile 7. For conventional nanomedicines, there are two strategies to achieve anti-cancer drug delivery. One traditional way is to encapsulate hydrophobic anti-cancer drugs by using amphiphilic block polymers. However, for such type of nanomedicine, the outer polymer carrier occupies the major component while the therapeutic drug content remains relatively low, barely more than 10%8–12. Besides, such approach may result in poor drug release profile such as pre-mature drug release during circulation. To improve drug loading efficiency and release profile, researchers directly conjugated hydrophobic anticancer drugs to the synthetic high molecular polymers, developing polymeric micelles or nanocapsules. Such strategy could increase the drug loading to some extent. However, the covalent conjugation might retard drug release, inducing long-term insufficient drug exposure, which might activate the metastatic or multi-drug resistance of tumors13–15. Therefore, design of new nanomedicines that could simultaneously achieving high drug loading capacity and controllable drug release is clearly of great significance.
In this regard, we designed a dimeric prodrug structure (as shown in Scheme 1), which introduce “structure defects” into the prodrug design forming less rigid molecules. Unlike most rigid hydrophobic anti-cancer drugs, which could easily form crystals or large aggregates during formulation due to the long-distance-order structure, our dimeric prodrug structure was deliberately designed by conjugating two molecules of anti-cancer drugs onto a phenol ring via σ bonds, which could freely rotate and elongate drug distance, substantially inhibiting long-distance-ordered drug molecule packing. Such less-rigid dimeric prodrug could significantly lower the precipitation rate of prototype drug, prevent amorphous aggregation, and therefore greatly increased the drug loading efficiency during formulation16. Apart from that, we directly conjugating dimeric prodrug with polyethylene glycol (PEG5000) forming drug containing amphiphilic polymer, which could spontaneously encapsulate hydrophobic dimeric prodrug during self-assembly. Such highly analogous structure between amphiphilic polymer and hydrophobic dimeric prodrug enables a much stronger molecular interaction, which further increase the drug loading content and also enhanced stability of polymeric nanoparticles at the same time.
Scheme 1.
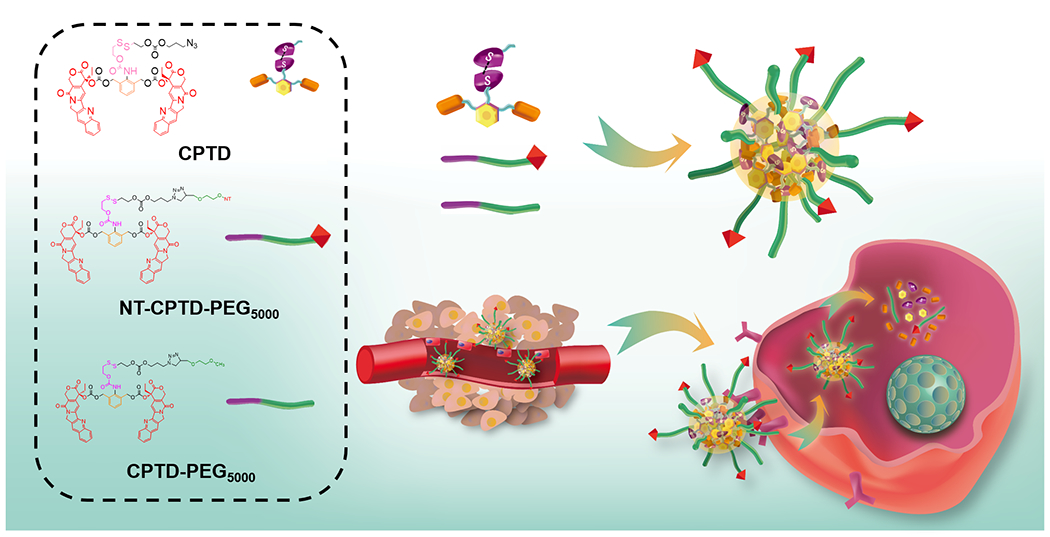
(a) NT-CPTD NPs preparation and TNBC intracellular redox-triggered drug release
In addition to high drug loading efficiency, a temporally and spatially controlled drug release in response to the triggers in tumor microenvironment is also desired to enhance safety profile of nanomedicines 17. Intracellular redox potential, which possesses approximately 3 orders of magnitude higher glutathione level (~10 mM) comparing to the extracellular environment (~ 10 μM), makes it an ideal trigger for nanomedicine design 11, 34, 36. In addition, with the co-presence of the specific reducing enzyme such as gamma interferon-inducible lysosomal thiol reductase (GILT) 18, general redox responsive materials containing disulfide bond can stay stable at oxidizing physiological conditions, while undergo thiol-disulfide exchange and rapid breakdown in tumor cells 34. Therefore, we intentionally embedded disulfide bond into the dimeric prodrug structure for controlled redox-responsive drug release.
Utilizing the huge redox potential difference between tumor extracellular and intracellular environment, we herein used disulfide bond as the trigger in prodrug design and constructed a redox-responsive dimeric prodrug (CPTD). By directly conjugating hydrophobic CPTD to hydrophilic PEG5000, we successfully synthesized a well-controlled drug-containing amphiphilic vehicle (CPTD-PEG5000) with capacity to spontaneously encapsulate CPTD with extremely high drug loading efficiency. Noting that neurotensin receptor type 1 (NTSR1) displays a major role in cancer progression, malignancy and metastasis19, 20, and also showed high expression in TNBC21. A NTSR1 ligand, neurotensin (NT), was further covalently anchored on the polymeric nanoparticles surface forming NT-CPTD NPs to achieve in vivo TNBC-targeting capability.
In this work, we deliberately designed the novel NT-CPTD NPs with the following features: (i) disulfide-containing dimeric CPTD structure achieved temporally and spatially controlled drug release behavior; (ii) dimeric structure of CPTD prodrug, highly analogous structure between CPTD-PEG5000 amphiphilic polymer and hydrophobic CPTD, enabled the extremely high drug loading content; (iii) modification of NT peptide could improve the chemodrug accumulation in TNBC. Detailed design, preparation and characterization of NT-CPTD NPs were performed. The in vitro and in vivo TNBC therapeutic efficiency were also investigated. We believe that NT-CPTD NPs could serve well as a promising platform for TNBC therapy.
2. RESULTS AND DISCUSSION
Efficient drug accumulation in tumor cells is essential in cancer therapy. To achieve such goal, we constructed a novel dimeric prodrug nanoparticle, NT-CPTD NPs. As shown in Scheme 1, NT-CPTD NPs contained a specially designed dimeric prodrug CPTD, which could assemble into sub-100 nm particles in the presence of the prodrug-based amphiphilic copolymer CPTD-PEG5000 to achieve ultra-high drug loading and excellent stability. By modification with NT peptide, NT-CPTD NPs could accumulate in tumor tissues via EPR effect and actively be endocytosed by TNBC cells through NTSR1 receptor. Additionally, chemotherapeutic drug release was well-controlled by the disulfide bond cleavage in dimeric prodrug structure, which could prevent immature extracellular drug leakage and ensure efficient drug release by reductive thiols in TNBC cells. Combining TNBC-specific recognition and controlled drug release, NT-CPTD NPs could deliver CPT into TNBC with higher efficiency and much less side effects.
2.1. Synthesis and characterization of CPTD prodrug, CPTD-PEG5000 and NT-PEG5000-CPTD polymer
In order to achieve precise drug release control and high drug loading capacity, we deliberately constructed the dimeric prodrug CPTD (Scheme 1), where two hydrophobic anticancer drug CPT were stably conjugated to a phenol backbone via freely rotatable carbonate linkage. Such dimeric structure performed less rigidity comparing to prototype CPT, which could highly retard long-distance-order packaging and large aggregates forming during nanoparticle formulation. Besides, we conjugated CPTD with PEG5000 forming prodrug-based amphiphilic copolymer CPTD-PEG5000 (Scheme 1), which could spontaneously encapsulate hydrophobic dimeric prodrug CPTD during self-assembly. Such highly analogous structure between CPTD-PEG5000 and CPTD enables a much stronger molecular interaction, which further increase the drug loading content and also enhanced stability of polymeric nanoparticles at the same time. To achieve controlled drug release in response to tumor microenvironment, we embedded disulfide bond into the side chain of CPTD. Encountered with high redox potential in tumor cells, disulfide bond in CPTD could be cleaved and induce self-cyclization and 1,6-elimination to release prototype CPT spontaneously (Scheme S3).
The synthetic routes for CPTD prodrug and prodrug-contained amphiphilic copolymers were illustrated in Scheme S1 and Scheme S2. Prodrug-based amphiphilic CPTD-PEG5000 copolymer was synthesized by conjugating CPTD prodrug to PEG (Mw = 5000) through click reaction between the azido group of CPTD and the alkyne group of alkyne-PEG5000. As shown in Figure S1–2, the disappearance of alkyne peak at 2.5 ppm of alkyne-PEG5000 and the new peaks between 7.50-8.50 ppm indicated the successful synthesis of CPTD-PEG5000. To enable the TNBC-targeting capability, NT-PEG5000-CPTD polymer was synthesized by bi-functional PEG5000 (MAL-PEG5000-ALK). The disappearance of the MAL peaks at 6.70 ppm and appearance of CPTD peaks between 7.50-8.50 ppm indicated the successful preparation of the NT-PEG5000-CPTD polymer (Figure S3). The MALD-TOF results of CPTD-PEG5000 and NT-PEG5000-CPTD were also measured (Figure S4).
2.2. Formulation and characterization of CPTD NPs and NT-CPTD NPs
Camptothecin (CPT) shows a broad range of anticancer activity via binding to topoisomerase-I and DNA complex22, however, the poor water solubility greatly hindered its application in clinical application23, 24. The strong π-π interaction of CPT leads to a fast aggregation during nanoformulation, resulting in uncontrollable drug formulations with very broad particle size distribution. Large precipitates were observed when CPT was co-precipitated with CPTD-PEG5000 polymer (Figure 1E, Entry 9&10). In contrast, CPTD prodrug could effectively disrupt the rapid intermolecular aggregation and prevent the formation of large aggregates 16, thus forming NPs with much smaller particle size. By adjusting the weight ratio between CPTD and CPTD-PEG5000 copolymer, we could form a series of NPs with different particle sizes and stability (Figure 1E, Entry 1-7). Stable CPTD nanoparticles could be prepared with CPTD/CPTD-PEG5000 ratio up to 16:1 with an average particle size of 103.1±1.6 nm (Figure 1E, entry1). Increasing CPTD-PEG5000 content resulted in decreased particle size, broader PDI, and slightly compromised stability (Figure 1E, Entry 7). CPTD-PEG5000 by itself couldn’t form stable nanostructures (Figure 1E, Entry 8), because of the relatively high hydrophilicity of CPTD-PEG5000 polymer.
Figure 1.
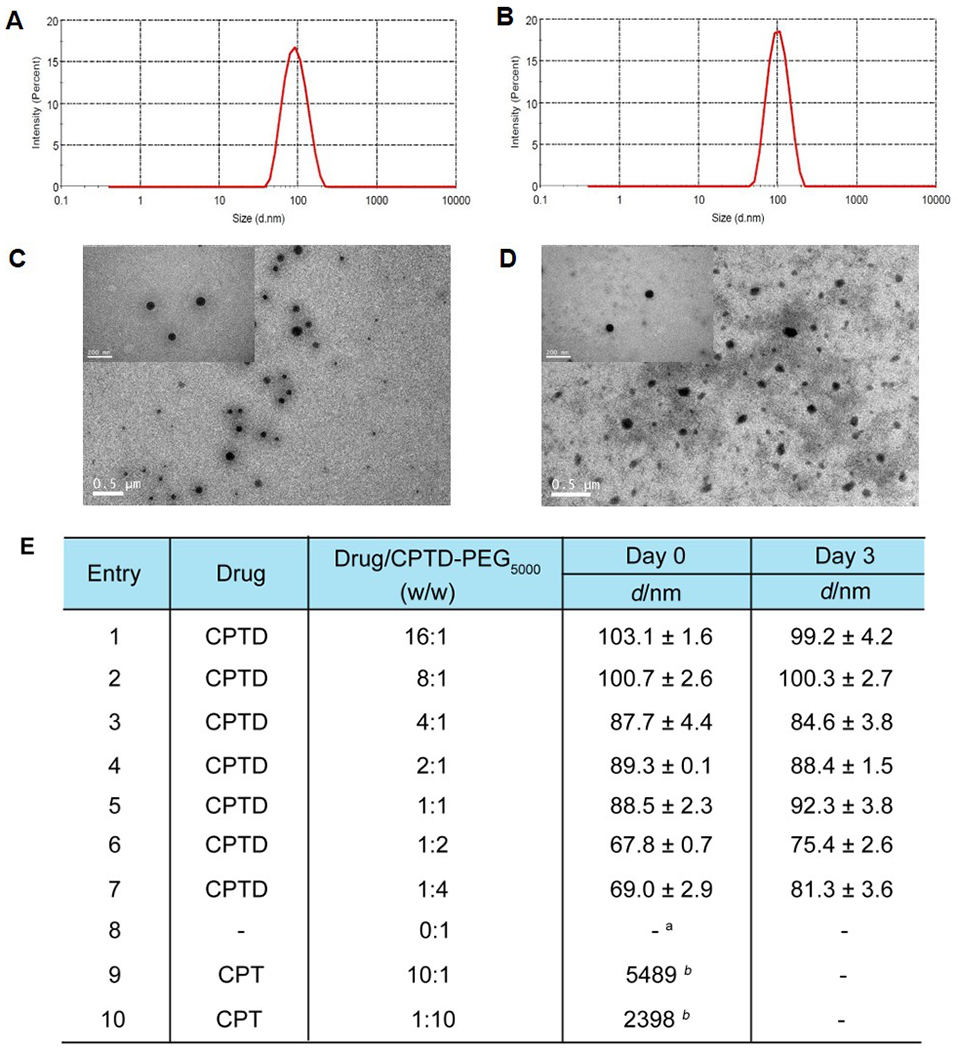
Size distribution of (A) CPTD NPs and (B) NT-CPTD NPs; TEM images of (C) CPTD NPs and (D) NT-CPTD NPs; (E) Formulation of CPTD and CPT with CPTD-PEG5000 via Nanoprecipitation.
a No parpticles were detected
b Large aggregates were detected
To further optimizing a CPTD NPs with relatively high drug loading efficiency and excellent stability. We tested the stability of formulation 1-7 (Figure 1E) against PBS and Fetal Bovine Serum (FBS). CPTD/CPTD-PEG5000 (16:1) NPs and CPTD/CPTD-PEG5000 (8:1) NPs formed precipitates instantly after diluted with PBS (data not shown). As shown in Figure S5, particle size of CPTD/CPTD-PEG5000 (4:1) NPs increased a lot after 72 h, while formulation 4-7 could stay stable against both PBS and FBS. Considering relatively high drug loading, we chose CPTD/CPTD-PEG5000 (2:1) NPs for further study. The particle size of CPTD NPs was 89.3±0.1 nm with PDI of 0.04±0.012 (Figure 1A). NT-CPTD NPs showed slightly increased size of 96.6±1.6 nm with PDI of 0.07±0.029 (Figure 1B), which may due to the conjugation with TNBC targeting NT peptide. The morphology of CPTD NPs and NT-CPTD NPs were captured by TEM, which showed an extremely dense drug aggregates core(Figure 1C, D). Such phenomenon was barely observed in most organic material-based nanoparticles, which also directly revealed the extremely high encapsulate efficiency of NPs. Both CPTD NPs and NT-CPTD NPs showed slightly negative zeta potential of −8.51±1.64 mV and −5.51±0.19 mV respectively. The drug encapsulation efficiency (EE) and drug loading (DL) of CPTD NPs was 99% and 38.3% respectively, exceeded most current anti-cancer nanomedicines whose DL barely exceed 20%23, 25, 26. More importantly, CPTD NPs showed extremely high stability against dilution effect. As shown in Figure S6, CPTD NPs remained stable at concentration as low as 0.75 μg/ml, which is well below the critical micelle concentration (CMC) of most micellar systems8, 27. To further evaluate the NPs stability during circulation, we incubated CPTD NPs and NT-CPTD NPs in PBS 7.4 and also 10 % FBS containing buffers, which mimicking the physiological pH and human serum conditions. As shown in Table S1, both CPTD NPs and NT-CPTD NPs stayed stably with slightly size change after 24 h incubation. We also investigated the nanoparticle stability in PBS 6.5, which mimicking the acidic tumor extracellular microenvironment. As shown in Table S1, both CPTD NPs and NT-CPTD NPs stayed stably with slightly size change after 24 h incubation. Therefore, our dimeric prodrug self-delivery nanoparticles showed satisfied stability in vitro.
2.3. In vitro drug release
To verify the controlled drug release, we next evaluated the in vitro drug release profile of non-targeting CPTD NPs and NT-CPTD NPs in various buffer solutions mimicking the tumor redox microenvironment. As shown in Figure 2B, with the presence of 10 mM dithiothreitol (DTT), which mimics tumor intracellular thiol environment, as high as 93% and 87% of prototype CPT were released over 48 hour from CPTD NPs and NT-CPTD NPs respectively, while no immature CPTD prodrug release was observed during the experiment (Figure 2B). As for 20 μM DTT, which represents tumor extracellular thiol environment, only about 11% and 5 % CPT were released from CPTD NPs and NT-CPTD NPs.
Figure 2.
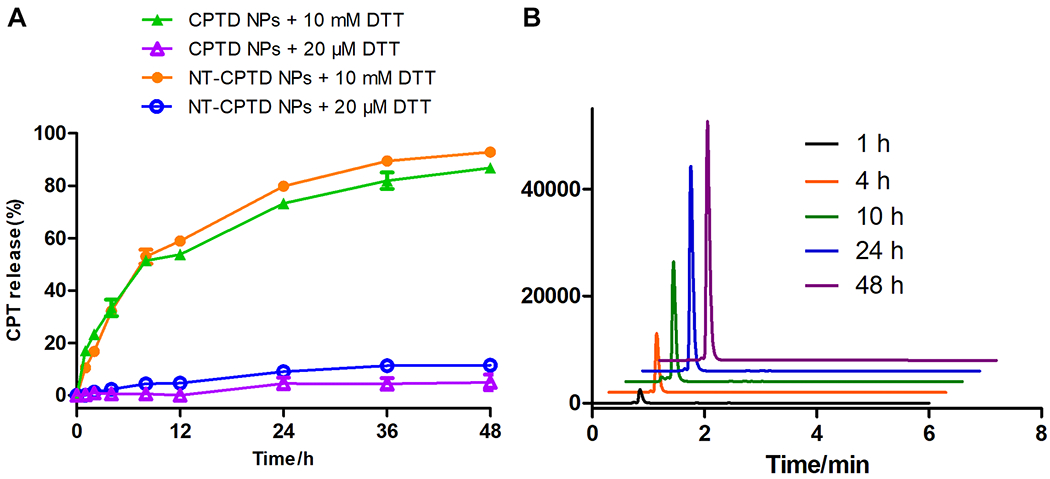
(A) In vitro CPT release from NT-CPTD NPs and CPTD NPs triggered by different concentrations of thiol trigger in PBS (pH = 7.4) at 37 °C. Data were presented as means ± SD (n = 3). (B) HPLC trace of CPT release from NT-CPTD NPs in the presence of 10 mM DTT.
The controlled CPT release was realized via the disulfide bond designed in the CPTD structure. Encountered with the high redox potential in cancer cell cytosol, the disulfide bond in CPTD structure could be cleaved through thiol-disulfide exchange. The exposed thiol group could go through cyclization toward the carbonyl bond and release an arylamine subunit that could spontaneously go through self-elimination to release carbonate-capped free CPT from the CPTD prodrug (Scheme S3).
2.4. Extraordinary stability of NPs during endocytosis
Several studies have indicated that hydrophobic cargos encapsulated in the polymeric micelle might go through pre-mature released during circulation28, 29. In our nanoplatform, the unique dimeric structure of CPTD and strong interaction between hydrophobic CPTD prodrug and prodrug-based amphiphilic copolymer CPTD-PEG5000 could ensure the ultra-high stability of NPs. To investigate the endocytosis stability of CPTD NPs, we encapsulated fluorescent probe, DiO (λex = 488 nm, λem = 500 nm) and DiI (λex = 500 nm, λem = 580 nm) as a pair of hydrophobic FRET probes in CPTD NPs. When the FRET pair is in vicinity with each other, only DiI fluorescent would be observed when being excited with 488 nm light due to energy transfer from DiO to DiI. A strong DiI fluorescence signal was observed from CPTD NPs in DI water (Figure 3A black curve) with a FRET ratio IR/(IG + IR) of 0.86, where IR and IG are fluorescence intensities at 580 nm and 500 nm, respectively. When the FRET NPs were disrupted with methane, the FRET phenomenon disappeared (Figure 3A red curve) resulting in a FRET ratio of 0.08. Therefore, by observing fluorescence status of FRET NPs, we could easily monitor the stability of NPs during endocytosis.
Figure 3.
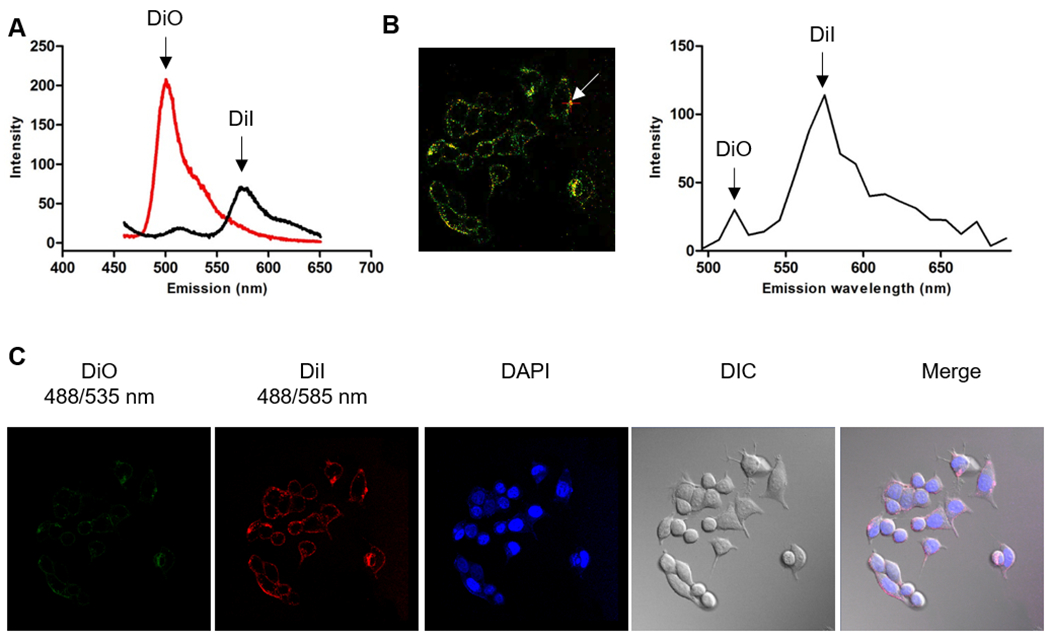
(A) Spectra of CPTD/CPTD-PEG5000 FRET NPs with DiO and DiI as the FRET probe diluted with 10× water (black curve, stable NPs) and 10× methanol (red curve, dissociated NPs). (B) Confocal fluorescence image of MDA-MB-231 cells incubated with 0.2 mg/ml CPTD/CPTD-PEG5000 FRET NPs for 1 h and spectra measured inside of cells. (C) Confocal images of CPTD/CPTD-PEG5000 FRET NPs in MDA-MB-231 cells for 1h. λex = 488 nm, λem (DiO) = 535 ± 20 nm, λem (DiI) = 585 ± 20 nm. Fluorescence of DiI could be observed only when DiO and DiI are co-encapsulated in the NPs. All images were visualized via 63×oil immersion lens.
As shown in Figure 3C, after MDA-MB-231 TNBC cells were incubated with CPTD FRET NPs for 1 h, barely no DiO fluorescence (green) was observed and the FRET signal was predominant in the cytoplasm, indicating that CPTD NPs could be taken into cells as an intact form and effectively prevented extracellular pre-release of anti-cancer drugs. To further confirm the status of intracellular CPTD NPs, fluorescence spectra of CPTD NPs inside of cells were measured by using a spectral detector, with 484 nm excitation and emission scan from 490 to 690 nm (Figure 3B). We collected the whole spectra data from 490 to 690 nm of the arrow pointed site after 488 nm excitation. As shown in Figure 3B, due to the FRET effect between DiO and DiI dyes, we observed the predominant the high peak at 585 nm and little signal of disassembled DiO dye at 535 nm, which also successfully confirmed integrity of CPTD NPs in cytoplasm. Therefore, unlike most micellar or polymer NPs, whose integrity is immensely compromised in the process of endocytosis 28. Our unique nanoplatform CPTD NPs exhibited extremely high stability during endocytosis, therefore successfully prevent pre-drug release and effectively avoid intrinsic systemic cytotoxicity of the anti-cancer drugs. We also observed the disassembly behavior of CPTD NPs by increasing the incubation time period of nanodrugs. As shown in Figure S10, with time increasing, DiO signals which represented the disassembly of NPs increased and after incubation for 4 h there showed no FRET signals, which indicated complete NPs disassembly due to the high redox potential in tumor cells. Such phenomenon suggested the on-demand drug release profile of our redox-responsive dimeric prodrug nanoplatform.
2.5. Cellular uptake and internalization mechanism study
To improve CPTD NPs accumulation in tumor site, TNBC targeting NT peptide was chosen to modify the polymer for targeted TNBC uptake. Coumarin-6 was encapsulated into CPTD NPs and NT-CPTD NPs for in vitro nanoparticle tracing. Cellular uptake mechanism and possible endocytosis pathway of NPs were investigated in MDA-MB-231 cells. To evaluate the optimized ligand modification ratio, 10, 20, 40 wt% NT peptide modified NPs were prepared. Compared with the non-targeting CPTD NPs, cellular uptake of NT peptide modified NPs were significantly enhanced (Figure 4A). However, there showed no significant cellular uptake difference between 20% and 40% NPs, which might due to the saturation transport capacity of NTSR1 expressed on the cell surface. Previous study also revealed a compromised systemic circulation time with high ligands modification nanoparticles 27. Therefore, we chose 20 % modified NT-CPTD NPs in following studies.
Figure 4.
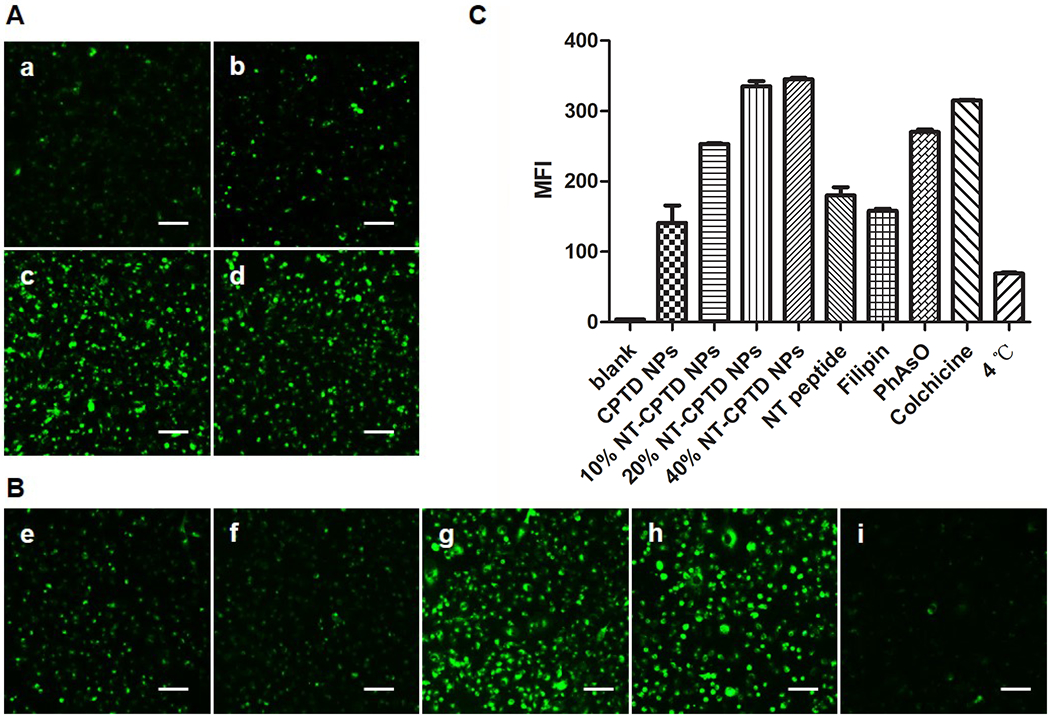
(A) Cellular uptake of (a) CPTD NPs and NT-CPTD NPs with different NT-PEG5000-CPTD ratios (b) 10, (c) 20, and (d) 40 wt% in MDA-MB-231 cells after 30 min incubation. (B) Possible endocytosis pathway of NT-CPTD NPs study. Cells were blocked by different inhibitors (e) 50 μM NT peptide (f) 1 μg/mL filipin (g) 0.3 μg/mL PhAsO (h) 1 μg/mL Colchicine (i) Cells were incubated at 4 °C. Scale bars represent 100 μm. (C) Quantitative results of (A) and (B) analyzed from Flow cytometry analysis.
To further elucidate the endocytosis pathway of NT-CPTD NPs, several internalization inhibitors were used including NT peptide (blocking NTSR1), colchicine (blocking macropinocytosis), Phenylarsine oxide (blocking clathrin-dependent pathway) and filipin (blocking caveolae-mediated pathway) (Figure 4B). The internalization of NT-CPTD NPs was inhibited significantly by NT peptide and filipin. Additionally, low temperature remarkably inhibited the cellular uptake as well (Figure 4B), demonstrating the NPs internalization was energy-dependent. Therefore, NT-CPTD NPs were mostly endocytosed into MDA-MB-231 cells via caveolae-mediated pathway. In addition, such unique endocytosis pathway could avoid localization of NPs in acidic organelle such as lysosomes, whose acidic environment could greatly hinder cleavage of disulfide bond and free CPT drug release profile (Figure S7). We also examined the cellular uptake of NT-CPTD NPs in NTR-positive MDA-MB-231 cells and NTR-negative 293 cells (Figure S8). NTR-negative 293 cells showed almost no signal of NT-CPTD NPs uptake, which also proved the specific active targeting effect of NT peptide.
2.6. In vitro antitumor efficacy
The in vitro antitumor efficacy of NPs was performed by MTT study and cellular apoptosis assay on MDA-MB-231 cell. As shown in Figure 5A, NT-CPTD NPs showed much lower IC50 values compared to CPTD NPs, because of the active targeting capacity between NT peptide and NTSR1 receptor expressed in MDA-MB-231. IC50 values of CPTD NPs and NT-CPTD NPs were slightly higher than free CPT. Such phenomenon was understandable, because hydrophobic drug CPT could passive diffuse into tumor cells, whereas NPs were mainly internalized through endocytosis pathway. Nevertheless, our designed high drug loading nanoparticles CPTD NPs and NT-CPTD NPs both presented significantly higher cytotoxicity than clinically used CPT derivative Irinotecan. We also performed the in vitro apoptosis assay on MDA-MB-231 cells by Annexin V-FITC and PI assay. As shown in Figure 5B, the apoptosis results shared consistent trend with the MTT study (Figure 5A), indicating the potential of NT-CPTD NPs as a novel drug delivery platform for TNBC treatment.
Figure 5.
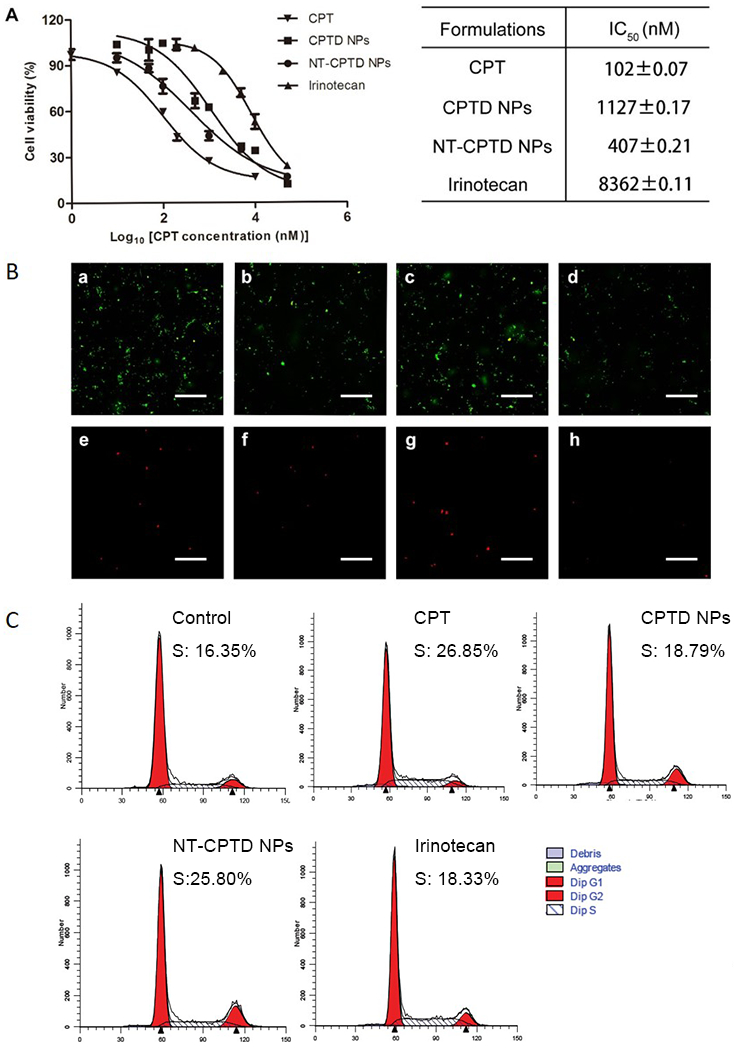
(A) Cytotoxicity of CPT, CPTD NPs, NT-CPTD NPs and Irinotecan against MDA-MB-231 cells. Data are presented as means ± SD (n=4). (B) Cellular apoptosis of MDA-MB-231 cells induced by CPT (a, e), CPTD NPs (b, f), NT-CPTD NPs (c, g), and Irinotecan (d, h) was examined by fluorescence microscopy. Red, PI labeled dead cells; Green, Annexin V-FITC labeled apoptotic cells. Scale bars represent 200 μm. (C) Representative cell cycle distribution in MDA-MB-231 after incubation of CPT, CPTD NPs, NT-CPTD NPs and Irinotecan.
CPT was reported to trigger cell cycle arrest on S phase and inducing cell apoptosis 25, 30. We investigated cell cycle arrest on MDA-MB-231 cells induced by CPT, CPTD NPs, NT-CPTD NPs and Irinotecan. As shown in Figure 5C, CPT, CPTD NPs and NT-CPTD NPs all showed significant S phase arrest (18.79 % ~ 26.85 %). Such phenomenon indicated that prototype CPT could successfully cleaved and released by intracellular thiols for cytotoxicity as we designed. The S phase delay percentage in each groups shared the consistant trend with previous MTT study and cell apoptosis assay.
2.7. Enhanced TNBC accumulation and reduced systemic toxicity in vivo
To further investigate NPs stability and anti-leaking signature confronting physiological conditions, we studied the NPs pharmacokinetics in healthy SD rats. Pharmacokinetics profiles of free CPT, CPTD NPs and NT-CPTD NPs were shown in Figure 6. Compared with free CPT, whose blood concentration decreased rapidly, CPTD NPs and NT-CPTD NPs showed enhanced systemic circulation time, which could still be detected after 48 h administration. There exhibited a significant differences of pharmacokinetic parameters between free CPT and NPs, where the area under the plasma-concentration curves (AUC) of CPTD NPs and NT-CPTD NPs were 5.7 fold and 7.6 fold higher than that of free CPT respectively. Corresponding mean residence time (MRT) of NPs were around 5.1 fold longer than that of free CPT, while clearance (CL) of both NPs were much shorter than free CPT. By conjugating CPT into dimer structured NPs, the circulation of CPT was prolonged and higher amount of CPT was detained in plasma, providing higher chance of drug accumulation in tumor tissues. Therefore, dimer structured NPs could provide a novel stable platform to improve the pharmacokinetic and pharmacodynamics profile of anti-cancer drugs.
Figure 6.
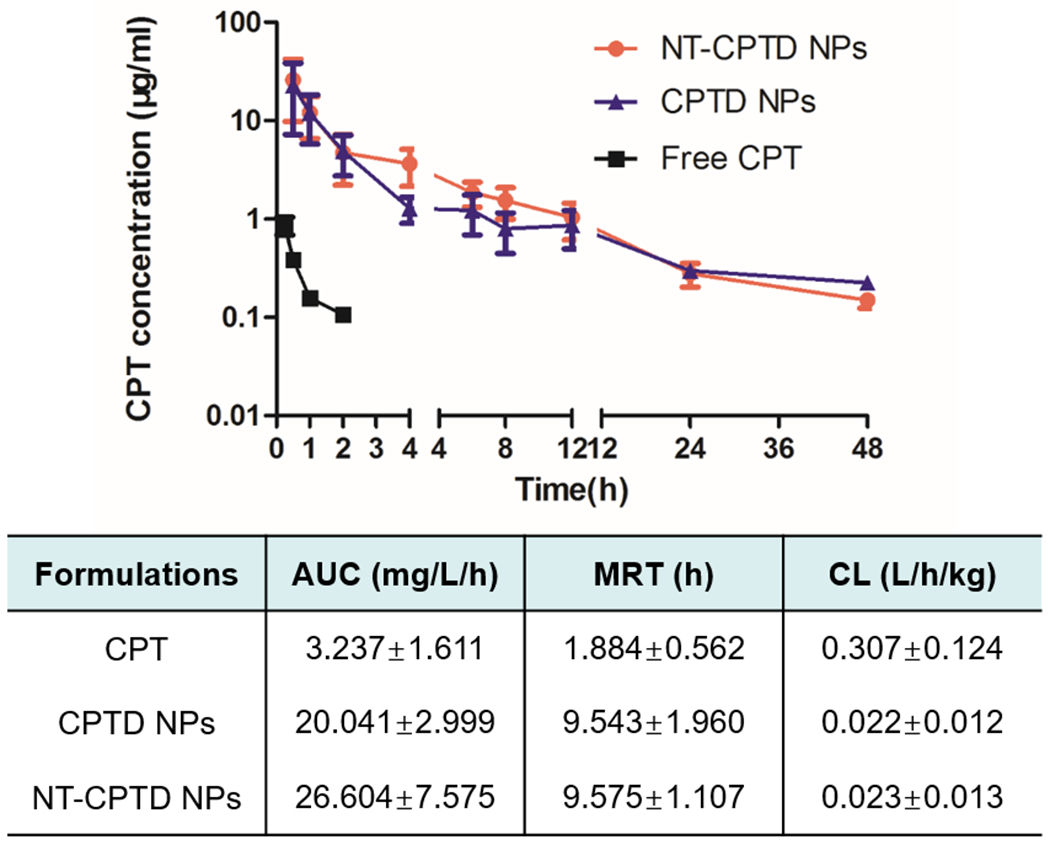
Pharmacokinetic profiles of CPTD NPs, NT-CPTD NPs and Free CPT in SD rats after i.v. injection of at a normalized CPT dose of 1 mg/kg. Data were presented as mean ± SD (n = 5).
To further evaluate the TNBC accumulation of NPs, we intravenously injected BODIPY loaded CPTD NPs and NT-CPTD NPs in orthotopic MDA-MB-231 tumor-bearing mice and tracked the biodistribution of NPs. As shown in Figure 7B, both NPs showed accumulation in TNBC due to the EPR effect 31. However, compared with CPTD NPs, NT-CPTD NPs exhibited much higher targeting efficacy in tumor area from 8 h to 24 h (Figure 7B). From the 3D image of NT-CPTD NPs treated mouse, a portion of NT-CPTD NPs were localized in the central region of tumor (Figure 7A). After 24 h administration, tumors and major organs of treated mice were excised for ex vivo imaging and tissue distribution quantification (Figure 7C, D). NT-CPTD NPs showed approximately 3-fold accumulation in tumor tissue compared to CPTD NPs. Moreover, NT-CPTD NPs exhibited similar accumulation in heart, spleen and kidney and slightly lower accumulation in liver and lung (Figure 7C). These results indicated that NT-CPTD NPs could maintain higher tumor accumulation in vivo while reducing tis accumulation in major organs.
Figure 7.
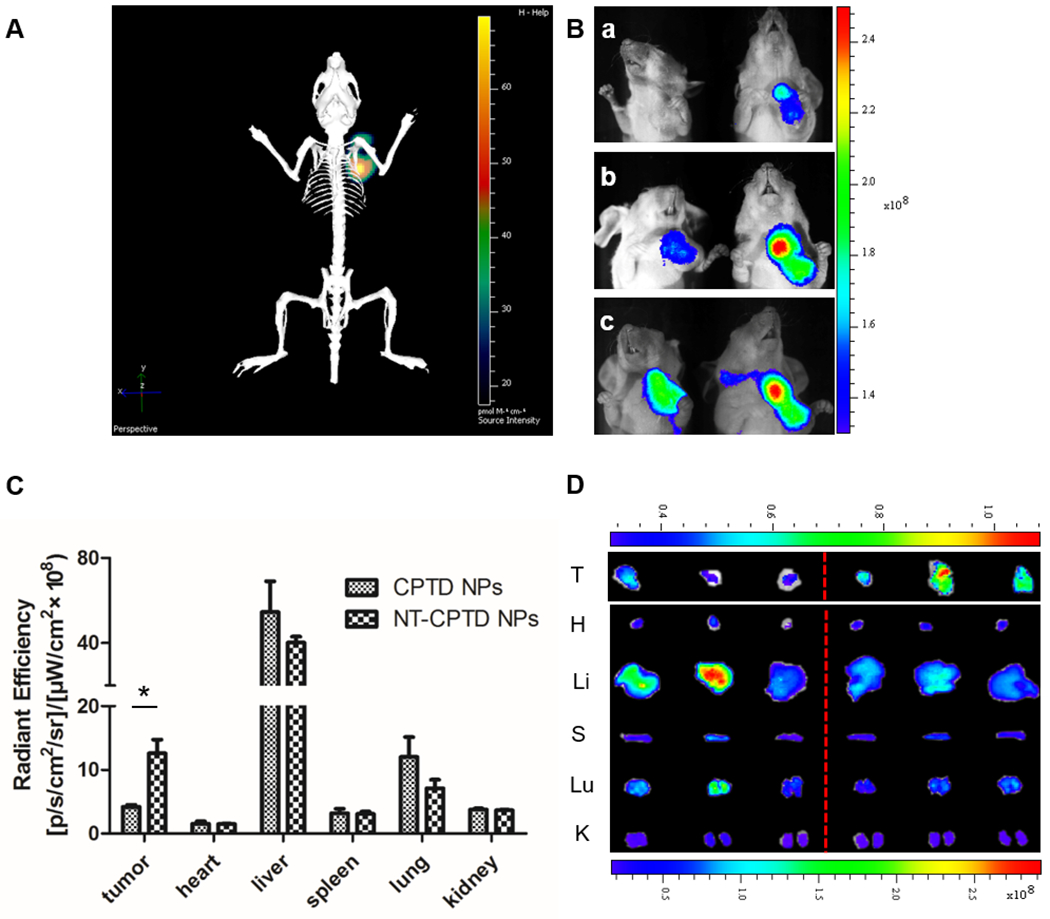
Biodistribution of CPTD NPs and NT-CPTD NPs. (A) In vivo 3D imaging of i.v. injection of NT-CPTD NPs after 24 h. (B) In vivo 2D imaging of i.v. injection of CPTD NPs (left) and NT-CPTD NPs (right) after 8 h (a), 12 h (b), 24 h (c). (C) Quantitative fluorescence intensities of tumors and organs after 24 h i.v. injection. Data were presented as mean ± SD (n=3). *P<0.05 (D) Ex vivo images of tumors and organs after 24 h i.v. injection of CPTD NPs (left) and NT-CPTD NPs (right).
As shown in ex vivo images (Figure 7C, D), there also exhibited a high liver accumulation of both CPTD NPs and NT-CPTD NPs. Such phenomenon is mainly caused by RES phagocytic cells, which could capture certain scale of NPs especially NPs with size over 100 nm35. To further confirm the biosafety of NPs, H&E staining of the major organs were investigated during the in vivo antitumor studies. As shown in Figure 8, CPTD NPs or NT-CPTD NPs didn’t induce severe toxicity to liver, whereas obvious inflammatory symptoms in the portal area of liver were observed in prototype CPT treated mice. Consequently, NT-CPTD NPs could effectively improve drug delivery efficacy in carcinoma tissues while reduce systemic toxicity.
Figure 8.
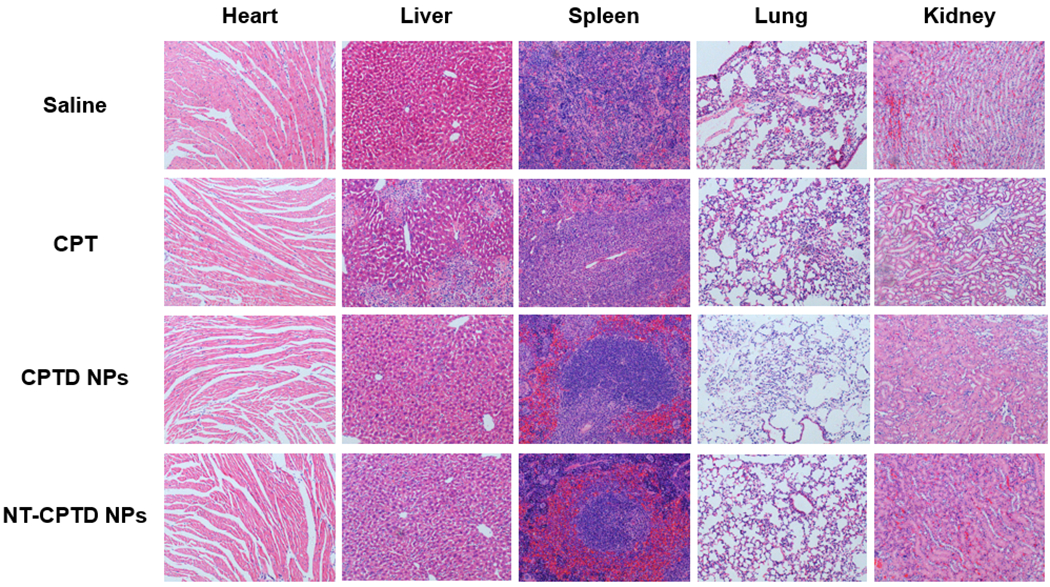
Pathological examination of organs resected from MDA-MB-231 tumor-bearing mice treated with different CPT formulations. Saline group was served as control. Nuclei were stained by hematoxylin (blue), and extracellular matrix and cytoplasm were doped by eosin (red).
2.8. In vivo antitumor efficacy
Antitumor efficacy of NPs were evaluated in TNBC orthotopic tumor model. Saline, free CPT, CPTD NPs and NT-CPTD NPs were i.v. injected on day 0, 4 and 8. As shown in Figure 9A, tumor growth was inhibited in prototype CPT, CPTD NPs and NT-CPTD NPs, comparing to control saline group, while NT-CPTD NPs showed most effective antitumor behavior. Limited tumor inhibition observed in prototype CPT, CPTD NPs groups were mainly because of the rapid clearance of CPT or lacking of TNBC targeting ligand. Body weight of TNBC orthotopic mice was recorded to evaluate general biosafety between different groups (Figure 9B), which showed no significant difference.
Figure 9.
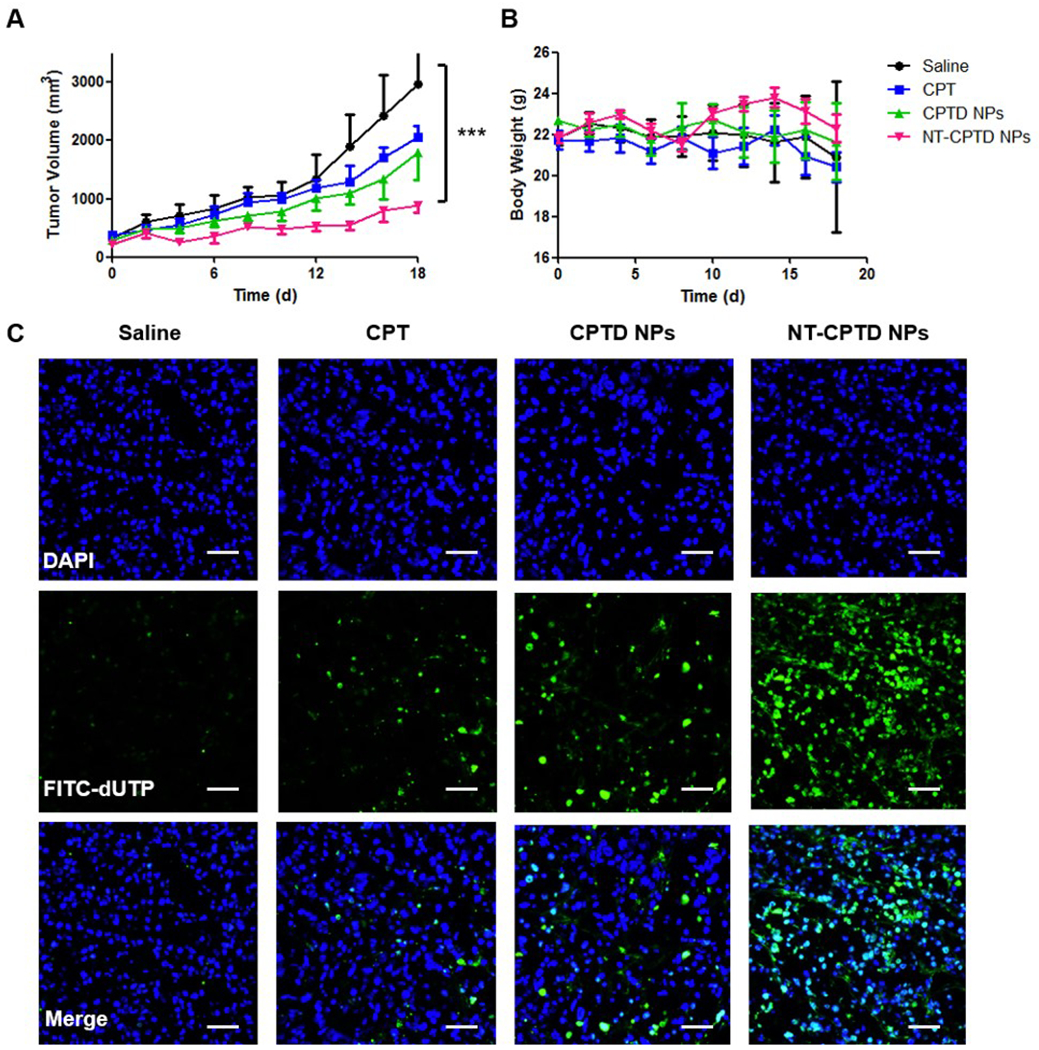
(A) Tumor volume changes and (B) Body weight changes of MDA-MB-231 tumor-bearing mice after i.v. injection three times with different CPT formulations at a normalized CPT dose of 10 mg/kg on day 0, 4 and 7. Saline group was served as control. Data were presented as mean ± SD (n=6). ***P<0.001. (C) TUNEL assay of MDA-MB-231 tumor xenografts of treated groups. Blue, DAPI; Green, apoptosis cells. Scale bars represent 100 μm.
After three times treatment, TNBC orthotopic mice were sacrificed. Excised tumors were sliced and stained with TUNEL to evaluate the apoptosis (Figure 9C). Compared with free CPT group, CPTD NPs treated group showed higher antitumor efficacy, which possibly due to the NPs accumulation caused by long circulation of drug formulation and EPR effect. NT-CPTD NPs treated group showed most apoptotic cells, revealing the most effective antitumor efficacy. Such results shared conformance with in vivo tumor growth inhibition and also in vitro MTT and apoptosis results, indicating the excellent antitumor efficacy of NT-CPTD NPs.
3. CONCLUSION
The key issue in cancer therapy lies in how to enhance the tumor specific drug delivery efficiency and decreasing side effect simultaneously. Although nanomedicines have been shown to greatly improve the therapeutic outcome of chemodrugs, the formulation challenge prohibit their further development and clinical translation. Our dimeric prodrug self-delivery nanoparticles NT-CPTD NPs exhibited extremely high drug loading, which could significantly reduce the bio-burden of polymer during drug administration. The prodrug-based amphiphilic copolymer design could relieve the concern of reproducibility of nanomedicines. The disulfide bond in NPs could achieve the tumor microenvironment redox responsive drug release, which effectively prevented pre-mature drug leakage during circulation. TNBC targeting capacity of NT-CPTD NPs successfully improved chemodrug accumulation, exhibited enhanced in vitro and in vivo anti-tumor efficacy. Therefore, NT-CPTD NPs was promising for TNBC therapy and such dimeric prodrug self-delivery platform could be widely applied for other drug delivery.
4. MATERIALS AND METHODS
4.1. Chemicals and Materials
2,6-bis(hydroxymethyl)aniline (BHA) was synthesized according to literature report 16. Anhydrous dimethylformamide (DMF), anhydrous tetrahydrofuran (THF), camptothecin (CPT), mPEG (Mw 5000), 3-chloro-1-propanol, sodium azide, tert-butyldimethylchlorosilane (TBSCl), 2-hydroxyethyl disulfide, N,N-dimethylamino pyridine, dithiothreitol(DTT), copper(I) iodide, propargyl bromide, triphosgene and MTT (3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide) were purchased from Sigma-Aldrich. Phosphate buffered saline (PBS) was purchased from Mediatech, Inc.
4.2. Cell lines
MDA-MB-231-luci cells were purchased from the American Type Culture Collection (Rockville, MD, USA). MDA-MB-231 cells and 293 cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences. All the cells were cultured according to previous report 17.
4.3. Synthesis of redox-responsive CPT dimeric prodrug (CPTD) and outer-shell component CPTD-PEG5000
The synthesis routes of CPTD, CPTD-PEG5000 and NT-PEG5000-CPTD were generally described in Scheme S1. Dimeric prodrug CPTD was synthesized according to literature 2,16.
4.3.1. Synthesis of alkyne-PEG5000
NaH (7.2 mg, 0.3 mmol, 3 equiv) was added slowly to an ice bath cooled THF solution of mPEG5000 (500mg, 0.1 mmol) and then warmed to room temperature gradually. After 1 h, 3-bromo-1-propyne (35.7 mg, 0.3 mmol, 3 equiv) was added and stirred overnight. The reaction was quenched by MeOH and concentrated via evaporation. The reaction solution was poured into diethylether and the crude white precipitate was isolated and purified by neutral alumina column chromatography (EtOAc: MeOH 3:1) and characterized by 1H-NMR.
4.3.2. Synthesis of CPTD-PEG5000
To 2 ml DMF solution of CPTD (1.2 mg, 0.001 mmol) and alkyne-PEG5000(5 mg, 0.001 mmol, 1 equiv), CuBr (2 mg) and PMDTA (2 ul) were added and stirred for 24 h. The product was purified by dialysis against 2,2′,2″,2‴-(Ethane-1,2-diyldinitrilo)tetraacetic acid and deionized water for 2 days. White powder of CPTD-PEG5000 was acquired through lyophilization and characterized by 1H-NMR.
4.3.3. Synthesis of NT-PEG5000-CPTD
NT peptide was reacted with MAL-PEG5000-alkyne (2:1 mol/mol) in PBS (pH 7.0) for 24 h at room temperature. NT-PEG5000-alkyne was purified by dialysis using 5 kDa molecular weight cutoff membrane, lyophilized and characterized by 1H-NMR.
To 2 ml DMF solution of CPTD (1.2 mg, 0.001 mmol) and NT-PEG5000-alkyne (5 mg, 0.001 mmol, 1 equiv), CuBr (2 mg) and PMDTA (2 ul) were added and stirred for 24 h. The product was purified by dialysis against 2,2′,2″,2‴-(Ethane-1,2-diyldinitrilo)tetraacetic acid and deionized water for 2 days. White powder of NT-PEG5000-CPTD was acquired through lyophilization and characterized by 1H-NMR.
4.4. Preparation of CPTD NPs and NT-CPTD NPs
CPTD NPs were prepared by nanoprecipitation method. Briefly, CPTD, CPTD-PEG5000 and NT-PEG5000-CPTD were dissolved in DMF with CPTD-PEG5000 or NT-PEG5000-CPTD concentration of 10 mg/ml if not specified. CPTD and CPTD-PEG5000 were mixed at certain ratio, then added dropwise into 2.0 ml of DI water with mild stirring using a magnetic bar. For NT-CPTD NPs, certain weight ratio of NT-PEG5000-CPTD was blended with CPTD-PEG5000 solution followed by the same preparing procedure as above.
CPTD FRET NPs were prepared by mixing CPTD and CPTD-PEG5000 at weight ratio of 2:1 containing 0.75% DiI and 0.75% DiO in DMF and prepared by the same procedure as above.
To trace the intracellular distribution of NPs, coumarin-6 (0.5 % w/w) and CPTD were co-loaded using the same method described as above. The concentration of coumarin-6 was measured by HPLC with standard calibration curve. To trace the in vivo distribution of NPs, BODIPY (0.5 % w/w) and CPTD were co-loaded using the same procedure described as above. The concentration of BODIPY was measured by fluorescent absorption at 700/730 nm (ex/em) with standard calibration curve.
4.5. Characterization of CPTD NPs and NT-CPTD NPs
Particle size and zeta-potential of NPs was measured by a ZetaPlus dynamic light scattering (DLS) detector (Brookhaven Instruments, Holtsville, NY, USA) or dynamic light scattering (DLS) (Zetasizer Nano-ZS, Malvern, U.K.). TEM samples were prepared on 200 mesh carbon film supported copper grids. One drop of the nanoparticles solution was placed on the grid and allowed to stand overnight. The morphology of NPs was observed using Biology Transmission Electron Microscope (B-TEM, Tecnai G2 spirit Biotwin, FEI, USA).
4.6. Nanoparticle stability test
Freshly prepared NPs solution was diluted with PBS to test its stability in the presence of salt, and diluted with DI water 10, 100 and 500 fold to test its stability against dilution. For serum stability test, 1.0 ml of NPs solution was diluted with 1.0 ml FBS. The size and distribution of NPs were characterized by DLS. For different pH stability, 100 μL NPs solution was diluted with PBS 7.4, PBS 6.5 or 10 % FBS-containing PBS 7.4 buffer for 10-fold and tested the size and distribution of NPs by DLS.
4.7. Drug loading efficiency measurement
To characterize the encapsulation efficiency (EE) of NPs, freshly prepared CPTD NPs was dissolved in DMF and CPTD content was quantified via HPLC.
To characterize the loading efficiency (LE) of CPT in NPs, formulations were cleaved with PBS 7.4 containing 100 mM DTT for 2 h. The amount of CPT was measured via HPLC. The weight of NPs were measured via lyophilization. The EE and LE were calculated according to the following equations:
4.8. In vitro CPT release from NPs
Freshly prepared CPTD NPs and NT-CPTD NPs were diluted with PBS to approximately 2 μg/ml. Then 10 mM or 20 μM DTT were added into NPs solution and incubated at 37 ℃ respectively. At specified time points, 500 μL solution was collected, centriguged at 15 krpm to remove particles and the supernatant was subject to HPLC analysis after 1:1 dilution with 0.1% TFA-H2O to quantify the drug release content (λex = 369 nm, λem = 441 nm).
4.9. In vitro NPs stability test via FRET confocal microscopy
MDA-MB-231 cells were seeded in 4-well confocal chamber (Corning, USA) at a density of 5000 cells/well and incubated at 37°C for 24 h. Cells were incubated with 0.2 mg/ml of CPTD FRET NPs for 1 h. Then cells were washed with PBS for 3 times, fixed with 4% formaldehyde solution for 30 min and stained the nucleus with Hoechst for 15 min. The images were obtained using Multiphoton Confocal Microscope (LSM 710, ZEISS, German) with a 63×oil objective. Images were recorded in the DiO channel (488 nm excitation, 535 ± 20 nm emission), DiI FRET channel (488 nm excitation, 585 ± 20 nm emission) and Hoechst channel (346 nm excitation, 460 ± 20 nm emission). Fluorescence spectra of CPTD FRET NPs inside of cells at pixels of interest were measured by using a spectral detector, with 484 nm excitation and emission scan from 490 to 690 nm.
4.10. Cellular uptake and internalization mechanism study
MDA-MB-231 cells were seeded in 6-well plates (Corning, USA) at a density of 3×104 cells/well and incubated at 37°C for 24h before reaching a confluence of 80% - 90%. Cells were incubated with CPTD NPs and NT-CPTD NPs with different NT modified ratios (10, 20, 40 wt %) at normalized coumarin-6 concentration of 0.5 μg/mL. Cells were washed and observed via fluorescent microscope (Leica, Wetzlar, Germany) after 30 min incubation. For flow cytometry analysis, cells were collected and analyzed using flow cytometer (BD biosciences, Bedford, MA, USA). Cells without any treatment were used as control.
For internalization mechanism studies, cells were pre-incubated with various inhibitor solutions including 50 μM NT peptide as NTSR1 receptor inhibitor, 1 μg/mL filipin, 0.3 μg/mL phenylarine oxide (PhAsO), 1 μg/mL colchicine respectively 17. After 15 min pre-incubation, coumarin-6 loaded NT-CPTD NPs were added and incubated for 30 min. After 30 min incubation, cells were washed and visualized under fluorescent microscope (Leica, Wetzlar, Germany).
4.11. In vitro antitumor efficacy study
Standard MTT protocol was followed to evaluate the cytotoxicity of NPs. Briefly, MDA-MB-231 cells were seeded in 96-well plate at a density of 3000 cells/well. When 60-70 % confluence was achieved, cells were incubated with free CPT, commercially available drug Irinotecan, CPTD NPs and NT-CPTD NPs at various concentrations at 37 °C for 72 h in DMEM. Afterwards, cell medium was removed and cell viability was tested according to previous MTT protocols 17. Cells incubated in DMEM were served as control.
As for cell apoptosis assay, MDA-MB-231 cells were seeded in 24-well plate at a density of 1×104 cells/well and incubated at 37°C for 24h before reaching a confluence of 70% - 80%. Cells were treated with free CPT, Irinotecan, CPTD NPs and NT-CPTD NPs at normalized CPT concentration of 10 μM for 12 h. Afterwards, cells were treated according to Annexin V-FITC Apoptosis Detection Kit (KeyGEN BioTECH, Nanjing, China) protocols and visualized.
4.12. Cell cycle determination
MDA-MB-231 cells were treated with free CPT, commercially available drug Irinotecan, CPTD NPs and NT-CPTD NPs at normalized CPT concentration of 10 μM for 1 h. Drug solutions were then replaced by cell culture medium and incubated for another 12 h. Afterwards, cells were collected and detected according to Cell Cycle Detection Kit (KeyGEN BioTECH, Nanjing, China) protocols. The percentage of cell cycle phases was analyzed with Flowjo 6.0.
4.13. Pharmacokinetics of NPs
SD rats were intravenously injected with free CPT, CPTD NPs and NT-CPTD NPs at a normalized CPT dose of 1 mg/kg (n=5). Free CPT solution preparation, blood sample treatment and methodology establishment were determined according to previously reported method with slightly modification32. Briefly, 500 μL blood samples of each group were collected from the tail vein into heparinized-eppendorf tubes at certain time points. After 3000 rpm centrifugation, 200 μL of plasma was collected. Then the biological samples were treated with 25 μL DMSO and 25 μL PBS 7.4 containing 300 mM DTT at 37 °C for 4 h, then added with 50 μL 0.2 N HCl and 200 μL MeOH. The mixture was vortex for 1 min and centrifuged (12,000 rpm, 10 min) to collect the supernatant. 20 μL of supernatant was injected into HPLC (Agilent ODS C18 column (4.6 × 250 mm, 5 μm particle size), gradient method (0.1% TFA-H2O/Acetonitrile, Figure S9), 1.0 mL/min, 25°C, Ex/Em = 369 nm/441 nm for CPT quantification.
4.14. Tumor implantation
All animal experiments were carried out in accordance with guidelines evaluated and approved by the ethics committee of Fudan University, Shanghai, China. The xenograft tumor model was established by orthotopic injection of 1×106 cells in 100 μL serum-free DMEM medium containing 5 mg/mL Matrigel into the right flank of the mice under the fat pad.
4.15. Nanoparticle distribution in TNBC orthotopic model
Near infrared fluorescence (NIR) optical imaging technology was utilized to monitor the in vivo 2D and 3D distribution. Briefly, TNBC orthotopic mice were i.v. injected with BODIPY-loaded NPs at normalized BODIPY dose of 0.1 mg/kg. At time point of 8, 12 and 24 h, mice were anaesthetized and monitored using IVIS Spectrum with Living Image software v 4.2 (Caliper Life Science). After 24 h in vivo imaging, mice were anesthetized and sacrificed. Tumors and organs were dissected and imaged.
4.16. In vivo anti-tumor efficacy study
TNBC orthotopic mice were divided into four groups (n=6) and intravenously injected with saline, free CPT, CPTD NPs and NT-CPTD NPs on day 0, 4, 7 at a normalized CPT dose of 10 mg/kg. Free CPT solution was prepared according to previous reference33. Body weight of TNBC orthotopic mice was recorded every other day. Tumor volume was calculated as a × b2/2, where a was the largest and b the smallest diameter.
TNBC orthotopic mice were sacrificed on day 18 and major organs (heart, liver, spleen, lung, kidney and tumor) were excised and fixed in 4 % neutral buffered formalin solution. H&E study staining and TUNEL assay were performed according to previous report 17.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the grants from National Science Fund for Distinguished Young Scholars (81425023) and Program of Shanghai Academic Research Leader (18XD1400500). J.C. acknowledges the support of NIH 1R01CA207584.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
Synthesis route of CPTD, Synthesis route of CPTD-PEG5000 and NT-PEG5000-CPTD, Proposed release mechanism of redox-responsive drug release, Particle size change of CPTD NPs with PBS or FBS, Particle size of CPTD NPs diluted with DI water, In vitro CPT release from NT-CPTD NPs in different pH, Cellular uptake of NT-CPTD in NTSR1-positive cells and NTSR1-negative cells, Representative cell cycle distribution of NPs, HPLC method for CPT analysis and standard curve.
The authors declare no competing financial interest.
REFERENCES
- [1].Jin G, He R, Liu Q, Dong Y, Lin M, Li W, Xu F. Theranostics of Triple-Negative Breast Cancer Based on Conjugated Polymer Nanoparticles[J]. ACS Appl. Mater. Interfaces, 2018, 10(13):10634–10646. [DOI] [PubMed] [Google Scholar]
- [2].Zhang F, Ni Q, Jacobson O, Cheng S, Liao A, Wang Z, He Z, Yu G, Song J, Ma Y, Niu G, Zhang L, Zhu G, Chen X. Polymeric Nanoparticles with Glutathione-Sensitive Heterodimeric Multifunctional Prodrug for In Vivo Drug Monitoring and Synergistic Cancer Therapy.[J]. Angew. Chem, 2018, 57(24):7066–7070. [DOI] [PubMed] [Google Scholar]
- [3].Araki T, Ogawara K I, Suzuki H, Kawai R, Watanabe T, Ono T, Higaki K. Augmented EPR effect by photo-triggered tumor vascular treatment improved therapeutic efficacy of liposomal paclitaxel in mice bearing tumors with low permeable vasculature[J]. J. Controlled Release, 2015, 200:106–114. [DOI] [PubMed] [Google Scholar]
- [4].Shi Y, Van d M R, Theek B, Oude B E, Pieters EH, Fens MH, Ehling J, Schiffelers RM, Storm G, Van N CF, Lammers T, Hennink WE. Complete Regression of Xenograft Tumors upon Targeted Delivery of Paclitaxel via π-π Stacking Stabilized Polymeric Micelles[J]. ACS Nano, 2014, 9(4):3740–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liu S, Ono RJ, Yang C, Gao S, Ming T JY, Hedrick JL, Yang YY. Dual pH-responsive Shell-cleavable Polycarbonate Micellar Nanoparticles for In Vivo Anticancer Drug Delivery[J]. ACS Appl. Mater. Interfaces, 2018, 10(23):19355–19364. [DOI] [PubMed] [Google Scholar]
- [6].Tang L, Yang X, Yin Q, Cai K, Wang H, Chaudhury I, Yao C, Zhou Q, Kwon M, Hartman JA, Dobrucki IT, Dobrucki LW, Borst LB, Lezmi S, Helferich WG, Ferguson AL, Fan TM, Cheng J. Investigating the optimal size of anticancer nanomedicine.[J]. Proc. Natl. Acad. Sci. U. S. A, 2014, 111(43):15344–15349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wagner E Programmed drug delivery: nanosystems for tumor targeting[J]. Expert Opin. Biol. Ther, 2007, 7(5):587. [DOI] [PubMed] [Google Scholar]
- [8].Liu L, Bi Y, Zhou M, Chen X, He X, Zhang Y, Sun T, Ruan C, Chen Q, Wang H, Jiang C. Biomimetic Human Serum Albumin Nanoparticle for Efficiently Targeting Therapy to Metastatic Breast Cancers[J]. ACS Appl. Mater. Interfaces, 2017, 9(8):7424–7435. [DOI] [PubMed] [Google Scholar]
- [9].Yao Q, Dai Z, Hoon C J, Kim D, Zhu L. Building stable MMP2-responsive multifunctional polymeric micelles by an all-in-one polymer-lipid conjugate for tumor-targeted intracellular drug delivery[J]. ACS Appl. Mater. Interfaces, 2017, 9(38):32520–32533. [DOI] [PubMed] [Google Scholar]
- [10].Maiti C, Parida S, Kayal S, Maiti S, Mandal M, Dhara D. Redox-Responsive Core Cross-Linked Block Copolymer Micelles for Overcoming Multidrug Resistance in Cancer Cells.[J]. ACS Appl. Mater. Interfaces, 2018, 10(6):5318–5330. [DOI] [PubMed] [Google Scholar]
- [11].Shao K, Ding N, Huang S, Ren S, Zhang Y, Kuang Y, Guo Y, Ma H, An S, Li Y, Jiang C. Smart nanodevice combined tumor-specific vector with cellular microenvironment-triggered property for highly effective antiglioma therapy.[J]. ACS Nano, 2014, 8(2):1191. [DOI] [PubMed] [Google Scholar]
- [12].Yin Q, Tang L, Cai K, Tong R, Sternberg R, Yang X, Dobrucki LW, Borst LB, Kamstock D, Song Z, Helferich WG, Cheng J, Fan TM. Pamidronate functionalized nanoconjugates for targeted therapy of focal skeletal malignant osteolysis.[J]. Proc. Natl. Acad. Sci. U. S. A, 2016, 113(32):E4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gottesman Michael M., Fojo Tito, Bates Susan E.. Multidrug resistance in cancer: role of ATP–dependent transporters[J]. Nat. Rev. Cancer, 2002, 2(1):48–58. [DOI] [PubMed] [Google Scholar]
- [14].Gillet JeanPierre, Gottesman Michael M.. Mechanisms of Multidrug Resistance in Cancer[J]. Methods Mol. Biol, 2010, 596:47–76.. [DOI] [PubMed] [Google Scholar]
- [15].Kunjachan S, Rychlik B, Storm G, Kiessling F, Lammers T. Multidrug resistance: Physiological principles and nanomedical solutions.[J]. Adv. Drug Delivery Rev, 2013, 65(13-14):1852–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cai K, He X, Song Z, Yin Q, Zhang Y, Uckun FM, Jiang C, Cheng J. Dimeric drug polymeric nanoparticles with exceptionally high drug loading and quantitative loading efficiency[J]. J. Am. Chem. Soc, 2015, 137(10):3458–3461. [DOI] [PubMed] [Google Scholar]
- [17].He X, Zhang J, Li C, Zhang Yu, Lu Y, Zhang Y, Liu L, Ruan C, Chen Q, Chen X, Guo Q, Sun T, Cheng J, Jiang C. Enhanced bioreduction-responsive diselenide-based dimeric prodrug nanoparticles for triple negative breast cancer therapy[J]. Theranostics, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Arunachalam B, Phan UT, Geuze HJ, Cresswell P. Enzymatic reduction of disulfide bonds in lysosomes: Characterization of a Gamma-interferon-inducible lysosomal thiol reductase (GILT)[J]. Proc. Natl. Acad. Sci. U. S. A, 2000, 97(2):745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Somaï S, Gompel A, Rostène W, Forgez P. Neurotensin counteracts apoptosis in breast cancer cells[J]. Biochem. Biophys. Res. Commun, 2002, 295(2):482–488. [DOI] [PubMed] [Google Scholar]
- [20].Wu Z, Martinez FD, Tredaniel J, Forgez P. Neurotensin and its high affinity receptor 1 as a potential pharmacological target in cancer therapy[J]. Front. Endocrinol, 2012, 3:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Castillo-Rodríguez RA, Arango-Rodríguez ML, Escobedo L, Hernandez-Baltazar D, Gompel A, Forgez P, Martinez FD. Suicide HSVtk gene delivery by neurotensin-polyplex nanoparticles via the bloodstream and GCV Treatment specifically inhibit the growth of human MDA-MB-231 triple negative breast cancer tumors xenografted in athymic mice[J]. Plos One, 2014, 9(5):e97151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I[J]. J. Biol. Chem, 1985, 260(27):14873–14878. [PubMed] [Google Scholar]
- [23].Lu J, Liu C, Wang P, Ghazwani M, Xu J, Huang Y, Ma X, Zhang P, Li S. The Self-Assembling Camptothecin-Tocopherol Prodrug: An Effective Approach for Formulating Camptothecin[J]. Biomaterials, 2015, 62:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang F, Zhu G, Jacobson O, Liu Y, Chen K, Yu G, Ni Q, Fan J, Yang Z, Xu F, Fu X, Wang Z, Ma Y, Niu G, Zhao X, Chen X. Transformative Nanomedicine of an Amphiphilic Camptothecin Prodrug for Long Circulation and High Tumor Uptake in Cancer Therapy[J]. ACS Nano, 2017, 11(9):8838. [DOI] [PubMed] [Google Scholar]
- [25].Liu YS, Cheng RY, Lo YL, Hsu C, Chen SH, Chiu CC, Wang LF. Distinct CPT-induced deaths in lung cancer cells caused by clathrin-mediated internalization of CP micelles.[J]. Nanoscale, 2016, 8(6):3510–3522. [DOI] [PubMed] [Google Scholar]
- [26].Dai L, Yu Y, Luo Z, Li M, Chen W, Shen X, Chen F, Sun Q, Zhang Q, Gu H, Cai K. Photosensitizer enhanced disassembly of amphiphilic micelle for ROS-response targeted tumor therapy in vivo.[J]. Biomaterials, 2016, 104:1–17. [DOI] [PubMed] [Google Scholar]
- [27].Li J, Jiang X, Guo Y, An S, Kuang Y, Ma H, He X, Jiang C. Linear-Dendritic Copolymer Composed of Polyethylene Glycol and All-trans-Retinoic Acid as Drug Delivery Platform for Paclitaxel against Breast Cancer.[J]. Bioconjug. Chem, 2015, 26(3):418–426. [DOI] [PubMed] [Google Scholar]
- [28].Chen H, Kim S, Li L, Wang S, Park K, Cheng JX. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Forster resonance energy transfer imaging.[J]. Proc. Natl. Acad. Sci. U. S. A, 2008, 105(18):6596–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee S Y, Tyler J Y, Kim S, Park K, Cheng JX. FRET imaging reveals different cellular entry routes of self-assembled and disulfide bonded polymeric micelles[J]. Mol. Pharmaceutics, 2013, 10(9):3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zuco V, Cesare M D, Zaffaroni N, Lanzi C, Cassinelli G. PLK1 is a critical determinant of tumor cell sensitivity to CPT11 and its inhibition enhances the drug antitumor efficacy in squamous cell carcinoma models sensitive and resistant to camptothecins[J]. Oncotarget, 2015, 6(11):8736–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu Y, He X, Kuang Y, An S, Wang C, Guo Y, Ma H, Lou J, Jiang C. A Bacteria Deriving Peptide Modified Dendrigraft Poly-l-lysines (DGL) Self-Assembling Nanoplatform for Targeted Gene Delivery[J]. Mol. Pharmaceutics, 2014, 11(10):3330–3341. [DOI] [PubMed] [Google Scholar]
- [32].Tang G, Zhu S, Hong M, Jiang Y, Pei Y. Study on tumor active targeting with Transferrin-PEG-PAMAM dendrimer-CPT conjugate[J]. Anti-tumor Pharmacy, 2012. [Google Scholar]
- [33].Schluep T, Cheng J, Khin KT, Davis ME. Pharmacokinetics and biodistribution of the camptothecin-polymer conjugate IT-101 in rats and tumor-bearing mice [J]. Cancer Chemother. Pharmacol, 2006, 57(5):654–662. [DOI] [PubMed] [Google Scholar]
- [34].He X, Chen X, Liu L, Zhang Y, Lu Y, Zhang Y, Chen Q, Ruan C, Guo Q, Li C, Sun T, Jiang C. Sequentially triggered nanoparticles with tumor penetration and intelligent drug release for pancreatic cancer therapy [J]. Adv. Sci, 2018, 5(5):1701070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang Y, Guo Q, An S, Lu Y, Li J, He X, Liu L, Zhang Y, Sun T, Jiang C. ROS-switchable polymeric nanoplatform with stimuli-responsive release for active targeted drug delivery to breast cancer [J]. ACS Appl. Mater. Interfaces, 2017, 9(14):12227. [DOI] [PubMed] [Google Scholar]
- [36].Cai K, Yen J, Yin Q, Liu Y, Song Z, Lezmi S, Zhang Y, Yang X, Helferich WG, Cheng J. Redox-Responsive Self-Assembled Chain-Shattering Polymeric Therapeutics.[J]. Biomater. Sci, 2015, 3(7):1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.