

Histone deacetylase 6 (HDAC6) is an emerging target for the treatment of various diseases such as cancer through selective inhibition. While the second catalytic domain (HDAC6 CD2) has been extensively studied, comparatively little research has focused on the first catalytic domain (HDAC6 CD1). This work provides new structural insights regarding inhibitor binding to HDAC6 CD1 enabled by the study of active-site mutants.
Keywords: zinc enzymes, hydrolases, enzyme inhibitors, drug design
Abstract
The zinc hydrolase histone deacetylase 6 (HDAC6) is unique among vertebrate deacetylases in that it contains two catalytic domains, designated CD1 and CD2. Both domains are fully functional as lysine deacetylases in vitro. However, the in vivo function of only the CD2 domain is well defined, whereas that of the CD1 domain is more enigmatic. Three X-ray crystal structures of HDAC6 CD1–inhibitor complexes are now reported to broaden the understanding of affinity determinants in the active site. Notably, cocrystallization with inhibitors was facilitated by using active-site mutants of zebrafish HDAC6 CD1. The first mutant studied, H82F/F202Y HDAC6 CD1, was designed to mimic the active site of human HDAC6 CD1. The structure of its complex with trichostatin A was generally identical to that with the wild-type zebrafish enzyme. The second mutant studied, K330L HDAC6 CD1, was prepared to mimic the active site of HDAC6 CD2. It has previously been demonstrated that this substitution does not perturb inhibitor binding conformations in HDAC6 CD1; here, this mutant facilitated cocrystallization with derivatives of the cancer chemotherapy drug suberoylanilide hydroxamic acid (SAHA). These crystal structures allow the mapping of inhibitor-binding regions in the outer active-site cleft, where one HDAC isozyme typically differs from another. It is expected that these structures will help to guide the structure-based design of inhibitors with selectivity against HDAC6 CD1, which in turn will enable new chemical biology approaches to probe its cellular function.
1. Introduction
The biological functions of many proteins are regulated by enzyme-catalyzed lysine acetylation–deacetylation cycles (Choudhary et al., 2009 ▸; Verdin & Ott, 2015 ▸), and histone deacetylases play a prominent role in this regulatory strategy (López et al., 2016 ▸; Porter & Christianson, 2019 ▸). These enzymes are named for their ability to catalyze the deacetylation of nuclear histone proteins, the acetylation of which was first discovered nearly 60 years ago (Allfrey et al., 1964 ▸). However, this enzyme nomenclature is somewhat misleading, in that the substrates of histone deacetylases include not only histone proteins but also thousands of other proteins in various subcellular locations (Hornbeck et al., 2015 ▸). For example, histone deacetylase 6 (HDAC6) operates predominantly in the cell cytosol (Verdel & Khochbin, 1999 ▸; Grozinger et al., 1999 ▸), where its substrates include α-tubulin (Hubbert et al., 2002 ▸; Zhang et al., 2003 ▸; Li & Yang, 2015 ▸) and tau (Min et al., 2010 ▸; Cohen et al., 2011 ▸; Noack et al., 2014 ▸; Carlomagno et al., 2017 ▸; Tseng et al., 2017 ▸). HDAC6-mediated deacetylation of α-tubulin is required for proper microtubule dynamics, and the deacetylation of tau influences phosphorylation and aggregation.
Human HDAC6 (UniProt Q9UBN7) is a 1215-residue zinc-dependent hydrolase containing two catalytic domains: CD1 and CD2 (Verdel & Khochbin, 1999 ▸; Grozinger et al., 1999 ▸; Zhang et al., 2006 ▸; Zou et al., 2006 ▸). The amino-acid side chains required for the chemistry of catalysis are strictly conserved between CD1 and CD2: a tyrosine side chain assists the zinc ion in polarizing the scissile carbonyl group of the substrate, and tandem histidine residues serve general base–general acid functions (Lombardi et al., 2011 ▸; López et al., 2016 ▸; Porter & Christianson, 2019 ▸). However, the substrate specificity of CD1 is much narrower than that of CD2 owing to amino-acid substitutions in the active site; moreover, the narrow substrate specificity of HDAC6 CD1 from Homo sapiens (human) is even more stringent than that of HDAC6 CD1 from Danio rerio (zebrafish) as measured using single-domain or di-domain enzyme constructs (Hai & Christianson, 2016 ▸; Osko & Christianson, 2019 ▸). Curiously, a recent study of full-length human HDAC6 suggests a wider substrate specificity for CD1 (Kutil et al., 2019 ▸), so further studies of both CD1 and CD2 are required in order to fully understand their structure–function relationships.
To enable structure-based inhibitor-design strategies, the X-ray crystal structures of human HDAC6 CD2 and zebrafish HDAC6 CD1 and CD2 have been reported, together with the structures of selected enzyme–inhibitor complexes (Hai & Christianson, 2016 ▸; Miyake et al., 2016 ▸). Human HDAC6 CD1 was refractory to crystallization; moreover, even though human HDAC6 CD2 yielded a crystal structure, the crystals were inferior to those of the zebrafish enzyme. Thus, X-ray crystallographic studies of HDAC6 CD1 and CD2 have relied on the zebrafish enzyme as a surrogate for the human enzyme. More than 50 structures of zebrafish HDAC6 CD2–inhibitor complexes have since appeared in the literature (Osko et al., 2020 ▸; Bhatia et al., 2018 ▸; Mackwitz et al., 2018 ▸; Porter et al., 2017 ▸; Porter, Osko et al., 2018 ▸; Porter, Shen et al., 2018 ▸; Porter, Wagner et al., 2018 ▸; Shen et al., 2020 ▸). In contrast, only seven additional crystal structures of zebrafish HDAC6 CD1–inhibitor complexes have since been reported (Osko & Christianson, 2019 ▸). These structures illuminate the active-site features that contribute to inhibitor affinity and selectivity. In light of the structural differences between human and zebrafish HDAC6 CD1, as well as the structural differences between HDAC6 CD1 and CD2, a deeper understanding of active-site binding determinants is needed to inform inhibitor-design strategies.
In order to broaden our understanding of inhibitor binding to HDAC6 CD1, we now report three new X-ray crystal structures of zebrafish HDAC6 CD1 mutants complexed with inhibitors containing hydroxamate zinc-binding groups (Fig. 1 ▸). Firstly, we report the crystal structure of H82F/F202Y HDAC6 CD1 complexed with the natural product inhibitor trichostatin A. The H82F and F202Y substitutions convert the active site of the zebrafish enzyme into a mimic of the active site of the human enzyme. Since the binding conformation of trichostatin A does not change significantly in complex with the mutant, the structure of this complex verifies that the zebrafish enzyme serves as a valid surrogate for the human enzyme. Finally, we report two crystal structures of K330L HDAC6 CD1 complexed with derivatives of suberoylanilide hydroxamic acid (SAHA), a drug currently in clinical use for cancer chemotherapy (Richon et al., 1996 ▸; Marks, 2007 ▸). These structures provide the first view of SAHA-based inhibitor binding in the active site of HDAC6 CD1, illuminating features of the protein landscape beyond the immediate active site that contribute to inhibitor binding.
Figure 1.

The inhibitors studied in complexes with the H82F/F202Y and K330L HDAC6 CD1 mutants.
2. Materials and methods
2.1. Reagents
All chemicals and buffers were purchased from Fisher, Millipore Sigma or Hampton Research and were used without further purification. The inhibitors trichostatin A, SAHA-BPyne and 4-iodo-SAHA were purchased from Caymen Chemicals and were used without further purification (each compound was listed as having >98% purity).
2.2. Preparation of D. rerio HDAC6 CD1 mutant plasmids
The HDAC6 gene from D. rerio (residues 60–798; UniProt F8W4B7) was synthesized by GenScript and the CD1 catalytic domain (residues 60–419) was recombinantly expressed in Escherichia coli using a pET-28a(+) vector. The gene sequence was optimized for E. coli expression. The genes for the H82F/F202Y HDAC6 CD1 and K330L HDAC6 CD1 mutants were similarly prepared by GenScript using PCR mutagenesis. Both of these constructs utilized an NdeI/BamHI cloning site and contained kanamycin bacterial resistance. The final constructs utilized for crystallization are summarized in Table 1 ▸.
Table 1. Macromolecule production.
| Source organism | D. rerio |
| DNA source | HDAC6 gene from D. rerio (UniProt F8W4B7; CD1 residues 61–419) |
| Cloning vector | pET-28a |
| Expression vector | pET-28a |
| Expression host | E. coli |
| Complete amino-acid sequence of the construct produced† | |
| K330L HDAC6 CD1 | MGSSHHHHHHSSGLVPRGSHM TGTGLVYVDAFTRFHCLWDASHPECPARVSTVMEMLETEGLLGRCVQVEARAVTEDELLLVHTKEYVELMKSTQNMTEEELKTLAEKYDSVYLHPGFFSSACLSVGSVLQLVDKVMTSQLRNGFSINRPPGHHAQADKMNGFCMFNNLAIAARYAQKRHRVQRVLIVDWDVHHGQGIQYIFEEDPSVLYFSVHRYEDGSFWPHLKESDSSSVGSGAGQGYNINLPWNKVGMESGDYITAFQQLLLPVAYEFQPQLVLVAAGFDAVIGDPLGGMQVSPECFSILTHMLKGVAQGRLVLALEGGYNLQSTAEGVCASMRSLLGDPCPHLPSSGAPCESALKSISKTISDLYPFWKSLQTFE |
| H82F/F202Y HDAC6 CD1 | MGSSHHHHHHSSGLVPRGSHM TGTGLVYVDAFTRFHCLWDASFPECPARVSTVMEMLETEGLLGRCVQVEARAVTEDELLLVHTKEYVELMKSTQNMTEEELKTLAEKYDSVYLHPGFFSSACLSVGSVLQLVDKVMTSQLRNGFSINRPPGHHAQADKMNGYCMFNNLAIAARYAQKRHRVQRVLIVDWDVHHGQGIQYIFEEDPSVLYFSVHRYEDGSFWPHLKESDSSSVGSGAGQGYNINLPWNKVGMESGDYITAFQQLLLPVAYEFQPQLVLVAAGFDAVIGDPKGGMQVSPECFSILTHMLKGVAQGRLVLALEGGYNLQSTAEGVCASMRSLLGDPCPHLPSSGAPCESALKSISKTISDLYPFWKSLQTFE |
The His-tag sequence is underlined. The CD1 sequence starts with Thr61 (bold). Mutated amino acids are shown in bold and underlined.
2.3. Expression and purification of D. rerio HDAC6 CD1 mutants
The expression and purification of D. rerio (zebrafish) HDAC6 CD1 was completed as recently described for the wild-type and mutant enzymes (Osko & Christianson, 2019 ▸). To briefly summarize, E. coli One Shot BL21(DE3) cells (Invitrogen) were used for protein expression and were grown in 2×YT medium with 50 µg ml−1 kanamycin. The cells were grown in an Innova 40 incubator shaker at 37°C and 250 rev min−1 until the OD600 reached approximately 0.80. The temperature was then decreased to 18°C until the OD600 reached 1.0. At an OD600 of 1.0, the cells were supplemented with 400 µM isopropyl β-d-1-thiogalactopyranoside (IPTG; Gold Biotechnology) and grown for an additional 18 h at 250 rev min−1. Finally, the cells were centrifuged using a Sorvall LYNX 6000 centrifuge at 5000 rev min−1 for 20 min. The cell pellets were stored at −80°C until further use.
Prior to purification, the cell pellet was thawed and resuspended in 100 ml buffer A [50 mM K2HPO4 pH 8.0, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 300 mM NaCl, 30 mM imidazole, 5% glycerol]. Two protease-inhibitor tablets, 0.1 mg ml−1 lysozyme and 50 µg ml−1 DNase were added to the solution. Sonification was used to lyse the cells and the cell lysate was centrifuged using a Sorval LYNX 6000 centrifuge for 1 h at 15 000 rev min−1. Once complete, the lysate was applied onto a 5 ml pre-equilibrated HisTrap HP column.
Buffer B (50 mM K2HPO4 pH 8.0, 1 mM TCEP, 300 mM NaCl, 300 mM imidazole, 5% glycerol) was used to elute the His-tagged HDAC6 CD1 protein bound to the HisTrap HP column. All CD1-containing fractions were concentrated to 5 ml using a 15 ml centrifugal filter unit with a molecular-weight cutoff of 10 kDa. The protein was then filtered using a 0.22 µm Millex-GV filter unit prior to being loaded onto a HiLoad 26/600 Superdex 200 pg column. The column was pre-equilibrated with 360 ml buffer C [50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) pH 7.5, 100 mM KCl, 1 mM TCEP, 5% glycerol]. The 5 ml sample of HDAC6 CD1 protein was injected at a rate of 1 ml min−1 and 5 ml fractions were collected. Pure ‘humanized’ H82F/F202Y and K330L HDAC6 CD1 proteins were confirmed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, concentrated to approximately 10 mg ml−1 and stored at −80°C.
2.4. Crystallization of HDAC6 CD1 mutant–inhibitor complexes
The crystallization of zebrafish HDAC6 CD1 mutants complexed with inhibitors was achieved by sitting-drop vapor diffusion as recently described for cocrystallization with a different series of inhibitors (Osko & Christianson, 2019 ▸). Briefly, a 100 nl drop of protein solution [10 mg ml−1 HDAC6 CD1 mutant, 50 mM HEPES pH 7.5, 100 mM KCl, 5%(v/v) glycerol, 1 mM TCEP, 2 mM inhibitor] was added to a 100 nl drop of precipitant solution and equilibrated against 80 µl precipitant solution in the well reservoir of a 96-well PS MRC crystallization plate using a Mosquito crystallization robot (TTP Labtech).
For cocrystallization of the H82F/F202Y HDAC6 CD1–trichostatin A complex, the precipitant solution was 0.2 M potassium acetate, 20% PEG 3350. For cocrystallization of the K330L HDAC6 CD1–SAHA-BPyne complex, the precipitant solution was 0.2 M potassium sodium tartrate tetrahydrate, 20% PEG 3350. For cocrystallization of the K330L HDAC6 CD1–4-iodo-SAHA complex, the precipitant solution was 0.2 M magnesium chloride hexahydrate, 20% PEG 3350. Crystals of each enzyme–inhibitor complex formed within 2–3 days at 21°C (room temperature) and were soaked in their respective mother liquors augmented with 20% ethylene glycol prior to flash-cooling. Crystallization parameters are summarized in Table 2 ▸.
Table 2. Crystallization.
| Method | Sitting-drop vapor diffusion |
| Plate type | PS MRC crystallization plate |
| Temperature (K) | 100 |
| Protein concentration (mg ml−1) | 10 |
| Buffer composition of protein solution | 50 mM HEPES pH 7.5, 100 mM KCl, 1 mM TCEP, 5% glycerol, 2 mM inhibitor |
| Composition of reservoir solution | |
| Trichostatin A complex | 0.2 M potassium acetate, 20% PEG 3350 |
| SAHA-BPyne complex | 0.2 M potassium sodium tartrate tetrahydrate, 20% PEG 3350 |
| 4-Iodo-SAHA complex | 0.2 M magnesium chloride hexahydrate, 20% PEG 3350 |
| Volume and ratio of drop | 1:1 ratio of protein:precipitant solution |
| Volume of reservoir (µl) | 80 |
2.5. Data collection, data reduction and crystal structure determinations
X-ray diffraction data were collected from crystals of the H82F/F202Y HDAC6 CD1–trichostatin A complex on Northeastern Collaborative Access Team (NE-CAT) beamline 24-ID-C at the Advanced Photon Source (APS). A PILATUS 6M-F detector, an incident X-ray beam wavelength of 0.98 Å and a temperature of 100 K were used for data collection. A full 180° of data were collected at a detector distance of 300 mm in 0.20° angle increments with 0.20 s exposure periods.
X-ray diffraction data were collected from crystals of the K330L HDAC6 CD1–SAHA-BPyne complex on beamline 17-ID-1 (AMX) at the National Synchrotron Light Source II (NSLS-II). A Dectris EIGER 9M detector, an incident X-ray beam wavelength of 0.92 Å and a temperature of 100 K were used for data collection. A full 180° of data were collected at a detector distance of 230 mm in 0.20° angle increments with 0.02 s exposure periods.
X-ray diffraction data from crystals of the K330L HDAC6 CD1–4-iodo-SAHA complex were collected on NE-CAT beamline 24-ID-E at APS. A Dectris EIGER 16M detector, an incident X-ray beam wavelength of 0.98 Å and a temperature of 100 K were used for data collection. A full 180° of data were collected at a detector distance of 150 mm in 0.20° angle increments with 0.20 s exposure periods. Although the completeness and mutiplicity of this data set were lower than expected, the structure of this complex ultimately refined satisfactorily and yielded excellent electron-density maps. All data-collection statistics are recorded in Table 3 ▸.
Table 3. Data-collection and refinement statistics for HDAC6 CD1 mutant–inhibitor complexes.
Values in parentheses are for the highest resolution shell.
| HDAC6 CD1 mutant | H82F/F202Y | K330L | |
|---|---|---|---|
| Inhibitor | Trichostatin A | SAHA-BPyne | 4-Iodo-SAHA |
| Data collection | |||
| Beamline | 24-ID-C, APS | 17-ID-1, NSLS-II | 24-ID-E, APS |
| Wavelength (Å) | 0.98 | 0.92 | 0.98 |
| Temperature (K) | 100 | 100 | 100 |
| Detector | PILATUS 6M-F | EIGER 9M | EIGER 16M |
| Wilson B factor (Å2) | 32 | 46 | 15 |
| Crystal-to-detector distance (mm) | 300 | 230 | 150 |
| Rotation range per image (°) | 0.20 | 0.20 | 0.20 |
| Total rotation range (°) | 180 | 180 | 180 |
| Exposure time per image (s) | 0.20 | 0.02 | 0.20 |
| Space group | P21 | C22121 | P21 |
| a, b, c (Å) | 53.1, 124.0, 55.0 | 66.0, 95.2, 119.7 | 53.1, 123.8, 55.1 |
| α, β, γ (°) | 90.0, 114.4, 90.0 | 90.0, 90.0, 90.0 | 90.0, 113.5, 90.0 |
| R merge † | 0.054 (0.418) | 0.190 (0.817) | 0.091 (0.271) |
| R p.i.m. ‡ | 0.052 (0.395) | 0.119 (0.507) | 0.071 (0.231) |
| CC1/2 § | 0.996 (0.792) | 0.971 (0.618) | 0.981 (0.829) |
| Multiplicity | 3.4 (3.2) | 6.4 (6.7) | 2.4 (2.3) |
| Completeness (%) | 98.7 (98.4) | 100.0 (100.0) | 77.9 (80.6) |
| 〈I/σ(I)〉 | 8.9 (1.9) | 4.8 (2.0) | 5.2 (2.0) |
| Refinement | |||
| Resolution (Å) | 48.34–2.30 (2.38–2.30) | 54.25–2.40 (2.49–2.40) | 39.14–1.90 (1.97–1.90) |
| No. of reflections | 28350 (2842) | 15104 (1483) | 39267 (3884) |
| R work/R free ¶ | 0.170/0.232 (0.210/0.256) | 0.204/0.243 (0.263/0.274) | 0.177/0.223 (0.206/0.270) |
| No. of atoms†† | |||
| Protein | 5288 | 2688 | 5338 |
| Ligand | 50 | 73 | 46 |
| Solvent | 116 | 47 | 268 |
| Average B factors (Å2) | |||
| Protein | 33 | 47 | 14 |
| Ligand | 29 | 44 | 17 |
| Solvent | 31 | 43 | 18 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.007 | 0.002 | 0.007 |
| Bond angles (°) | 0.9 | 0.6 | 0.8 |
| Ramachandran plot‡‡ | |||
| Favored | 95.53 | 96.63 | 96.30 |
| Allowed | 4.47 | 3.37 | 3.70 |
| Outliers | 0.00 | 0.00 | 0.00 |
| PDB code | 6wyo | 6wyp | 6wyq |
R
merge = 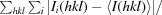
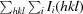 , where 〈I(hkl)〉 is the average intensity calculated for reflection hkl from replicate measurements.
, where 〈I(hkl)〉 is the average intensity calculated for reflection hkl from replicate measurements.
R
p.i.m. = 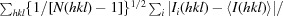 , where 〈I(hkl)〉 is the average intensity calculated for reflection hkl from replicate measurements and N(hkl) is the number of reflections.
, where 〈I(hkl)〉 is the average intensity calculated for reflection hkl from replicate measurements and N(hkl) is the number of reflections.
Pearson correlation coefficient between random half data sets.
R
work = 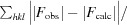
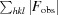 for reflections contained in the working set. |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively. R
free is calculated using the same expression for reflections contained in the test set that was held aside during refinement.
for reflections contained in the working set. |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively. R
free is calculated using the same expression for reflections contained in the test set that was held aside during refinement.
Per asymmetric unit.
Calculated with MolProbity.
The CCP4 program suite (Winn et al., 2011 ▸) was used for data reduction for all three structures. All data were indexed using the CCP4 program iMosflm (Battye et al., 2011 ▸) and were scaled using AIMLESS (Evans & Murshudov, 2013 ▸). The initial electron-density map of each enzyme–inhibitor complex was phased by molecular replacement using the atomic coordinates of HDAC6 CD1 (PDB entry 5eef; Hai & Christianson, 2016 ▸) as a search probe in Phaser (McCoy et al., 2007 ▸). The interactive graphics program Coot (Emsley et al., 2010 ▸) was used to build and manipulate atomic models of each enzyme–inhibitor complex. Crystallographic refinement was performed using Phenix (Liebschner et al., 2019 ▸). Final refined structures were validated using MolProbity (Chen et al., 2010 ▸) prior to deposition in the Protein Data Bank (PDB; http://www.rcsb.org). All data-reduction and refinement statistics are recorded in Table 3 ▸.
In order to improve the PDB statistical sliders for the HDAC6 CD1 enzyme–inhibitor complexes, the resolutions were adjusted during the final phases of refinement. The resolution of the H82F/F202Y HDAC6 CD1–trichostatin A complex was adjusted from 1.90 to 2.30 Å, the resolution of the K330L HDAC6 CD1–SAHA-BPyne complex was adjusted from 2.15 to 2.40 Å and the resolution of the K330L HDAC6 CD1–4-iodo-SAHA complex was adjusted from 1.74 to 1.90 Å. The electron density for each inhibitor is unambiguous.
3. Results and discussion
3.1. Crystal structure of the H82F/F202Y HDAC6 CD1–trichostatin A complex
The active-site residues in zebrafish and human HDAC6 CD1 are generally conserved, with the exception of His82 and Phe202 in the zebrafish enzyme, which appear as Phe105 and Tyr225, respectively, in human HDAC6 CD1. Therefore, the double mutant H82F/F202Y was prepared to ‘humanize’ zebrafish HDAC6 CD1 and provide a view of how these residues might influence inhibitor binding.
The 2.30 Å resolution crystal structure of the H82F/F202Y HDAC6 CD1–trichostatin A complex contains two monomers in the asymmetric unit. The inhibitor hydroxamate chelates the catalytic Zn2+ ion with bidentate coordination in each monomer (Figs. 2 ▸ a and 2 ▸ b). The Zn2+-bound hydroxamate C=O group accepts a hydrogen bond from Tyr363, the Zn2+-bound hydroxamate NO− group accepts a hydrogen bond from His192, and the hydroxamate NH group donates a hydrogen bond to His193. The linker resides in an aromatic crevice defined by Tyr202 and Trp261. The ketone carbonyl of the capping group accepts a hydrogen bond from Lys330. No major conformational changes are triggered by the H82F or F202Y substitutions (Fig. 2 ▸ c), and the root-mean-square deviation (r.m.s.d.) is 0.14 Å for 301 Cα atoms between the H82F/F202Y and wild-type HDAC6 CD1–trichostatin A complexes.
Figure 2.

Stereoviews of the H82F/F202Y HDAC6 CD1–trichostatin A complex (PDB entry 6wyo). (a) Polder OMIT maps (Liebschner et al., 2017 ▸) showing trichostatin A bound to monomer A (contoured at 3.5σ), Phe82 (contoured at 2.0σ) and Tyr202 (contoured at 2.5σ). Atoms are color-coded as follows: C, light blue (monomer A), light gray (monomer B) or wheat (inhibitor); N, blue; O, red; Zn2+, gray sphere. Metal-coordination and hydrogen-bond interactions are indicated by solid and dashed black lines, respectively. (b) Polder OMIT maps showing trichostatin A bound to monomer B (contoured at 3.5σ), Phe82 (contoured at 2.0σ) and Tyr202 (contoured at 2.5σ). Atoms are color-coded as in (a). (c) Superposition of the trichostatin A complexes with wild-type HDAC6 CD1 (PDB entry 6uo2; monomer B) and H82F/F202Y HDAC6 CD1 (PDB entry 6wyo; monomer A). Residue 82 displays slight flexibility, consistent with the weaker electron density observed for Phe82 in (a) and (b). Atoms are color-coded as follows: C, light blue (H82F/F202Y HDAC6 CD1), light gray (HDAC6 CD1), wheat (trichostatin A bound to H82F/F202Y HDAC6 CD1) or orange (trichostatin A bound to wild-type HDAC6 CD1); N, blue, O, red; Zn2+, gray sphere.
Overall, there are no major conformational changes between monomers A and B, and the r.m.s.d. deviation is 0.19 Å for 286 Cα atoms. However, residues 74–87 in monomer A appear to be predominately disordered, with the exception of Phe82 and Pro83; in contrast, these residues are well ordered in monomer B. In both monomers, structural changes of 0.3–1.3 Å are observed for Phe82, Pro83 and Tyr202 compared with the wild-type HDAC6 CD1–trichostatin A complex (PDB entry 6uo2). The remaining active-site residues adopt similar conformations to those observed in the wild-type HDAC6 CD1–trichostatin A complex.
3.2. Crystal structure of the K330L HDAC6 CD1–SAHA-BPyne complex
The pan-HDAC inhibitor SAHA-BPyne contains a SAHA scaffold derivatized with a benzophenone linker and an alkyne tag. As such, this inhibitor can be used to profile cellular HDAC through click chemistry (Salisbury & Cravatt, 2007 ▸). Until now, no crystal structure has been reported for a complex of HDAC6 CD1 with SAHA or any SAHA derivative. Some HDAC6 CD1–inhibitor complexes crystallize more readily using K330L HDAC6 CD1 (Osko & Christianson, 2019 ▸), and we found this to be the case in cocrystallization trials with SAHA-BPyne.
The 2.40 Å resolution crystal structure of the K330L HDAC6 CD1–SAHA-BPyne complex contains one monomer in the asymmetric unit. The hydroxamate group of the inhibitor chelates the catalytic Zn2+ ion in a bidentate fashion (Fig. 3 ▸). The amide group of SAHA forms water-mediated hydrogen bonds to Ser150 and the zinc ligand His232. Similar hydrogen-bond interactions are observed to the corresponding residues Ser531 and His614 in several HDAC6 CD2–inhibitor complexes (Osko & Christianson, 2020 ▸; Hai & Christianson, 2016 ▸; Porter et al., 2017 ▸; Bhatia et al., 2018 ▸; Mackwitz et al., 2018 ▸). The benzophenone carbonyl group forms a hydrogen bond to a water molecule, which in turn forms hydrogen bonds to Asp79 and Ser81. The interaction with Ser81 has not been observed previously for inhibitor binding to HDAC6 CD1. This residue is conserved only in human HDAC6 CD1 as Ser104; the corresponding residue is a histidine in human and zebrafish HDAC6 CD2. Thus, interactions with Ser81 can be targeted in future inhibitor designs for the selective inhibition of HDAC6 CD1.
Figure 3.

Stereoview of the K330L HDAC6 CD1–SAHA-BPyne complex (PDB entry 6wyp). Polder OMIT maps showing SAHA-BPyne bound to monomer A and Leu330 (each contoured at 2.5σ). Atoms are color-coded as follows: C, light blue (monomer A), light gray (symmetry mate) or wheat (inhibitor); N, blue; O, red; Zn2+, gray sphere; solvent, small red spheres. Metal-coordination and hydrogen-bond interactions are indicated by solid and dashed black lines, respectively.
Finally, the alkyne tag of SAHA-BPyne is disordered between two conformations, and electron density is weak or absent toward the end of the tag (Fig. 3 ▸). This portion of the inhibitor makes no hydrogen-bond interactions with protein residues or ordered solvent molecules.
3.3. Crystal structure of the K330L HDAC6 CD1–4-iodo-SAHA complex
The pan-HDAC inhibitor 4-iodo-SAHA is essentially isosteric to SAHA apart from the substitution of a bulky I atom for an H atom at the para position of the phenyl ring in the capping group. The 1.90 Å resolution crystal structure of the K330L HDAC6 CD1–4-iodo-SAHA complex contains two monomers in the asymmetric unit. There are no major conformational changes between monomers A and B, and the r.m.s.d. is 0.16 Å for 293 Cα atoms. The hydroxamate of the inhibitor chelates the catalytic Zn2+ ion with bidentate coordination, and the aliphatic linker resides in an aromatic crevice defined by Phe202 and Trp261 (Figs. 4 ▸ a and 4 ▸ b). As observed in the binding of SAHA-BPyne to K330L HDAC6 CD1, the amide moiety of the capping group forms water-mediated hydrogen bonds to Ser150 and the zinc ligand His232.
Figure 4.

Stereoviews of the K330L HDAC6 CD1–4-iodo-SAHA complex (PDB entry 6wyq). (a) Polder OMIT maps showing 4-iodo-SAHA bound to monomer A and Leu330 (each contoured at 2.5σ). Atoms are color-coded as follows: C, light blue (monomer A), light gray (monomer B) or wheat (inhibitor); N, blue; O, red; I−, magenta; Zn2+, gray sphere; solvent, small red spheres. Metal-coordination and hydrogen-bond interactions are indicated by solid and dashed black lines, respectively. The halogen bond between the inhibitor I atom and Asp79 is indicated by a dashed magenta line. (b) Polder OMIT maps showing 4-iodo-SAHA bound to monomer B (contoured at 3.5σ) and Leu330 (contoured at 3.0σ). Atoms are color-coded as in (a), except that C atoms are in light blue for monomer B and light gray for monomer A. (c) Superposition of the K330L HDAC6 CD1 complexes with 4-iodo-SAHA (monomer A and inhibitor are in blue and wheat, respectively; monomer B and inhibitor are in gray and light gray, respectively) and SAHA-BPyne (protein and inhibitor are in orange and dark orange, respectively) reveal that the SAHA moieties of each inhibitor bind with generally similar conformations, with slight variations in capping-group conformations.
Interestingly, the I atom of the inhibitor capping group makes a halogen bond with the carboxylate side chain of Asp79 in the outer active-site cleft of monomer A (I⋯O separation of 3.1 Å). Owing to conformational differences of the inhibitor capping group in monomers A and B involving a 3.3 Å shift of the I atom, this enzyme–inhibitor halogen-bond interaction is not observed in monomer B. Electron density for the capping group is broken in monomer A and somewhat weak in monomer B, consistent with disorder. Even so, the refinements were better fitted with an iodo group than a hydroxyl group at the para position of the aromatic capping group. Residual negative density on the I atom in the final |F o| – |F c| map is likely to be the consequence of disorder, i.e. a less than full occcupancy of the electron-rich I atom.
Overall, there are no major conformational changes for protein atoms between the complexes of HDAC CD1 with 4-iodo-SAHA and SAHA-BPyne, and the r.m.s.d. is 0.22 Å for 329 Cα atoms between these two complexes. Superposition of these two complexes reveals that the binding of the SAHA moieties are generally similar, with slight variations in capping group conformations (Fig. 4 ▸ c).
3.4. Analysis of inhibitor binding
The use of histone deacetylase inhibitors to modulate acetylation–deacetylation cycles is a validated strategy in cancer chemotherapy and is also being explored for the treatment of other diseases (Arrowsmith et al., 2012 ▸; Falkenberg & Johnstone, 2014 ▸; Dokmanovic et al., 2007 ▸; West & Johnstone, 2014 ▸). The first HDAC inhibitor to be approved for clinical use was SAHA (formulated as vorinostat, trade name Zolinza) for the treatment of refractory cutaneous T-cell lymphoma (Richon et al., 1996 ▸; Marks, 2007 ▸; Mann et al., 2007 ▸). However, SAHA is a pan-HDAC inhibitor, in that it inhibits most HDAC isozymes nearly equally well; thus, its use can lead to unwanted off-target effects (Duvic et al., 2007 ▸).
HDAC6 CD2 is a particularly important target for the design of selective inhibitors for treating cancer and neurodegenerative diseases owing to its biological function as a tubulin deacetylase (Hubbert et al., 2002 ▸; Zhang et al., 2003 ▸; Haggarty et al., 2003 ▸) and as a tau deacetylase (Min et al., 2010 ▸; Cohen et al., 2011 ▸; Noack et al., 2014 ▸). The design of selective inhibitors has been guided by numerous X-ray crystallographic studies that have illuminated distinctive structural features contributing to inhibitor affinity and selectivity (recently reviewed by Osko & Christianson, 2020 ▸). However, as increasing numbers of inhibitors are being developed for the inhibition of HDAC6 CD2, it is critical to understand the impact that these inhibitors may have on the catalytic domain CD1 of HDAC6.
The in vivo function of HDAC6 CD1 is not fully understood. Measurements made with cell extracts suggest that CD1 serves as an E3 ubiquitin ligase (Zhang et al., 2014 ▸) as well as an RNA helicase DDX3X deacetylase (Saito et al., 2019 ▸). However, the only activity that has been confirmed in vitro for a single-domain HDAC6 CD1 construct is as a lysine deacetylase with much narrower substrate specificity than CD2 (Hai & Christianson, 2016 ▸; Osko & Christianson, 2019 ▸), although the CD1 substrate specificity is reported to be wider in the full-length enzyme (Kutil et al., 2019 ▸). Regardless of the elusive biological functions of HDAC6 CD1, it is important to understand how this catalytic domain accommodates bound inhibitors. The crystal structures of complexes of HDAC6 CD1 mutants with pan-HDAC inhibitors reported here represent the first step in understanding how different active-site features can be taken into account to guide the design of domain-specific inhibitors.
How valid is the ‘humanized’ zebrafish enzyme, H82F/F202Y HDAC6 CD1, as a surrogate for the actual human enzyme HDAC6 CD1? A definitive answer may be elusive owing to the lack of a crystal structure of the human enzyme, but useful inferences may nonetheless be drawn from the current study. The binding conformation of the pan-HDAC inhibitor trichostatin A in the active site of the zebrafish enzyme is unperturbed by the H82F/F202Y substitutions (Fig. 2 ▸ c). In studies with another pan-HDAC inhibitor, a SAHA derivative bearing a para-substituted fluorescein tag designated ‘fl-SAHA’ (Kim et al., 2015 ▸), the inhibitor dissociation constants changed very little from wild-type zebrafish HDAC6 CD1 (K d = 1.6 ± 0.3 µM) to zebrafish H82F/F202Y HDAC6 CD1 (K d = 2.9 ± 0.7 µM) (Hai & Christianson, 2016 ▸). The binding of fl-SAHA to a di-domain construct of the human enzyme in which CD2 is deactivated by a mutation, H651V CD12, indicates an approximately tenfold weaker binding (K d = 30 ± 20 µM; Hai & Christianson, 2016 ▸). Given the general conservation of the amino acids that define the active-site contours of the zebrafish and human HDAC6 CD1 enzymes, the similar binding conformations of trichostatin A, and the slightly perturbed fl-SAHA binding affinities, we suggest that both the wild-type and ‘humanized’ zebrafish H82F/F202Y HDAC6 CD1 serve as valid and readily studied surrogates for human HDAC6 CD1.
We have previously observed that the K330L mutant of zebrafish HDAC6 CD1 can be more amenable to crystallization with certain inhibitors than the wild-type enzyme (Osko & Christianson, 2019 ▸). Even though this amino-acid substitution makes the CD1 active site more like that of CD2, inhibitor binding is identical in the active sites of both the wild-type and mutant zebrafish CD1 enzymes. Here, the use of K330L HDAC6 CD1 enabled cocrystallization with two different SAHA derivatives, which represent the first structures of CD1 complexed with analogs of an approved drug for cancer chemotherapy. The SAHA core of each inhibitor studied binds in a similar fashion (Fig. 4 ▸ c); intriguingly, the benzophenone proteomics tag of SAHA-BPyne maps out a region of the outer active-site cleft that could be targeted for the design of a CD1-selective inhibitor, including interactions with Ser81 (Fig. 3 ▸ b), a residue that is unique to CD1 in the zebrafish and human enzymes. The SAHA-BPyne inhibitor is the largest inhibitor studied to date in complex with HDAC6 CD1, so the structure of this complex reveals new information regarding the accessibility of the protein surface surrounding the active-site cleft that can be targeted by a bound inhibitor.
4. Conclusions
The current work demonstrates that active-site mutants of zebrafish HDAC6 CD1 enable cocrystallization with inhibitors to map out intermolecular interactions in the active site that contribute to enzyme–inhibitor affinity. The active site of H82F/F202Y zebrafish HDAC6 CD1 is a more faithful mimic of the active site of human HDAC6 CD1, yet the inhibitor binding conformation and affinity are quite similar between the wild-type and H82F/F202Y HDAC6 enzymes. Even so, an interaction with the side chain of Tyr202 in human HDAC6 CD1 might confer isozyme selectivity, since this tyrosine residue is unique to human HDAC6 CD1 and the phenolic hydroxyl group protrudes into the active site.
The inhibitor binding conformations are generally identical between wild-type and K330L HDAC6 CD1, even though the K330L substitution mimics HDAC6 CD2. Interestingly, cocrystallization of wild-type zebrafish HDAC6 CD1 sometimes does not yield crystalline enzyme–inhibitor complexes, whereas cocrystallization with the active-site mutants described here yields high-quality crystals and high-resolution crystal structures. We expect that these mutants will facilitate future X-ray crystal structure determinations of HDAC6 CD1 complexes with inhibitors designed to selectively block its deacetylase function.
Supplementary Material
PDB reference: HDAC6 CD1, H82F/F202Y mutant, complex with trichostatin A, 6wyo
PDB reference: K330L mutant, complex with SAHA-BPyne, 6wyp
PDB reference: K330L mutant, complex with 4-iodo-SAHA, 6wyq
Acknowledgments
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences (NIGMS) from the National Institutes of Health (P30 GM124165). The EIGER 16M detector on beamline 24-ID-E is funded by an NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Additionally, this work is based on research conducted on beamline 17-ID-1 (AMX) at the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the National Institutes of Health, NIGMS through a Biomedical Technology Research Resource P41 grant (P41GM111244) and by the DOE Office of Biological and Environmental Research (KP1605010). All authors contributed equally to this work. The authors declare no competing financial interests.
Funding Statement
This work was funded by National Institutes of Health grant GM49758.
References
- Allfrey, V. G., Faulkner, R. & Mirsky, A. E. (1964). Proc. Natl Acad. Sci. USA, 51, 786–794. [DOI] [PMC free article] [PubMed]
- Arrowsmith, C. H., Bountra, C., Fish, P. V., Lee, K. & Schapira, M. (2012). Nat. Rev. Drug Discov. 11, 384–400. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Bhatia, S., Krieger, V., Groll, M., Osko, J. D., Ressing, N., Ahlert, H., Borkhardt, A., Kurz, T., Christianson, D. W., Hauer, J. & Hansen, F. K. (2018). J. Med. Chem. 61, 10299–10309. [DOI] [PMC free article] [PubMed]
- Carlomagno, Y., Chung, D. C., Yue, M., Castanedes-Casey, M., Madden, B. J., Dunmore, J., Tong, J., DeTure, M., Dickson, D. W., Petrucelli, L. & Cook, C. (2017). J. Biol. Chem. 292, 15277–15286. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., Olsen, J. V. & Mann, M. (2009). Science, 325, 834–840. [DOI] [PubMed]
- Cohen, T. J., Guo, J. L., Hurtado, D. E., Kwong, L. K., Mills, I. P., Trojanowski, J. Q. & Lee, V. M. Y. (2011). Nat. Commun. 2, 252. [DOI] [PMC free article] [PubMed]
- Dokmanovic, M., Clarke, C. & Marks, P. A. (2007). Mol. Cancer Res. 5, 981–989. [DOI] [PubMed]
- Duvic, M., Talpur, R., Ni, X., Zhang, C., Hazarika, P., Kelly, C., Chiao, J. H., Reilly, J. F., Ricker, J. L., Richon, V. M. & Frankel, S. R. (2007). Blood, 109, 31–39. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Falkenberg, K. J. & Johnstone, R. W. (2014). Nat. Rev. Drug Discov. 13, 673–691. [DOI] [PubMed]
- Grozinger, C. M., Hassig, C. A. & Schreiber, S. L. (1999). Proc. Natl Acad. Sci. USA, 96, 4868–4873. [DOI] [PMC free article] [PubMed]
- Haggarty, S. J., Koeller, K. M., Wong, J. C., Grozinger, C. M. & Schreiber, S. L. (2003). Proc. Natl Acad. Sci. USA, 100, 4389–4394. [DOI] [PMC free article] [PubMed]
- Hai, Y. & Christianson, D. W. (2016). Nat. Chem. Biol. 12, 741–747. [DOI] [PMC free article] [PubMed]
- Hornbeck, P. V., Zhang, B., Murray, B., Kornhauser, J. M., Latham, V. & Skrzypek, E. (2015). Nucleic Acids Res. 43, D512–D520. [DOI] [PMC free article] [PubMed]
- Hubbert, C., Guardiola, A., Shao, R., Kawaguchi, Y., Ito, A., Nixon, A., Yoshida, M., Wang, X. F. & Yao, T. P. (2002). Nature, 417, 455–458. [DOI] [PubMed]
- Kim, B., Pithadia, A. S. & Fierke, C. A. (2015). Protein Sci. 24, 354–365. [DOI] [PMC free article] [PubMed]
- Kutil, Z., Skultetyova, L., Rauh, D., Meleshin, M., Snajdr, I., Novakova, Z., Mikesova, J., Pavlicek, J., Hadzima, M., Baranova, P., Havlinova, B., Majer, P., Schutkowski, M. & Barinka, C. (2019). FASEB J. 33, 4035–4045. [DOI] [PubMed]
- Li, L. & Yang, X. J. (2015). Cell. Mol. Life Sci. 72, 4237–4255. [DOI] [PMC free article] [PubMed]
- Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L.-W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O. V., Stockwell, D. H., Terwilliger, T. C., Urzhumtsev, A. G., Videau, L. L., Williams, C. J. & Adams, P. D. (2019). Acta Cryst. D75, 861–877.
- Liebschner, D., Afonine, P. V., Moriarty, N. W., Poon, B. K., Sobolev, O. V., Terwilliger, T. C. & Adams, P. D. (2017). Acta Cryst. D73, 148–157. [DOI] [PMC free article] [PubMed]
- Lombardi, P. M., Cole, K. E., Dowling, D. P. & Christianson, D. W. (2011). Curr. Opin. Struct. Biol. 21, 735–743. [DOI] [PMC free article] [PubMed]
- López, J. E., Sullivan, E. D. & Fierke, C. A. (2016). ACS Chem. Biol. 11, 706–716. [DOI] [PMC free article] [PubMed]
- Mackwitz, M. K. W., Hamacher, A., Osko, J. D., Held, J., Schöler, A., Christianson, D. W., Kassack, M. U. & Hansen, F. K. (2018). Org. Lett. 20, 3255–3258. [DOI] [PMC free article] [PubMed]
- Mann, B. S., Johnson, J. R., Cohen, M. H., Justice, R. & Pazdur, R. (2007). Oncologist, 12, 1247–1252. [DOI] [PubMed]
- Marks, P. A. (2007). Oncogene, 26, 1351–1356. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Min, S.-W., Cho, S.-H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., Huang, E. J., Shen, Y., Masliah, E., Mukherjee, C., Meyers, D., Cole, P. A., Ott, M. & Gan, L. (2010). Neuron, 67, 953–966. [DOI] [PMC free article] [PubMed]
- Miyake, Y., Keusch, J. J., Wang, L., Saito, M., Hess, D., Wang, X., Melancon, B. J., Helquist, P., Gut, H. & Matthias, P. (2016). Nat. Chem. Biol. 12, 748–754. [DOI] [PubMed]
- Noack, M., Leyk, J. & Richter-Landsberg, C. (2014). Glia, 62, 535–547. [DOI] [PubMed]
- Osko, J. D. & Christianson, D. W. (2019). Biochemistry, 58, 4912–4924. [DOI] [PMC free article] [PubMed]
- Osko, J. D. & Christianson, D. W. (2020). Bioorg. Med. Chem. Lett. 30, 127023. [DOI] [PMC free article] [PubMed]
- Osko, J. D., Porter, N. J., Narayana Reddy, P. A., Xiao, Y.-C., Rokka, J., Jung, M., Hooker, J. M., Salvino, J. M. & Christianson, D. W. (2020). J. Med. Chem. 63, 295–308. [DOI] [PMC free article] [PubMed]
- Porter, N. J. & Christianson, D. W. (2019). Curr. Opin. Struct. Biol. 59, 9–18. [DOI] [PMC free article] [PubMed]
- Porter, N. J., Mahendran, A., Breslow, R. & Christianson, D. W. (2017). Proc. Natl Acad. Sci. USA, 114, 13459–13464. [DOI] [PMC free article] [PubMed]
- Porter, N. J., Osko, J. D., Diedrich, D., Kurz, T., Hooker, J. M., Hansen, F. K. & Christianson, D. W. (2018). J. Med. Chem. 61, 8054–8060. [DOI] [PMC free article] [PubMed]
- Porter, N. J., Shen, S., Barinka, C., Kozikowski, A. P. & Christianson, D. W. (2018). ACS Med. Chem. Lett. 9, 1301–1305. [DOI] [PMC free article] [PubMed]
- Porter, N. J., Wagner, F. F. & Christianson, D. W. (2018). Biochemistry, 57, 3916–3924. [DOI] [PMC free article] [PubMed]
- Richon, V. M., Webb, Y., Merger, R., Sheppard, T., Jursic, B., Ngo, L., Civoli, F., Breslow, R., Rifkind, R. A. & Marks, P. A. (1996). Proc. Natl Acad. Sci. USA, 93, 5705–5708. [DOI] [PMC free article] [PubMed]
- Saito, M., Hess, D., Eglinger, J., Fritsch, A. W., Kreysing, M., Weinert, B. T., Choudhary, C. & Matthias, P. (2019). Nat. Chem. Biol. 15, 51–61. [DOI] [PubMed]
- Salisbury, C. M. & Cravatt, B. F. (2007). Proc. Natl Acad. Sci. USA, 104, 1171–1176. [DOI] [PMC free article] [PubMed]
- Shen, S., Svoboda, M., Zhang, G., Cavasin, M. A., Motlova, L., McKinsey, T. A., Eubanks, J. H., Bařinka, C. & Kozikowski, A. P. (2020). ACS Med. Chem. Lett. 11, 706–712. [DOI] [PMC free article] [PubMed]
- Tseng, J.-H., Xie, L., Song, S., Xie, Y., Allen, L., Ajit, D., Hong, J.-S., Chen, X., Meeker, R. B. & Cohen, T. J. (2017). Cell. Rep. 20, 2169–2183. [DOI] [PMC free article] [PubMed]
- Verdel, A. & Khochbin, S. (1999). J. Biol. Chem. 274, 2440–2445. [DOI] [PubMed]
- Verdin, E. & Ott, M. (2015). Nat. Rev. Mol. Cell Biol. 16, 258–264. [DOI] [PubMed]
- West, A. C. & Johnstone, R. W. (2014). J. Clin. Invest. 124, 30–39. [DOI] [PMC free article] [PubMed]
- Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011). Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed]
- Zhang, M., Xiang, S., Joo, H.-Y., Wang, L., Williams, K. A., Liu, W., Hu, C., Tong, D., Haakenson, J., Wang, C., Zhang, S., Pavlovicz, R. E., Jones, A., Schmidt, K. H., Tang, J., Dong, H., Shan, B., Fang, B., Radhakrishnan, R., Glazer, P. M., Matthias, P., Koomen, J., Seto, E., Bepler, G., Nicosia, S. V., Chen, J., Li, C., Gu, L., Li, G.-M., Bai, W., Wang, H. & Zhang, X. (2014). Mol. Cell, 55, 31–46. [DOI] [PMC free article] [PubMed]
- Zhang, Y., Gilquin, B., Khochbin, S. & Matthias, P. (2006). J. Biol. Chem. 281, 2401–2404. [DOI] [PubMed]
- Zhang, Y., Li, N., Caron, C., Matthias, G., Hess, D., Khochbin, S. & Matthias, P. (2003). EMBO J. 22, 1168–1179. [DOI] [PMC free article] [PubMed]
- Zou, H., Wu, Y., Navre, M. & Sang, B.-C. (2006). Biochem. Biophys. Res. Commun. 341, 45–50. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: HDAC6 CD1, H82F/F202Y mutant, complex with trichostatin A, 6wyo
PDB reference: K330L mutant, complex with SAHA-BPyne, 6wyp
PDB reference: K330L mutant, complex with 4-iodo-SAHA, 6wyq