

Abstract
A light-mediated method for the facile removal of polymer end groups that are common to controlled radical polymerization techniques is presented. This metal-free strategy is general, being effective for chlorine, bromine, and thiocarbonylthio moieties as well as a number of different polymer families (styrenic, acrylic, and methacrylic). In addition to solution reactions, this process is readily translated to thin films, where light mediation allows the straightforward fabrication of hierarchically patterned polymer brushes.
Graphical Abstract
INTRODUCTION
The ability to control molecular weight, architecture, and comonomer incorporation in the production of polymers has facilitated access to functional materials with diverse and targeted properties. Controlled radical polymerization techniques1–4 such as atom transfer radical polymerization (ATRP)1 and reversible addition–fragmentation chain transfer (RAFT)5 polymerization have been successfully used to commercially produce polymers1,6–8 from readily available starting materials under mild conditions. Both ATRP and RAFT processes require the presence of chain ends that can undergo reversible activation to give propagating radicals as a prerequisite for control. These chain ends are chemically reactive and thermally unstable, which negatively influences long-term stability and is problematic for polymer processing.9–14 For example, the elimination of toxic and corrosive hydrobromic (HBr) or hydrochloric acid (HCl) during thermal decomposition of ATRP polymer chain ends leads to a range of issues, including acid-catalyzed ester degradation and corrosion.10 Challenges related to polymerization-active chain ends also extend beyond solution-processed materials. The fabrication of patterned, functional polymer surfaces serves as an example where chain end removal with spatial control is often desirable for surface passivation and long-term stability.15–17
In previous work, spatial control has been achieved in the production of surface-grafted polymer brushes through deactivation by particle beams,18 UV irradiation,19–22 or light-mediated atom transfer radical addition (ATRA).23,24 However, the former methods are considered high cost, low throughput, and destructive while the latter method (ATRA) does not remove the halogen, merely changing its reactivity.
Our group has recently described the use of a reducing photoredox catalyst—10-phenylphenothiazine (PTH)—for the dehalogenation of small molecule halides.25,26 In this contribution, we apply this approach to efficiently remove the chain ends that are common to controlled radical polymerizations (see Figure 1). This mild, metal-free, and operationally simple method is applicable to a number of different polymer classes and is compatible with diverse chemical functionalities. Rigorous deoxygenation, anhydrous conditions, and elevated temperatures are all rendered unnecessary. In addition, the use of visible light as an external stimulus for catalyst activation allows spatial control and localized dehalogenation of surface-grafted polymer brushes. As a consequence, a much greater range of materials systems can be used in the preparation of soluble and grafted polymer chains.
Figure 1.
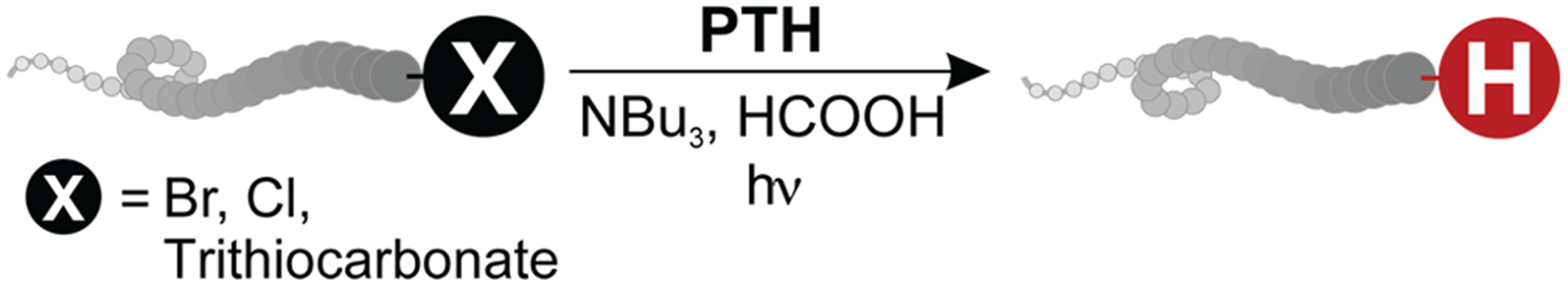
Graphical representation of the metal-free removal of terminal polymer chain end groups common to controlled radical polymerizations.
DISCUSSION
To initially demonstrate this metal-free chain end removal strategy, PTH in the presence of formic acid and tributylamine was used to debrominate a linear polystyrene–Br derivative (PS-Br, Figure 2a). Efficient chain end removal was confirmed via analysis of the characteristic 1H NMR signal at δ(CH-Br) = 4.5 ppm. As evident from Figure 2b,c, this key resonance, corresponding to the methine proton on the terminal styrene unit adjacent to the ω-end C–Br bond, was no longer detectable after 6 h of reaction. Size exclusion chromatography (SEC) showed no appreciable changes in molecular weight or polydispersity (see Figure S1), indicating the absence of undesired side reactions (e.g., polymer degradation, chain scission, chain–chain coupling) with field-desorption mass spectrometry (FD-MS) providing additional evidence for quantitative hydrogenation (see Figure 3). Prior to dehalogenation (Figure 3a), the absolute mass of each peak corresponds to a polystyrene chain with ethyl isobutyrate at the α-end and bromine at the ω-end. After reaction (Figure 3b), the spectrum was shifted by m/z = 78.9 (loss of Br and addition of H), reaffirming the successful removal of the bromine chain end and full retention of polymer structure. Control experiments under the same conditions, in the absence of either PTH or light, showed no dehalogenation, even after 24 h with analysis by 1H NMR revealing only starting material (see Supporting Information).
Figure 2.
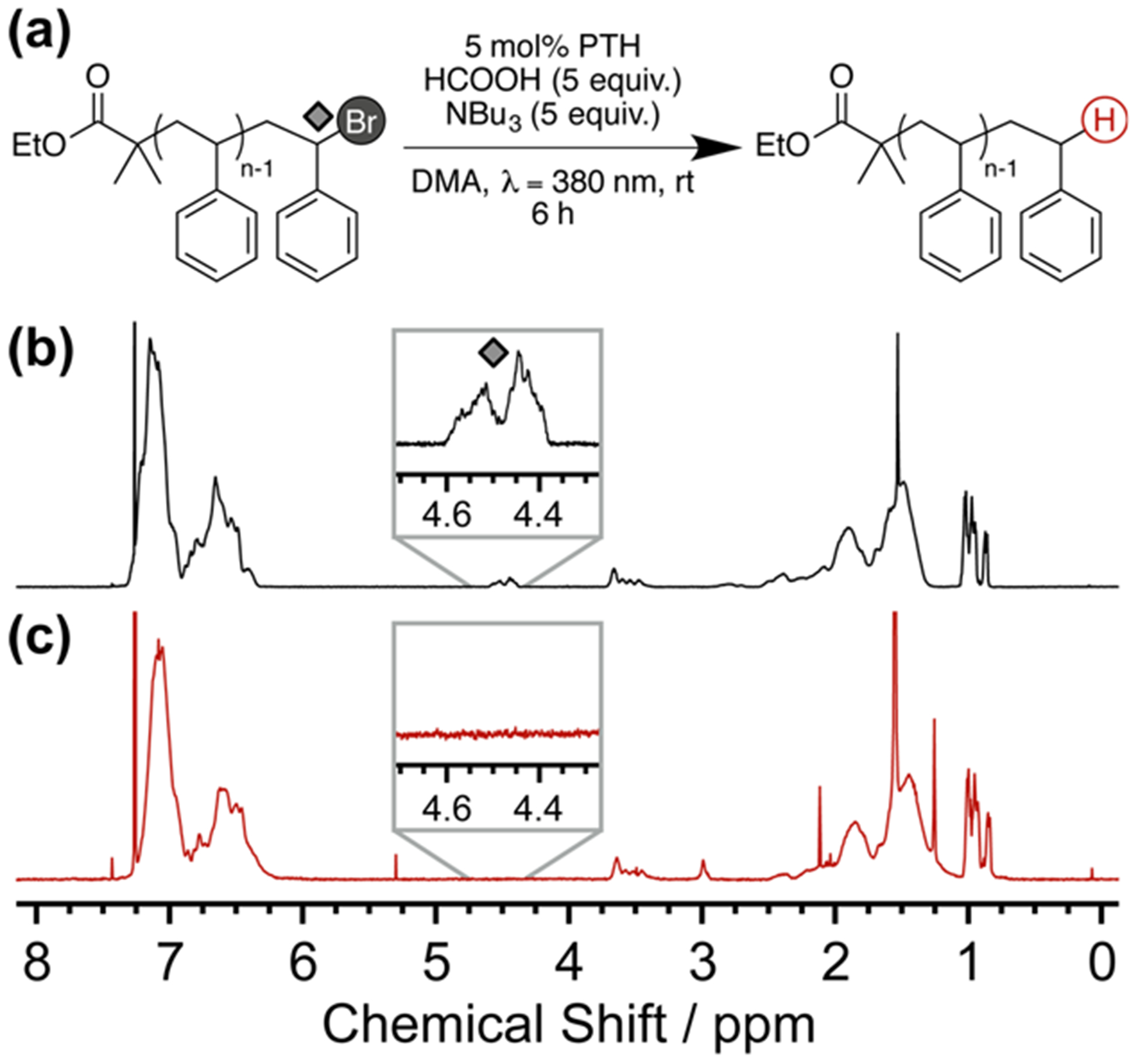
(a) Synthetic strategy for debromination of linear polystyrene. (b) 1H NMR spectrum of polystyrene-Br and (c) 1H NMR spectrum of the dehalogenated polymer. Insets emphasize the loss of the α-Br proton signal, indicative of successful dehalogenation.
Figure 3.
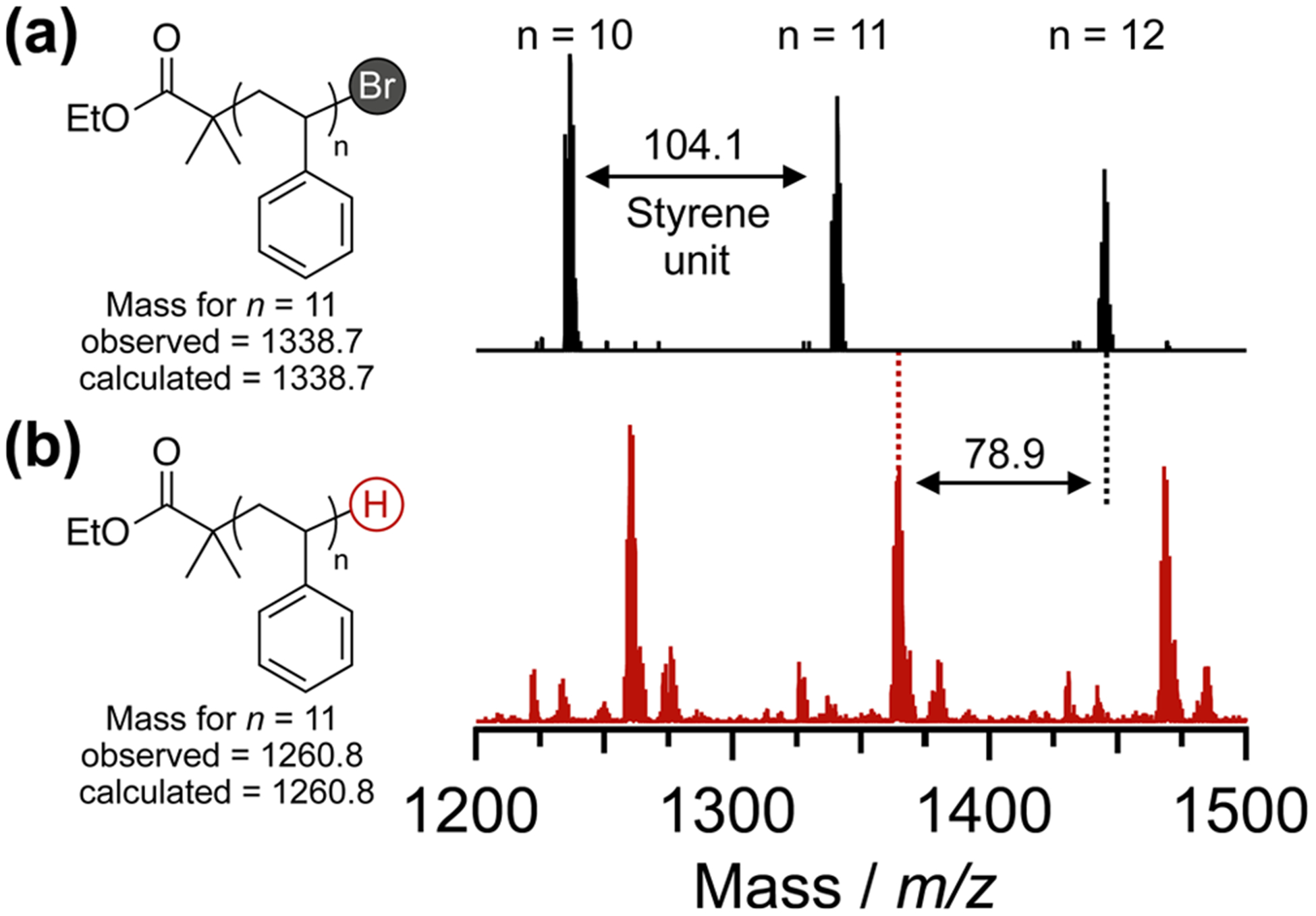
FD-MS spectra of (a) the unmodified PS-Br polymer and (b) dehalogenated PS-H confirmed the loss of bromine chain ends.
The removal of dormant Br chain ends is not only of considerable importance for chemical but also for thermal stability and related polymer processing (vide supra). Indeed, thermogravimetric analysis (Figure 4) showed increased thermal stability for the dehalogenated PS-H when compared to the initial PS-Br. In direct contrast to PS-Br, which lost 7 wt % at T = 215 °C (corresponding to HBr) with the onset of complete decomposition at T = 390 °C, the dehalogenated PSH polymer was thermally stable even above 325 °C with the onset of degradation for PS-H being delayed until ~400–420 °C.
Figure 4.
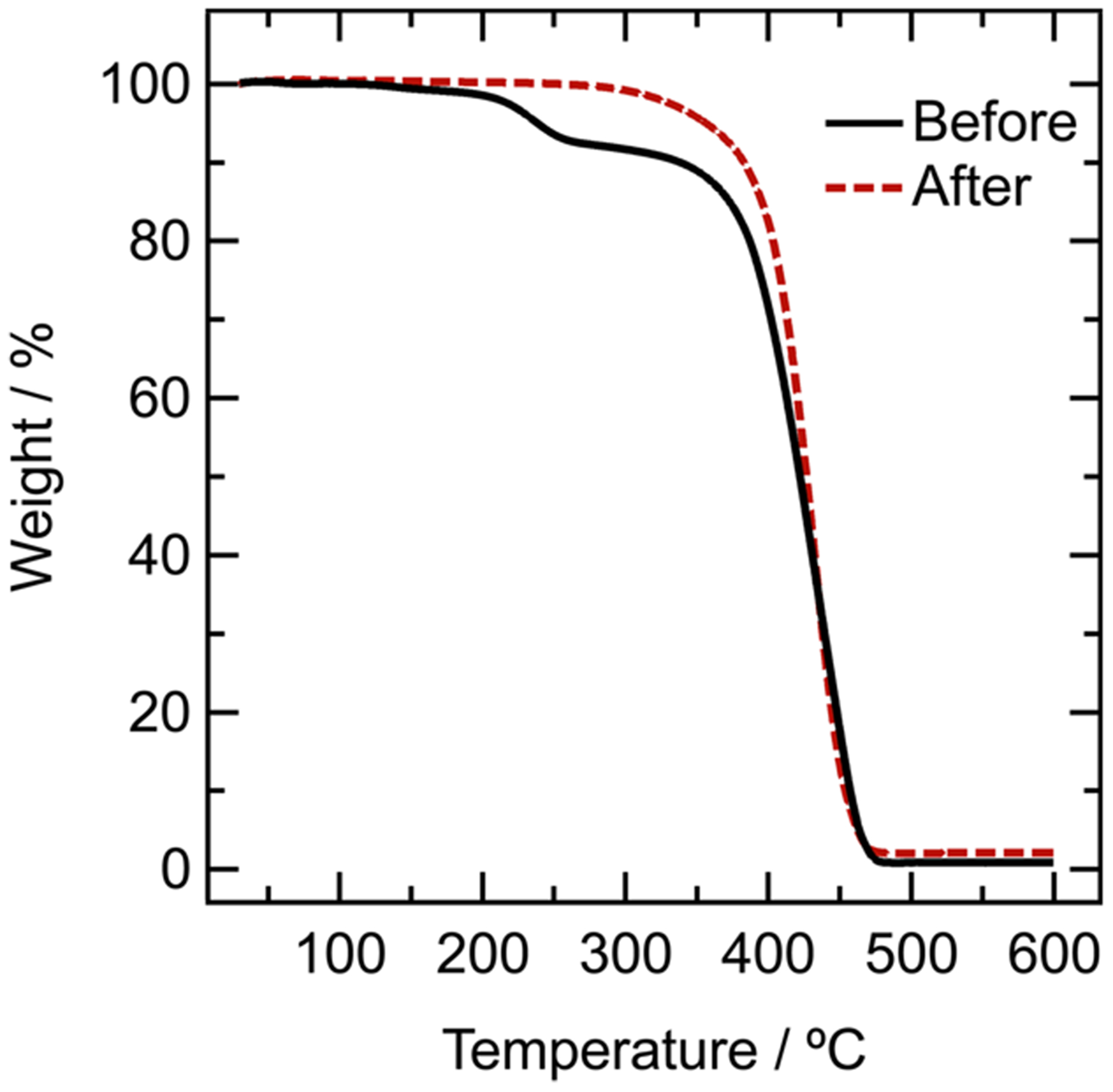
Thermogravimetric analysis (TGA) of polystyrene with bromine chain end (solid black line) and after dehalogenation (red dashed line).
The versatility of PTH-based chemistry for chain end removal was reinforced by its successful implementation with other polymer families. For the dehalogenation of poly(tert-butyl acrylate)-Br, loss of the halogen chain end was confirmed by the disappearance of the 1H NMR signal at δ = 4.4 ppm (CH-Br) (Figure S4). Again, no precautions were taken to ensure an inert environment during the reaction. Similar reactivity was observed for the removal of chloro-based chain ends (Figure S6). Significantly, macromolecules synthesized via RAFT polymerization with thiocarbonylthio RAFT chain ends also underwent facile deactivation and conversion to hydrogen (see Scheme 1 and Figure S7). In the latter case, the UV activity of the thiocarbonylthio moiety (absorption maximum, λmax = 310 nm) served as a marker to monitor the progress of this reaction via SEC (Figure S8). Again, SEC indicated no appreciable change in molecular weight or dispersity for all samples studied, reaffirming the mild and nondestructive nature of this process.
Scheme 1.
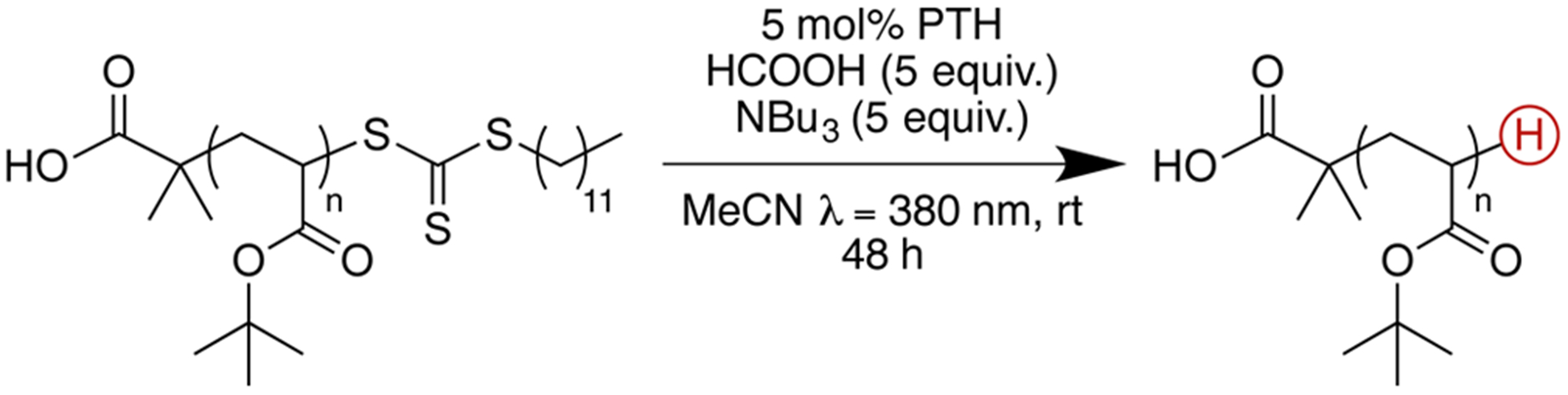
Synthetic Strategy for the Facile Deactivation and Conversion of Thiocarbonylthio RAFT Chain Ends to Hydrogen
Having demonstrated applicability to a wide range of polymeric starting materials in solution, our attention was drawn to the passivation of polymer brushes. The ability to pattern surfaces and remove reactive chains ends is useful for a variety of applications, and therefore we envisioned the translation of this chemistry to surfaces would be further enabled by the robust reaction conditions.
For the formation of complex 2-D and 3-D grafted polymer brushes, the selective termination of polymer chain ends allows for hierarchical patterning via surface-initiated ATRP (SIATRP).16,17,23,27–29 Figure 5 summarizes the advantages and disadvantages of common techniques for modulating the reactivity of polymer brushes through nucleophilic substitution15,30–32 and ATRA.23,24 This comparison with the PTH-based dehalogenation strategy described above clearly illustrates the potential advantages of this robust and spatially controlled alternative for polymer brush deactivation.
Figure 5.
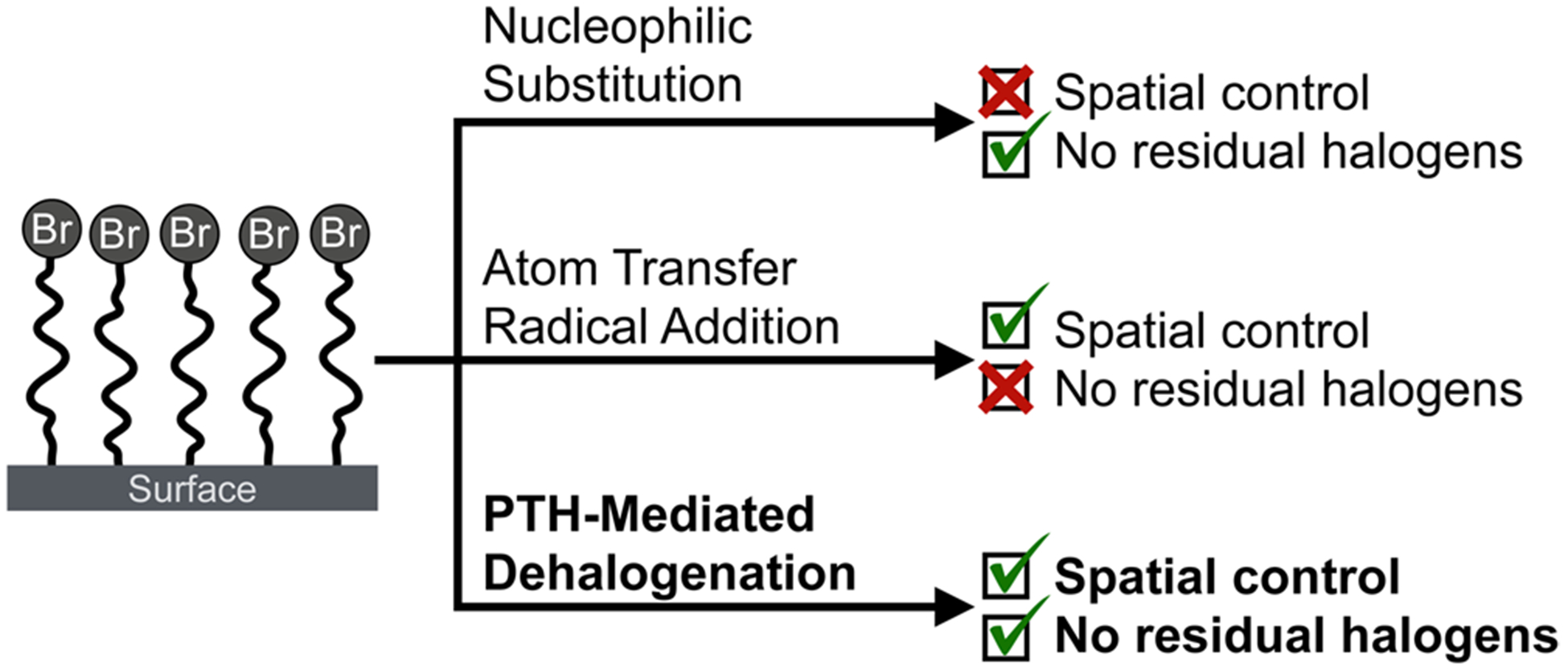
Comparison of the visible-light-mediated dehalogenation strategy, which combines both spatially and chemically controlled removal of reactive halogens, with traditional approaches based on nucleophilic substitution and ATRA chemistry.
Initial experiments were directed toward the uniform debromination of a silicon oxide substrate functionalized with the ATRP initiator undecyl 2-bromoisobutyrate (see Figure 6). X-ray photoelectron spectroscopy (XPS) was employed to detect the presence of bromine-containing functional groups on the substrate. For the substrate functionalized with immobilized ATRP initiators (Figure 6c), the bromine 3d orbital contains two components: the 3d5/2 and 3d3/2 doublet at binding energies BE = 69 eV and BE = 70 eV, respectively, as a result of spin–orbit splitting. Figure 6d illustrates disappearance of this Br 3d peak, confirming that irradiation at λ = 405 nm in the presence of PTH was capable of removing the halogen from the ATRP initiating layer.
Figure 6.
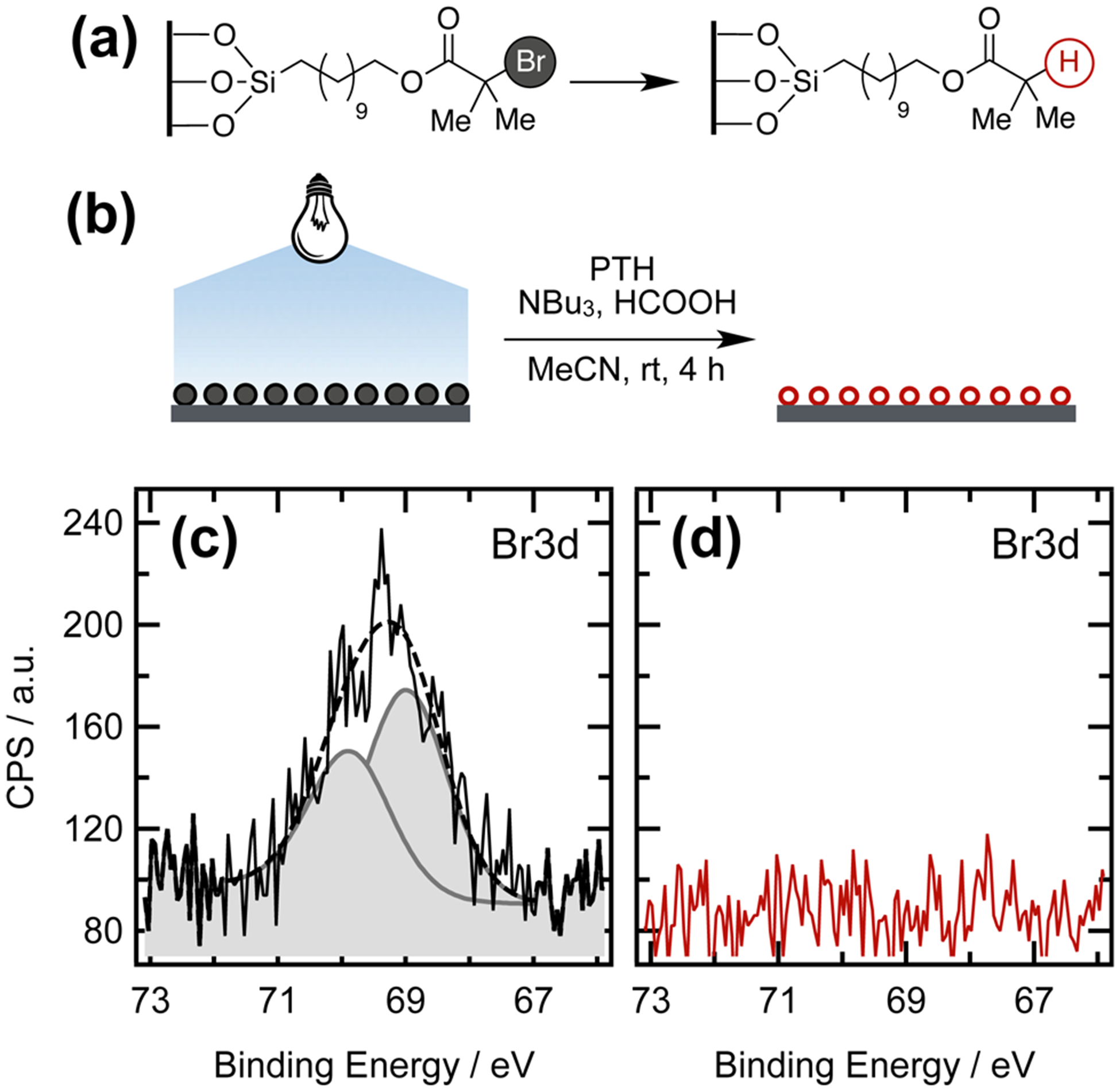
(a) Synthetic strategy and (b) schematic representation showing the uniform dehalogenation of α-bromoisobutyrate functionalized silicon surfaces. High-resolution XPS scans provided evidence for (c) the brominated ATRP initiator for the untreated substrates and (d) the absence of Br signals after PTH-catalyzed dehalogenation. Gaussian bell curves (c) correspond to the Br 3d5/2 and Br 3d3/2 orbitals. The dashed line represents the sum of both bell curves.
To illustrate the spatial fidelity of this light-mediated process, localized debromination of ATRP-initiator functionalized wafers was investigated by irradiation through a photomask with 20 × 200 μm2 clear rectangles. The photomask was subsequently removed, and poly(methyl methacrylate) (PMMA) polymer brushes were grown via light-mediated radical polymerization.33,34 Optical microscopy, secondary ion mass spectrometry (SIMS), and atomic force microscopy (AFM) confirmed spatially confined growth of polymer brushes exclusively in areas which were not previously irradiated/dehalogenated in the presence of PTH (Figure 7).
Figure 7.
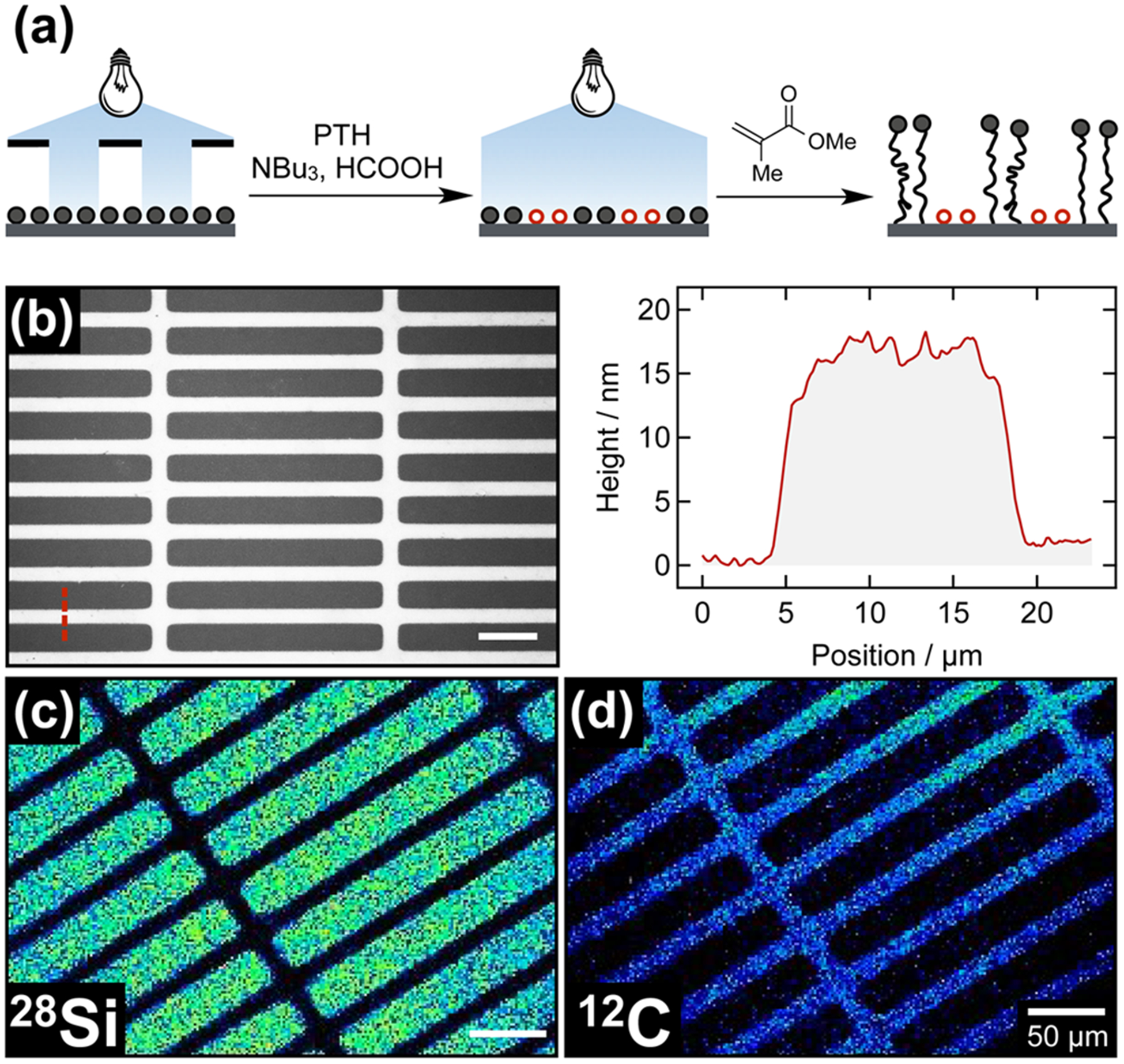
(a) Spatially confined dehalogenation of α-bromoisobuty-rate-functionalized silicon substrates by irradiation through a binary photomask with 20 × 200 μm2 clear rectangles, followed by subsequent homogeneous irradiation and light-mediated polymerization of MMA. (b) Optical micrograph (left) of the resulting patterned PMMA brushes confirmed the absence of polymer growth within the dehalogenated rectangles. AFM (right) indicated 15 nm polymer brush height and provided additional evidence for the absence of polymer in dehalogenated areas. SIMS chemical maps for (c) silicon and (d) carbon fragments confirmed spatially confined polymerization. All scale bars are 50 μm.
The utility of this process was further demonstrated by transitioning from the debromination of monolayers to the dehalogenation of surface-grafted polymer brush chain ends. As illustrated in Figure 8a, a uniform PMMA brush layer was initially grown (thickness 39 ± 2 nm), followed by spatially controlled dehalogenation by irradiation through a binary photomask with 2.5 × 25 μm2 clear rectangles. Removal of the photomask and homogeneous polymerization of a second monomer, 2,2,2-trifluoroethyl methacrylate (TFEMA), resulted in a chemically patterned surface composed of PMMA homopolymer and PMMA-b-PTFEMA diblock brushes. Both optical microscopy (Figure 8b) and SIMS (Figure 8c) confirmed successful patterning via spatially controlled dehalogenation and subsequent polymer brush extension. Detection of fluorine (19F) and carbon (12C) fragments in SIMS confirmed the spatial confinement of TFEMA exclusively in areas with PMMA-b-PTFEMA diblock copolymer brushes (Figure 8c).
Figure 8.
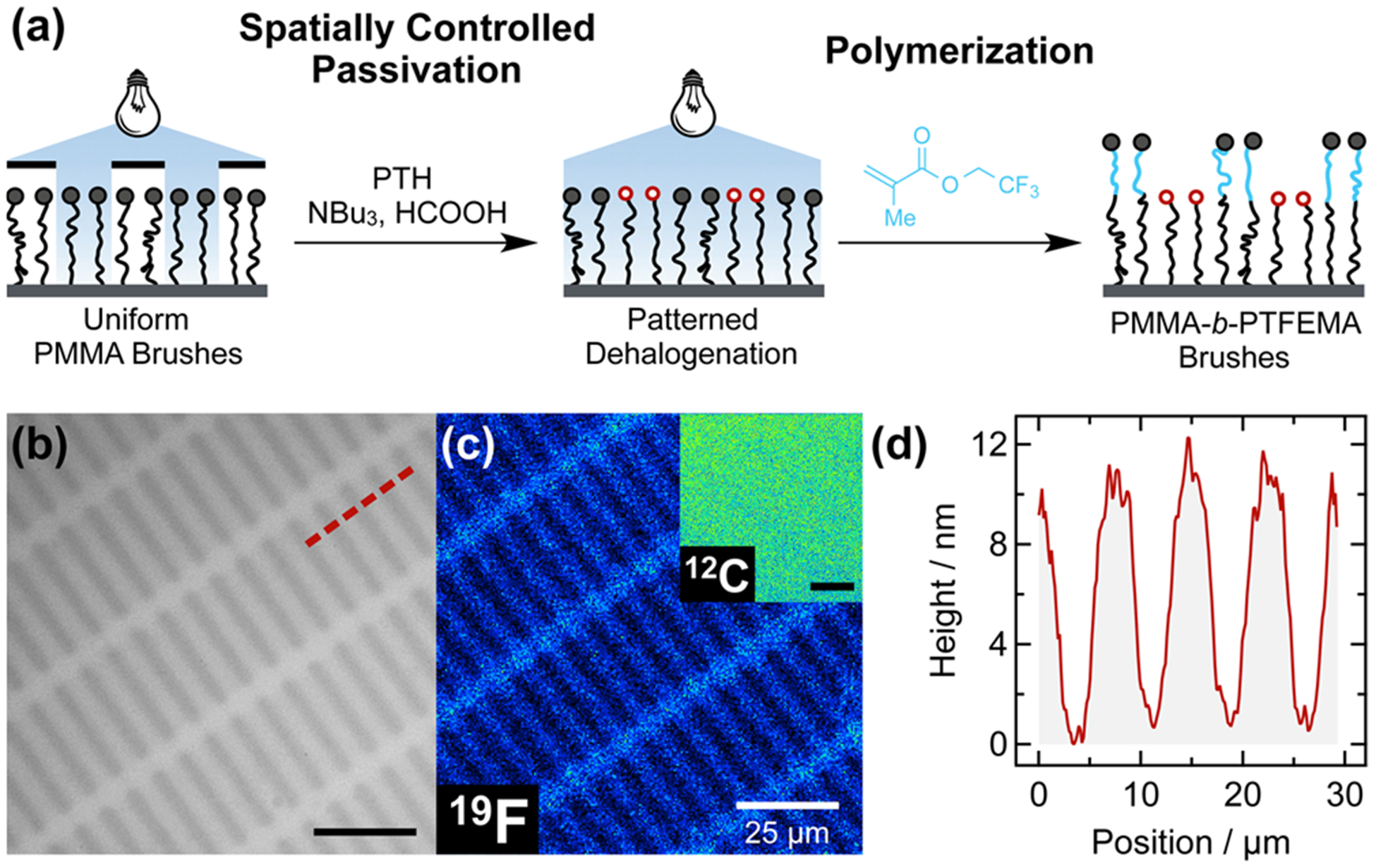
(a) Patterned diblock copolymer brush formation via spatially controlled dehalogenation and subsequent uniform polymerization of TFEMA. (b) Optical micrograph of the patterned polymer brushes. (c) 19F fluorine and (inset) 12C carbon SIMS scans confirmed localized presence of fluorine (PMMA-b-PTFEMA brushes) atop a uniform MMA polymer brush layer. (d) AFM height profile of the patterned PTFEMA brushes along the dashed line in (b) confirmed patterning and allowed quantification of brush height increase after block copolymerization. Scale bars in (b, c) are 25 μm.
CONCLUSIONS
This report describes a fully organic, light-mediated approach for the efficient removal of ATRP (Br, Cl) or RAFT (thiocarbonylthio) polymer chain ends from a variety of different polymer backbones. The facile and robust nature of this approach does not require elevated temperatures or inert reaction conditions, occurring with high efficiency under ambient conditions. Further, dehalogenation of both ATRP initiator monolayers and surface-grafted polymer brushes affords a novel and versatile procedure for surface patterning. Soluble and supported polymers with improved thermal and chemical stability can now be obtainable under mild, metal-free conditions and with external regulation.
Supplementary Material
ACKNOWLEDGMENTS
We thank the MRSEC program of the National Science Foundation (DMR 1121053, C.J.H.) and The Dow Chemical Company through the Dow Materials Institute at UCSB (K.M.M., W.R.G., E.H.D., Y.L., A.J.M., C.J.H.) for financial support. K.M.M. and C.J.H. also acknowledge the PREM program of the National Science Foundation (DMR-1205194) for support. K.M.M. and E.H.D. thank the NSF Graduate Research Fellowship for funding, and C.W.P. acknowledges the Alexander von Humboldt Foundation for financial support. W.R.G. thanks the NIH for a postdoctoral fellowship (F32GM108323). B.V.K.J.S. was supported by a fellowship within the Postdoc-Program of the German Academic Exchange Service (DAAD). A.T.H. acknowledges support through the Research Internships in Science and Engineering (RISE-UCSB MRSEC) program. We thank Dr. Thomas E. Mates for help with acquisition of SIMS data.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.6b01894..
Experimental procedures for preparation of all compounds and characterization data for all compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Matyjaszewski K Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45 (10), 4015–4039. [Google Scholar]
- (2).Braunecker WA; Matyjaszewski K Controlled/Living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci 2007, 32 (1), 93–146. [Google Scholar]
- (3).Stehling UM; Malmstrom EE; Waymouth RM; Hawker CJ Synthesis of poly(olefin) graft copolymers by a combination of metallocene and “living” free radical polymerization techniques. Macromolecules 1998, 31 (13), 4396–4398. [Google Scholar]
- (4).Moad G; Rizzardo E; Thang SH Radical Addition-Fragmentation Chemistry in Polymer Synthesis. Polymer 2008, 49 (5), 1079–1131. [Google Scholar]
- (5).Chiefari J; Chong YKB; Ercole F; Krstina J; Jeffery J; Le TPT; Mayadunne RTA; Meijs GF; Moad CL; Moad G; Rizzardo E; Thang SH Living Free-Radical Polymerization by Reversible Addition-Fragmentation Chain Transfer: the RAFT Process. Macromolecules 1998, 31 (16), 5559–5562. [Google Scholar]
- (6).Destarac M Controlled Radical Polymerization: Industrial Stakes, Obstacles and Achievements. Macromol. React. Eng 2010, 4 (3–4), 165–179. [Google Scholar]
- (7).Moad G; Rizzardo E; Thang SH Living Radical Polymerization by the RAFT Process – a Third Update. Aust. J. Chem 2012, 65 (8), 985–1076. [Google Scholar]
- (8).Matyjaszewski K; Spanswick J Controlled/Living Radical Polymerization. Mater. Today 2005, 8 (3), 26–33. [Google Scholar]
- (9).Perrier S; Takolpuckdee P Macromolecular Design via Reversible Addition-Fragmentation Chain Transfer (RAFT)/Xanthates (MADIX) Polymerization. J. Polym. Sci., Part A: Polym. Chem 2005, 43 (22), 5347–5393. [Google Scholar]
- (10).Altintas O; Josse T; Abbasi M; De Winter J; Trouillet V; Gerbaux P; Wilhelm M; Barner-Kowollik C ATRP-Based Polymers with Modular Ligation Points Under Thermal and Thermomechanical Stress. Polym. Chem 2015, 6 (15), 2854–2868. [Google Scholar]
- (11).Chong YK; Moad G; Rizzardo E; Thang SH Thiocarbonylthio End Group Removal From RAFT-Synthesized Polymers by Radical-Induced Reduction. Macromolecules 2007, 40(13), 4446–4455. [Google Scholar]
- (12).Willcock H; O’Reilly RK End Group Removal and Modification of RAFT Polymers. Polym. Chem 2010, 1 (2), 149–157. [Google Scholar]
- (13).Bibiao J; Jianbo F; Yang Y; Qiang R; Wenyun W; Jianjun H Modification of the Halogen End Groups of Polystyrene Prepared by ATRP. Eur. Polym. J 2006, 42 (1), 179–187. [Google Scholar]
- (14).Coessens V; Matyjaszewski K Dehalogenation of Polymers Prepared by Atom Transfer Radical Polymerization. Macromol. Rapid Commun 1999, 20 (2), 66–70. [Google Scholar]
- (15).Barbey R; Lavanant L; Paripovic D; Schüwer N; Sugnaux C; Tugulu S; Klok H-A Polymer Brushes via Surface-Initiated Controlled Radical Polymerization: Synthesis, Characterization, Properties, and Applications. Chem. Rev 2009, 109 (11), 5437–5527. [DOI] [PubMed] [Google Scholar]
- (16).Zhou X; Liu X; Xie Z; Zheng Z 3D-Patterned Polymer Brush Surfaces. Nanoscale 2011, 3 (12), 4929–4939. [Google Scholar]
- (17).Chen T; Amin I; Jordan R Patterned Polymer Brushes. Chem. Soc. Rev 2012, 41 (8), 3280–3296. [DOI] [PubMed] [Google Scholar]
- (18).Rastogi A; Paik MY; Tanaka M; Ober CK Direct Patterning of Intrinsically Electron Beam Sensitive Polymer Brushes. ACS Nano 2010, 4 (2), 771–780. [DOI] [PubMed] [Google Scholar]
- (19).Tugulu S; Harms M; Fricke M; Volkmer D; Klok H-A Polymer Brushes as Ionotropic Matrices for the Directed Fabrication of Microstructured Calcite Thin Films. Angew. Chem., Int. Ed 2006, 45(44), 7458–7461. [DOI] [PubMed] [Google Scholar]
- (20).Prucker O; Schimmel M; Tovar G; Knoll W; Rühe J Microstructuring of Molecularly Thin Polymer Layers by Photo-lithography. Adv. Mater 1998, 10 (14), 1073–1077. [Google Scholar]
- (21).Bang J; Bae J; Lowenhielm P; Spiessberger C; Given-Beck SA; Russell TP; Hawker CJ Facile routes to patterned surface neutralization layers for block copolymer lithography. Adv. Mater 2007, 19 (24), 4552–4556. [Google Scholar]
- (22).Zhou F; Jiang L; Liu W; Xue Q Fabrication of Chemically Tethered Binary Polymer-Brush Pattern Through Two-Step Surface-Initiated Atomic-Transfer Radical Polymerization. Macromol. Rapid Commun 2004, 25 (23), 1979–1983. [Google Scholar]
- (23).Fors BP; Poelma JE; Menyo MS; Robb MJ; Spokoyny DM; Kramer JW; Waite JH; Hawker CJ Fabrication of Unique Chemical Patterns and Concentration Gradients with Visible Light. J. Am. Chem. Soc 2013, 135 (38), 14106–14109. [DOI] [PubMed] [Google Scholar]
- (24).Pester CW; Poelma JE; Narupai B; Patel SN; Su GM; Mates TE; Luo Y; Ober CK; Hawker CJ; Kramer EJ Ambiguous Anti-Fouling Surfaces: Facile Synthesis by Light-Mediated Radical Polymerization. J. Polym. Sci., Part A: Polym. Chem 2016, 54(2), 253–262. [Google Scholar]
- (25).Discekici EH; Treat NJ; Poelma SO; Mattson KM; Hudson ZM; Luo Y; Hawker CJ; de Alaniz JR A Highly Reducing Metal-Free Photoredox Catalyst: Design and Application in Radical Dehalogenations. Chem. Commun 2015, 51 (58), 11705–11708. [DOI] [PubMed] [Google Scholar]
- (26).Poelma SO; Burnett GL; Discekici EH; Mattson KM; Treat NJ; Luo Y; Hudson ZM; Shankel SL; Clark PG; Kramer JW; Hawker CJ; Read de Alaniz J Chemoselective Radical Dehalogenation and C–C Bond Formation on Aryl Halide Substrates Using Organic Photoredox Catalysts. J. Org. Chem 2016, 81(16), 7155–7160. [DOI] [PubMed] [Google Scholar]
- (27).Discekici EH; Pester CW; Treat NJ; Lawrence J; Mattson KM; Narupai B; Toumayan EP; Luo Y; McGrath AJ; Clark PG; Read de Alaniz J; Hawker CJ Simple Benchtop Approach to Polymer Brush Nanostructures Using Visible-Light-Mediated Metal-Free Atom Transfer Radical Polymerization. ACS Macro Lett. 2016, 5 (2), 258–262. [DOI] [PubMed] [Google Scholar]
- (28).Huang X; Wirth MJ Surface-Initiated Radical Polymerization on Porous Silica. Anal. Chem 1997, 69 (22), 4577–4580. [Google Scholar]
- (29).Pester CW; Narupai B; Mattson KM; Bothman DP; Klinger D; Lee KW; Discekici EH; Hawker CJ Engineering Surfaces Through Sequential Stop-Flow Photopatterning. Adv. Mater 2016, DOI: 10.1002/adma.201602900. [DOI] [PubMed] [Google Scholar]
- (30).Zhou F; Zheng Z; Yu B; Liu W; Huck WTS Multicomponent Polymer Brushes. J. Am. Chem. Soc 2006, 128 (50), 16253–16258. [DOI] [PubMed] [Google Scholar]
- (31).Chen T; Jordan R; Zauscher S Extending Micro-Contact Printing for Patterning Complex Polymer Brush Microstructures. Polymer 2011, 52 (12), 2461–2467. [Google Scholar]
- (32).Balachander N; Sukenik CN Monolayer Transformation by Nucleophilic Substitution: Applications to the Creation of New Monolayer Assemblies. Langmuir 1990, 6 (11), 1621–1627. [Google Scholar]
- (33).Fors BP; Hawker CJ Control of a Living Radical Polymerization of Methacrylates by Light. Angew. Chem., Int. Ed 2012, 51 (35), 8850–8853. [DOI] [PubMed] [Google Scholar]
- (34).Poelma JE; Fors BP; Meyers GF; Kramer JW; Hawker CJ Fabrication of Complex Three-Dimensional Polymer Brush Nanostructures Through Light-Mediated Living Radical Polymerization. Angew. Chem., Int. Ed 2013, 52 (27), 6844–6848. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.