

Abstract
The present study addresses existing data on the affinity and conjugation of sulfhydryl (thiol; -SH) groups of low- and high-molecular-weight biological ligands with mercury (Hg). The consequences of these interactions with special emphasis on pathways of Hg toxicity are highlighted. Cysteine (Cys) is considered the primary target of Hg, and link its sensitivity with thiol groups and cellular damage. In vivo, Hg complexes play a key role in Hg metabolism. Due to the increased affinity of Hg to SH groups in Cys residues, glutathione (GSH) is reactive. The geometry of Hg(II) glutathionates is less understood than that with Cys. Both Cys and GSH Hg-conjugates are important in Hg transport. The binding of Hg to Cys mediates multiple toxic effects of Hg, especially inhibitory effects on enzymes and other proteins that contain free Cys residues. In blood plasma, albumin is the main Hg-binding (Hg2+, CH3Hg+, C2H5Hg+, C6H5Hg+) protein. At the Cys34 residue, Hg2+ binds to albumin, whereas other metals likely are bound at the N-terminal site and multi-metal binding sites. In addition to albumin, Hg binds to multiple Cys-containing enzymes (including manganese-superoxide dismutase (Mn-SOD), arginase I, sorbitol dehydrogenase, and δ-aminolevulinate dehydratase, etc.) involved in multiple processes. The affinity of Hg for thiol groups may also underlie the pathways of Hg toxicity. In particular, Hg-SH may contribute to apoptosis modulation by interfering with Akt/CREB, Keap1/Nrf2, NF-κB, and mitochondrial pathways. Mercury-induced oxidative stress may ensue from Cys-Hg binding and inhibition of Mn-SOD (Cys196), thioredoxin reductase (TrxR) (Cys497) activity, as well as limiting GSH (GS-HgCH3) and Trx (Cys32, 35, 62, 65, 73) availability. Moreover, Hg-thiol interaction also is crucial in the neurotoxicity of Hg by modulating the cytoskeleton and neuronal receptors, to name a few. However, existing data on the role of Hg-SH binding in the Hg toxicity remains poorly defined. Therefore, more research is needed to understand better the role of Hg-thiol binding in the molecular pathways of Hg toxicology and the critical role of thiols to counteract negative effects of Hg overload.
Keywords: Mercury, Cysteine, Apoptosis, S-mercuration, Conjugates
1. Introduction
World Health Organization (WHO) has listed mercury (Hg) as one of the ten pollutants of particular concern for public health [1]. Despite the Minamata Convention agreement on Hg emission reduction, human exposure, and its adverse health effects persist [2], with global Hg emissions increasing by 1.8%/year in 2010–2015 [3]. Southeastern Asia is considered the main contributor to Hg emissions [4]. Small-scale gold mining and artisanal work represent a key source of anthropogenic Hg emissions accounting for 37% of all Hg emitted into the environment. Other significant (>10%) sources of Hg emission include coal and fossil fuel combustion (25%) and non-ferrous metal production (10%) [5]. Wildfires also have a critical effect on the global Hg emissions [6]. Although the projections for 2050 demonstrate a sustained trend for increased Hg emissions, it is hopeful that legislative and technological efforts will reverse and abate this trend [7].
Adverse health effects at current Hg levels of exposure vary related to the geographic area [8], with arctic and tropical communities being at particularly high risk of Hg overexposure [9]. Mercury toxicity may aggravate neurological and neurodegenerative [10], metabolic [11], renal [12], and cardiovascular diseases [13]. For elemental Hg, the brain and kidneys are considered as the main targets [14]. The overall costs of current adverse Hg effects have been estimated to be in the range of 23 thousand to 52 thousand EUR/kg predominantly due to cardiovascular morbidity and mortality [15].
Multiple mechanisms of Hg toxicity underlie the wide spectrum of detrimental health effects due to Hg exposure. Particularly, Hg was shown to cause enzyme inactivation [16], oxidative stress [17], inflammation, and autoimmunity [18]. However, the particular molecular mechanisms have yet not been fully disclosed.
The high affinity of Hg-compounds to sulfhydryl (–SH) groups has been reported in numerous studies [19]. For example, a detailed proteomic analysis of murine liver revealed 7682 Hg-reactive cysteine (Cys) residues from 5664 proteins [20]. However, data on a particular involvement of Hg-thiol interaction in mechanisms of Hg toxicity are still insufficient.
Therefore, we discuss in this review the present data on affinity and conjugation of SH groups of low- and high-molecular-weight biological ligands with Hg, as well as the outcome of such interactions with a special emphasis on pathways of Hg toxicity.
2. Forms of mercury
Exposure to Hg occurs in different forms, in principle, either as elemental (Hg0), inorganic, or organic Hgn+. Metallic or elemental Hg (Hg0) as a liquid metal has low absorption by dermal and gastrointestinal routes [21,22]. Although it has a high vapor pressure, and at saturation, a cubic meter of air holds 20 mg Hg at room temperature. Upon inhalation, this colorless, odorless vapor is highly toxic [23]. Mercury vapor, which also is released from dental amalgams, is absorbed by the lungs and mucous membranes [22,24]. Mercury vapor is a monatomic gas, which is lipophilic and highly diffusible. It can pass through biological barriers, including the blood–brain barrier (BBB), the placenta, and cell membranes [21]. Also, proteins for the transport of small uncharged molecules may be involved [22]. Each Hg form has a particular type of toxicity. The specific toxicity is not only dependent upon the chemical form but is mainly associated with specific metabolic and distribution fractions. In inorganic salts, Hg can occur in two oxidation states: +1(I) for the mercurous cation; and +2(II) for the mercuric cation [22]. The transitory cation is known as mercurous form, which easily dissociates into Hg0 and Hg2+ [22,25]. Mercurous chloride (Hg(I) chloride; Cl-Hg-Hg-Cl), described as calomel, was a component in teething powders until about 1950 when it was recognized to cause acrodynia and was also used as a medicinal purgative and diuretic during the 17th and 18th centuries [26,27]. The end-product of the full oxidation of Hg is the mercuric form with a +2-oxidation state. Hg(II) may exist both as inorganic Hg2+ salt and as organomercurials, including the widespread compounds methylmercury (MeHg, CH3Hg+), dimethylmercury ((CH3)2Hg+), ethylmercury (EtHg, C2H5Hg+), and phenylmercury (C6H5Hg+) [28]. The capabilities of these mercurials to bind amino acids, peptides, and proteins result in a high number of Hg species in the living organisms. Mercury(II)-compounds are the most reactive forms of Hg and are related to different toxic effects [23]. Although certain studies demonstrate lipophilicity of MeHg, the complexes MeHg-S-R molecules are much less lipophilic. The compounds of organic Hg are those in which Hg2+ (highly reactive diva-lent) is covalently linked to carbon. The main source of EtHg (H3CH2C-Hg+ X−, in which X is often Cys) is thiomersal, which is used as a vaccine preservative. Methylmercury (H3C-Hg+ X) is found in dietary fish. The proposed pathways for MeHg transportation may include mimicking the amino acid, methionine, utilizing the large neutral amino acid transporter (LAT1) on the cell membrane [22]. As regards thimerosal, the EtHg moiety may gain intracellular access in exchange reactions with endogenous thiols such as glutathione (GSH). In contrast to Hg0, which is characterized by high lipophilicity, lipophilic diffusion across biological membranes is unlikely to be a significant mechanism of MeHg transport in vivo. At normal physiological pH in gut and blood (near 6.8–7.4), the vast majority of MeHg is expected to be bound to thiol groups. Methylmercury can cross the BBB, although less effective in comparison with elemental Hg vapor, but it is readily absorbed via the gut, and it accumulates in various tissues [24]. With their high reactivity and high mobility, organic compounds of Hg are highly neurotoxic [22]. Special attention should be paid to toxicokinetics of thiomersal (EtHg) yet recognizing that it may be quite different from that observed for MeHg, reflective of differential mechanisms of toxicity, including the particular patterns of thiol group binding [29].
In addition to different physicochemical properties, organic species and inorganic Hg(II) species are characterized by different bioavailability. Particularly, comparative analysis of MeHg and Hg(II) absorption rates have reported that the overall absorption of the studied Hg compounds in humans was estimated as 12%–79% and 49%–69%, respectively [30]. Although earlier estimated to be about 70 days, the human body half-life (t1/2) of MeHg has later been found to vary between 42.5 and 128.3 days in healthy volunteers. The shorter estimated value for t1/2 (about 40 days) and faster MeHg elimination was associated with complete demethylation as assessed by fecal Hg content [31].
Following exposure, the ions of Hg are absorbed and aggravated in different organs such as the placenta, brain, liver, kidneys, and intestine [32]. Notably, the accumulation patterns, as well as the target organs, are partially different in primates compared with rodents [33]. Among various exposure routes, humans can be exposed to high Hg levels through the ingestion of organic Hg forms in contaminated food (i.e., MeHg+). Regarding sea-food, high MeHg+ levels are found in the muscle of large predatory saltwater and freshwater fish, including pike, salmon, shark swordfish, and tuna [21]. The accumulation of Hg in these tissues is related primarily to their inability to efficiently eliminate Hg, their ingestion of other contaminated fish, and their longevity in contaminated waters [34]. After the contaminated food ingestion, MeHg+ can be absorbed easily through the gastrointestinal tract and transported to various organs. However, it is critical to evaluate the chemical properties and metabolism of mercuric ions along the pathways in view of the importance of correct Hg speciation in vivo.
3. Sulfhydryl groups and mercury
It has been reported that sulfhydryl (SH) is ubiquitous in peptides and proteins throughout the body. Molecules with SH groups are referred to as thiols or mercaptans (from Lat. mercurium captans, meaning ‘capturing mercury’) [35]. Proteins containing Cys residues are abundant throughout the body, both in enzymes, organelles, and in intracellular and extracellular membranes. Since most SH groups are important for the function or structure of numerous proteins [36], the particular goals for Hg, have still to be fully recognized. Many of the sulfhydryl targets are non-important. Therefore, restricted duration with low-content exposures to Hg may induce at least toxicity. The Hg binding affinity to sulfhydryl is higher in comparison with that of cadmium (Cd), arsenic (As), and lead (Pb) [37], being indicative of the higher role of thiol reactivity in Hg toxicity.
Furthermore, an Hg-S- bond is not necessarily stable in vivo [19], as the Hg-ion may move readily between free –SH groups according to their availability and reactivity. The process is described by Rabenstein’s ligand exchange reaction [38]. Particularly, the exchange of R-Hg-S-R1 with another R2-SH is extremely rapid (controlled by diffusion, first-order reaction in the order of 108 M−1 sec−1) and normally occurs when the free thiol group (R2-SH), which will attack the -Hg-S- bond, has similar nucleophilicity to the –SH group that is participating in the R1-S-Hg-bond (Fig. 1).
Fig. 1.
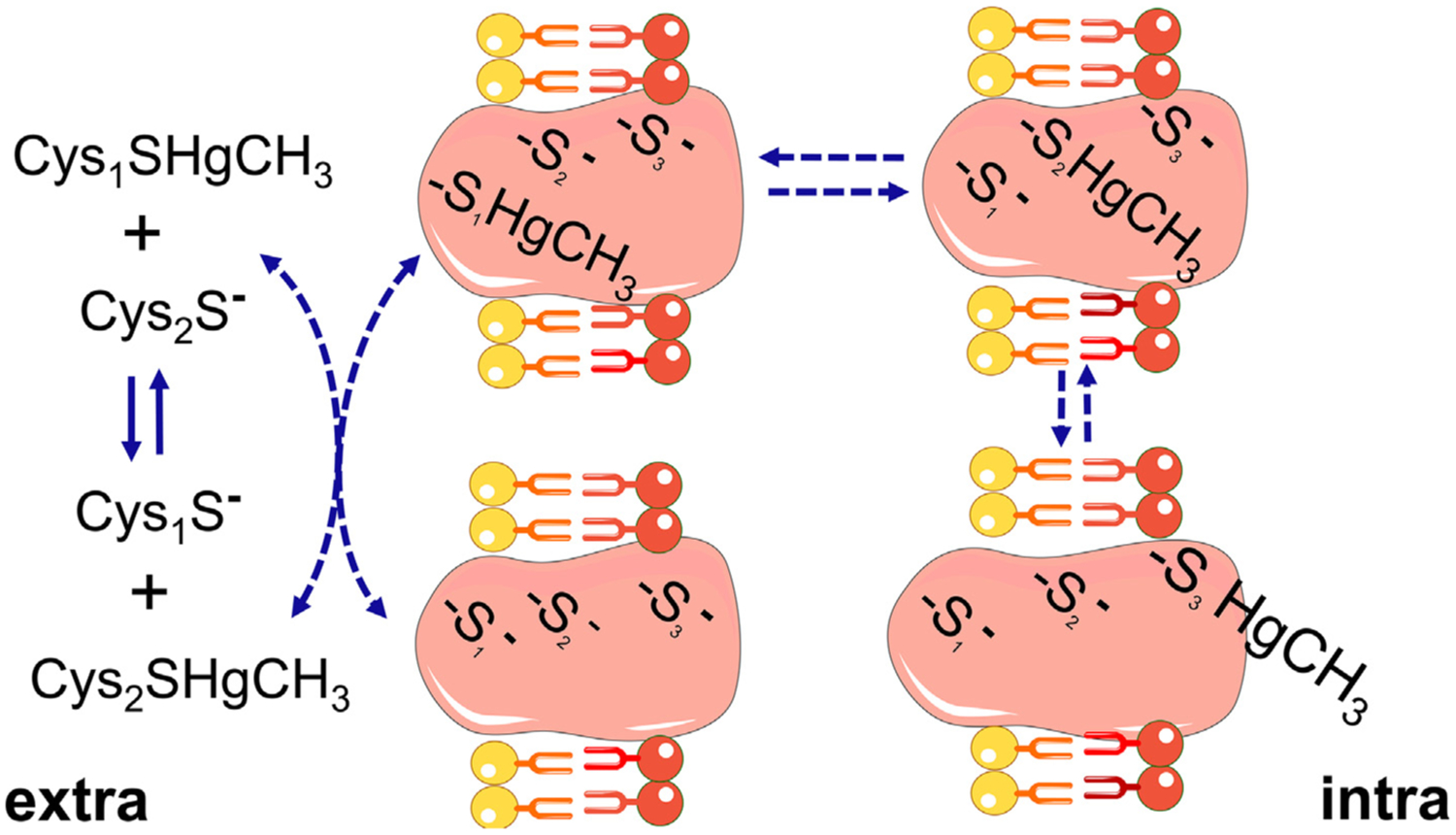
Movement of methylmercury (MeHg) from one thiol to other thiol groups. The exchange of MeHg from one thiol to other of similar reactivity can occur between low molecular mass thiols (LMM-SH, represented by to free cysteine (Cys) molecules, Cys1-SH or Cys2-SH) or between LMM-SH and thiol-containing proteins (HMM-SH; in the figure the integral transmembrane protein has three thiol groups located in different topological places of the membrane. The first thiol or thiolate (–S1−) is accessible to the extracellular space, which allows this group to exchange with the complex Cys2-S-HgMe. The complex protein-S1-HgMe can then exchange with a more buried free thiol group (–S2−) to form the protein-S2-HgMe complex. The second complex can react with a third thiol/thiolate group (–S3−) facing the intracellular surface of the membrane. The complex protein-S3-HgMe can exchange either with thiol-containing proteins or with cysteine or reduced glutathione molecules found inside the cell. The scheme is based on the exchange reactions of MeHg and thiol groups first described by Rabenstein et al. The migration of MeHg from the extracellular to the intracellular side of the membrane is based on indirect experimental data [74,79], but can explain additional mechanisms of MeHg transport other than those carried out by the LAT-1 transporter [132,152,234].
Shortly after exposure to Hg0 vapor, most of the Hg is converted to mercuric ions in the blood. And Hg2+ in plasma likely binds to SH-containing proteins such as albumin [39]. Notably, the Hg content of blood rapidly decreases after Hg exposure, indicating that Hg ions are readily being absorbed by the cells. So far, evidence suggests that Hg–albumin complexes do not appear to be absorbed readily by target cells, such as proximal tubular cells [40]. Apparently, the binding of Hg to plasma proteins (e.g., Hg–albumin) is reversible and less relevant in the presence of other (small) low molecular weight thiols. This allows Hg ions to conjugate with numerous non-protein SH-containing metabolites, that may form transportable mercuric complexes being taken up more quickly into target cells [41].
Circulating Hg is known to be protein-bound, whereas low-molecular-weight transporters interact only with a small fraction of the Hg ions [42]. It is known that Hg predominantly binds inorganic and organic S (HS−, sulfide or S2−, and thiol/thiolate) and Se (HSe−, selenide or Se2−, and selenol/selenolate) compounds, being soft nucleophiles in agreement with Pearson’s acid-base theory. In turn, electrophilic forms of S or Se (in the +4 or +6 redox state) do not have a high affinity for Hg.
It was with speciation analysis of Hg in blood proteins using MALDI-TOF-MS found that Hg is bound mainly to albumin and metallothionein (MT) in MeHg-exposed rats treated with Se (as sodium selenite) [43]. However, the reported molecular weight values raise the possibility of other species being involved. In corroboration with this, a recent detailed study by Li et al. (2018) demonstrated that hemoglobin, glutathione peroxidase 3 (GPX3), selenoprotein P (SELENOP), as well as albumin, are major Hg-binding molecules following Hg2+ exposure both in vivo (139.7–778.4 mg/L HgCl2) and in vitro (100–1000 mg/L Hg) [43]. In the case of GPX3 and SELENOP, Hg binding may occur through selenocysteine residues. At increasing HgCl2 concentrations in vitro to 10 mg/L, ApoA-I, ApoE, ApoA-IV, and transferrin were also found to bind Hg [43]. Cysteine, selenocysteine (found exclusively in the active center of selenoproteins), glutathione (GSH), albumin, hemoglobin, as well as MT are major binding sites for Hg in vivo. All the ligands mentioned above, except Hb and MT, bind both Hg2+ and CH3Hg+ through Cys thiols (–SH) or selenol groups (–SeH) of selenocysteine in selenoproteins [44].
However, predicting Hg species behavior in vivo must be pursued with caution. Feroci et al. (2005) stated that the processes of Hg complex formation are significantly affected by chemical constituents of biological fluids [45]. Particularly, in physiological medium rich in chloride ions, Hg cannot exist as simple Hg2+ cations, but the dominating species are [HgCl4]2− compounds [46], whereas in the presence of free thiols Hg predominantly binds –SH groups even despite the higher selenol group reactivity [47].
Mercury is bound to biological ligands through sulfur with an oxidation state of −1 and −2. In the majority of cases, these sites are thiol groups of Cys residue as a part of a more complex structure. The affinity of Hg to amino acids is relatively well studied. The outcomes of such investigations have been applied in the improvement of heavy metal chelators for detoxification therapy [48].
These data provide a basis for understanding the binding and transport kinetics of Hg by more complex molecules. Divalent Hg ions have a high affinity to reduced mercapto-groups. Mercury(II) complexes with specific low-molecular-weight ligands determine Hg distribution in the organism. Unfortunately, due to methodological limitations, the reported constants for complexes of Hg with biological thiols are rather contradictory [49]. The latter significantly affects the evaluation of newly formed Hg(II) species and their pathways of transformation.
3.1. Cysteine
Cysteine, one of the most ubiquitous and biochemically active amino acids in the body, is an important active site for functional proteins such as receptors, signaling molecules, enzymes, membrane transport channels, and transcription factors [50]. Cysteine is considered as the primary target for Hg, providing a link between its reactivity with thiol groups and cellular damage.
In Hg metabolism in vivo, Hg-S-Cys complexes play a key role. In parallel with Hg(HL)2 and HgL2 forms (L–ligand (Cys), HL–protonated ligand (HCys-)), Hg and Cys may form HgL3, HgL4, Hg2L2, Hg2(HL)LCl, Hg3L2Cl6 species [51–53]. The HgL3 and HgL4 species are prevalent in solutions at a M:L ratio of 1:5 [53]. The simultaneous production of species with 1:1 composition is still questionable. Hg(HL)2 and HgL2 complexes are linear, although at M:L ratio different from 1:2, the formation of species with linear, as well as tri- and tetragonal geometry, may occur [54]. It is also notable that chloride ions may also participate in the formation of Hg-Cys species, including Hg2L2HCl, Hg3L2Cl2, Hg3L2Cl6 [55].
The stability of Hg-Cys complexes is rather high. According to Stary and Kratzer (1988), stability constant of [HgHL2]0 complex is estimated as 40, whereas that for [HgL2]2− is 42.7 (H2L = NH2-CH(CH2SH)-COOH) [56]. Mercury(II) cysteinates are more stable than similar complexes of Cys with Zn(II), Pb(II), or Cd(II), but is less stable than copper (II) cysteinates [57]. Lenz and Martell (1964) indicate that stability constants for metal complexes with S-methylcysteine are estimated as 7.88 for Cu(II), 7.20 for Hg(II), 4.43 for Pb(II), 4.46 for Zn(II), and 3.77 for Cd(II) [57]. According to Berthon [58], the β values for M:L 1:1 compounds vary depending on the metal, being estimated as 14.21–37.8 for Hg2+, 11.39–13.58 for Pb2+, 6.45–12.88 for Cd2+, 8.06–9.87 for Zn2+, and 11.38–19.19 for Cu+. The respective values for M:L 1:2 complexes are estimated as 41.20–44.0, 15.90–19.30, 14.20–21.72, 17.54–19.39, and 16.0, respectively. High variability of the constants may be related to different methods of evaluation, as well as differences in background electrolyte characteristics and temperature (20 and 25 °C). In this particular case, the stability of Hg cysteinates is significantly higher than that for Cd, Pb, Zn, and even Cu, being in contrast with the earlier data by Lenz and Martell [57]. However, it should be mentioned that Cu(II) ions are bound to Cu2Ln complexes (β = 14.00–45.65) with different protonation degree. Therefore, one cannot definitely determine which cation will predominantly bind the ligand in vivo [58], though in vitro Hg2+ typically displace Cu2+, Cd2+ and Pb2+ from thiol-containing ligands [59].
Although the exact electronic and atomic properties determining the higher affinity of Hg2+ for thiols than Cu2+, Pb2+ and Cd2+ are not known, the highest softness of Hg in relation the other elements can explain the general trend of affinity (Hg(II) > Cu(II) > Pb(II) [60].
Reid and Rabenstein stated that similar to Hg2+, MeHg also form complexes of linear geometry with Cys in a wide interval of pH (<1 to >13). The stability of MeHg cysteinates (CH3HgHL, CH3HgL) is much lower than that of similar Hg2+ complexes with β = 15.38–15.56 and 16.45–16.58 according to Lenz and Martell (1964) [57] or β = 17.41 and 26.72 according to Cardiano et al. (2011) [61], respectively. The latter may be explained by the lack of complex formation with a M:L ratio of 1:2 in contrast to mercuric ions. Other metallorganic Hg compounds (EtHg) also possess the ability of 1:1 ratio complex formation with Cys [62].
Mercury(II) ion binds amino acid through donor atom S of thiol groups and does not interact with oxygen atoms from COOH groups [55]. In this particular case, Cys is not a chelator, as it provides only one coordination bond. Such behavior is atypical for amino acids and may be explained by the low affinity of Hg atoms to N− or especially O-donor centers (Fig. 2). At the same time, while studying L-Cysteinato(methyl)mercury(II) monohydrate using X-ray structure analysis, Taylor et al. (1975) noted weak intramolecular interactions between Hg and oxygen atoms of the carboxyl group [63]. These findings are corroborated by the study by Liem-Nguyen et al. (2017) [49]. These authors have performed density functional theory (DFT) calculations demonstrating that electron-donor carboxyl and carbonyl groups have a stabilizing effect on Hg(II)-LMM thiol complexes. At the same time, electron-acceptor protonated primary amino-groups possess a destabilizing effect [49]. In turn, data obtained by Mah and Jalilehvand (2008) have demonstrated that in alkaline solutions deprotonated amino-group of Cys takes part in the formation of [HgL2]2− chelate, as evidenced by a reduced moiety of complexes with high L:M ratio [64].
Fig. 2.
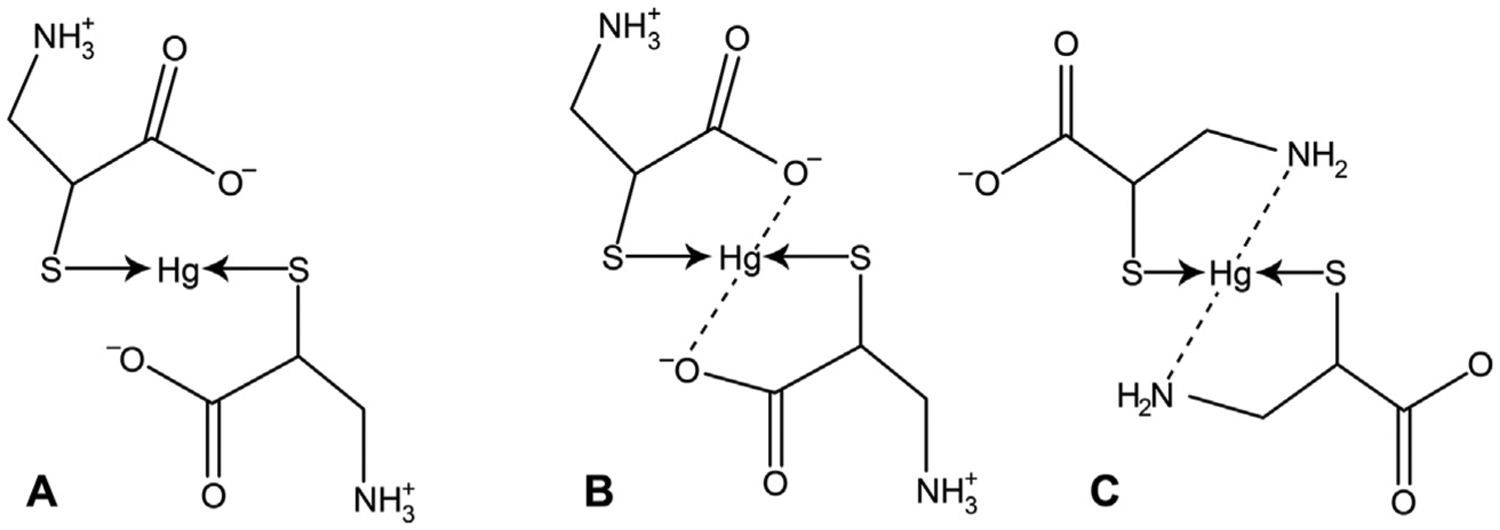
The proposed coordination of mercury(II) ion with cysteine molecules. A – Non-chelate complex formed using only sulfur atoms; B – The role of sulfur and oxygen atoms in metal–ligand coordination; C – The potential involvement of sulfur and nitrogen atoms in coordination between Hg2+ and cysteine molecules.
Complex formation between Cys and Hg(I) is less studied than that for Hg(II). Particularly, the formation of Hg2L2-type Hg (I) cysteinate is postulated by Heyrovský et al. (1997) [65]. Addition of two Cys residues to such a complex result in changes of Hg oxidation state from +1 to +2 ([Hg2L2] + 2HL = 2[HgL2] + 2e− + 2H+).
Given the significant role of Cys in the structure and functioning of biomolecules, Hg binding to Cys mediates multiple toxic effects of Hg, especially its inhibitory effects on enzymes and other proteins containing free Cys residues. The biological outcome of such interaction-based only on data on direct Hg-Cys interaction will be reviewed further.
3.2. Glutathione
Glutathione (GSH) is known as the most common compound with low molecular weight and SH groups in mammalian cells, which is present in millimolar amounts in different cells [66]. The reactivity of GSH with Hg is related to Hg’s affinity for the SH group of the Cys residue.
The reactivity between Hg(II) and GSH ((2S)-2-Amino-4-{[(1R)-1-[(carboxymethyl)carbamoyl]-2-sulfanylethyl]carbamoyl}buta noic acid) was studied by several researchers [67–71]. Stoichiometric analysis and formation constant assessment performed in these studies demonstrated that depending on metal and ligand concentration, the M:L particles of 1:1, 1:2. 1:3, 1:4, 2:2, 3:2 composition with different protonation may occur. According to Burford et al. (2005) based on the GSH-binding properties, all metals may be divided into two groups: i) metals capable of binding more than one GSH molecules (Hg, As, Sb, and Bi) with the formation of GSH rich complexes; ii) metals capable of binding only one GSH molecule (metal-rich complexes) [72]. The formation of 1:2 and 1:3 complexes of Hg(II) and GSH was demonstrated by Mah and Jalilehvand (2008), who revealed a predominance of the diagonal [HgL2]4− and trigonal [HgL3]7− particles in the solution [64]. The complex of [HgL4]10− composition is present in solutions with excess GSH, although its moiety does not exceed 30% [64].
The geometry of Hg(II) glutathionates is less clear than that in the case of Cys. Particularly, Fuhr and Rabenstein (1973), using the 13C NMR method, demonstrated that in a 1:2 Hg-Cys complex, Hg(II) atoms are coordinated with GSH only through S atoms of Cys residues.[71] The same type of Hg(II) binding has been mentioned by Krężel and Bal (1999) [73]. Complexes of the linear geometry are formed during the formation of -S-Hg-S or CH3Hg-S-bonds [71,74].
Other studies stipulate that interaction between Hg(II) and GSH may result in chelate formation. Neville and Drakenburg (1974) have demonstrated that in Hg-GSH, complex metal coordination occurs through the S atom of the mercapto-group as well as nitrogen atom of glycine residue [70]. However, other authors have proposed that donor atoms of the chelate ring are SH-group S atom as well as carbonyl group oxygen atom of the Cys residue. Hypothetically, the particular type of coordination depends on metal:ligand ratio. Particularly, under conditions of two- or three-fold excess of GSH, Hg(II) binding occurs through S atoms [47], whereas, at equimolar conditions, an additional metal–ligand bond is formed through oxygen or nitrogen atoms. This hypothesis generally corroborates earlier observations on the ability of Hg to form complexes with rather low coordination numbers (2, 3, 4).
The constants formation is especially important when considering the particular features of Hg(II) mobilization and distribution in biological systems. GSH binds Hg(II) at pH of 1–12 forming strong complexes with Hg2+ and CH3Hg+ [75], is more stable than Cd(II), Zn(II), Pb(II), or Sn(IV) glutathionates. The stability constants of M:L 1:1 complexes with different protonation degree were earlier estimated as β(HgHL) = 32.49; β(CH3HgHL) = 25.24; β(PbHL) = 17.14; β(ZnHL) = 14.76; β(CdHL) = 17.02; β((CH3)3SnL) = 14.17; β(HgL) = 35.68; β(CH3HgL) = 15.99; β(PbL) = 10.57; β(ZnL) = 8.57; β(CdL) = 10.18 [73]. Cheesman et al. (1988) proposed that despite the high stability of Hg(II)-GSH complexes, the bonds between Hg(II) and SH groups are rather labile [47]. These data are at least partly in agreement with Shoukry et al. (1988) [76]. The latter used polarimetric analysis to confirm the data obtained by 13C NMR. The values of 1:3 M:L complex formation from Hg(SG)2 and GSH reported that the third ligand is bound more weakly than the first two. Oram et al. (1996) proposed that the stability of Hg(II)-GSH complexes may be overestimated [67]. Based on the first three ionization constants of GSH the authors provided the following production constants for Hg(II)-GSH complex: 15.80 for [HgH3SG]4+, 26.04 for [HgH3L]3+, 32.49 for [HgH2L]2+, 35.68 for [HgHL]+, 33.40 for [Hg(H3L)2]4+, 42.40 for [Hg(H2L)(H3L)]3+, 52.29 for [Hg(H2L)2]2+, and 55.28 for [Hg(HL)(H2SL)]+. In contrast, the earlier data reported by Stricks and Kolthoff (1953) who demonstrated higher values for complexes of the same composition (41.58 for [Hg(H3L)2]4+, 41.92 for [Hg(H2L)(H3L)]3+, 40.96 for [Hg(H2L)2]2+) (L-GSH) [68]. The observed inconsistencies in the data may relate to different conditions of Hg(II)-GSH stability assessment. It is proposed that in the study by Stricks and Kolthoff (1953), the potential of Hg electrode in relation to the calomel reference electrode was affected not only by Hg2+ but also other electrolytes being present in the solution [67].
In contrast to Hg(II)’s interaction with GSH, the interaction of other forms of Hg, including Hg(I) and CH3Hg2+, has been insufficiently studied. The existing data demonstrate the ability of MeHg to form coordinate compounds with GSH with the composition of 1:1, 1:2, 1:3, and different protonation [77,78]. According to Rabenstein (1978), the formation constant of a completely deprotonated compound is 15.9 [79]. The formation constants for [CH3-HgH3L]2+, [CH3HgH2L]+, [CH3HgHL]0 are estimated as 28.68, 25.24, and 15.99, is an order of magnitude less than the respective complexes of Hg2+ except for completely protonated form [80]. However, the stability constants for MeHg-GSH complexes is much higher than that for similar Pb(II), Cd(II), and Zn(II) compounds. As in the case of Hg2+, MeHg interacts with GSH at a wide pH interval (0.5–13). Onyido et al. (2004) demonstrate that in an acidic medium CH3Hg+ ion forms with GSH, a particle of 2:1 composition due to the interaction of MeHg cation with mercapto-group [77]. Increasing pH results in the gradual cleavage of CH3Hg+ with subsequent formation of CH3HgOH in the alkaline medium (pH > 10). Despite a lack of data on Hg(I)-GSH complex formation, Miller and Woods (1993) have shown direct interaction between monovalent Hg and oxidized glutathione (GSSG) [81].
In contrast to Cys and other low-molecular-weight ligands, GSH is characterized by the ability to bind several different metals (heteronuclear complexes) due to the presence of various functional groups, as well as conformational flexibility of the molecule. Investigation of complex formation in the systems Hg2+-Mz+-GSH (M = Co, Ni) demonstrated the formation of HgML2 compounds where Hg binding (coordination number = 2) occurs through SH-groups of two different molecules, whereas Co2+ and Ni2+ (coordination number = 4) ions are bound through amino- and carboxyl-groups [82].
Multiple studies have reported that Hg exposure is associated with GSH dysregulation due to the protective role of the latter. Specifically, prenatal Hg exposure was shown to reduce cerebral GSH levels in mice [83]. GSH depletion has been shown to increase MeHg accumulation in both neurons and astrocytes [84]. It is also noteworthy that the earlier cloned Hg-resistant cell line is characterized by five times higher GSH levels in these cells in comparison to the original parent cells [85].
However, it is unclear whether these effects of physiological GSH-Hg antagonism may be mediated through direct Hg-thiol interaction. In view of the findings of a detailed study by Hansen et al. (2006) demonstrating lower susceptibility of GSH to Hg-induced oxidation in comparison to Trx1 and Trx2 [86], one can propose that protective effect of GSH in Hg toxicity may be more directly related to its antioxidant effect rather than Hg binding to -SH group.
3.3. Proteins
Proteins contribute significantly to total plasma thiol levels, with albumin being the most abundant plasma thiol donor [87,88]. Metallothionein is considered as one of the most cysteine-rich proteins [89]. Other cysteine-rich proteins (>5% cysteine residues) include representatives of 21 protein families, including hepcidin, G-protein-coupled receptors-targeting proteins (e.g., oxytocin, somatostatin, interleukin-8), enzyme-coupled receptor-targeting proteins (e.g., insulin-like growth factor, epidermal growth factor), extracellular enzyme inhibitors, antimicrobial peptides [90].
The ability of particular proteins to bind Hg is dependent on the presence of cysteine residues. Therefore, we propose that the ability of protein molecules to Hg binding should be discussed in view of the structural similarity of particular Hg-binding sites. The geometry of Hg-cysteinates is usually linear, corresponding to 1:1 and 1:2 composition of the compounds. Trigonal or tetrahedral geometry is observed only in three-fold and higher excess of cysteine. Given the rather high stability of 1:1 complexes, as well as non-chelate binding of the metal ion by amino acid residues, a spatial approximation of cysteine residues in the Hg-binding center is not required. Notably, the Hg-binging center in albumin molecule contains only one cysteine residue. At the same time, binding from 7 to 12 Hg(II) cations by metallothionein containing 20 cysteine residues is also indicative of linear geometry. The particular characteristics of Hg binding to albumin and metallothionein molecules are discussed lower.
3.3.1. Albumin
Albumin is considered as the most important metal transporter in the blood [91]. In blood plasma, albumin accounts for 90% of all protein-bound Hg in the form of Hg2+, CH3Hg+, C2H5Hg+, C6H5Hg [92,93].
Li et al. (2007) estimated the stoichiometry and stability of albumin complexes with various forms of Hg, demonstrating that in the systems M – albumin (M = Hg2+, CH3Hg+, C2H5Hg+, C6H5Hg+) metal to ligand ratio is estimated as 6:1, 4:1, 4:1, and 3:1, respectively [93]. Binding of inorganic and organic Hg occurred at two different sites, although such a high ratio may be explained by a non-specific Hg binding to albumin after Cys34 saturation [17]. According to Simpson (1961) and Yasutake et al. (1990), the stability constants for CH3Hg+ - HSA systems are estimated at 12–17 [94,95]. Although these values are 2–4 orders of magnitude lower as compared to the complexes with Cys and GSH, albumin-bound Hg is the predominant form in the blood. The latter may be explained by a higher concentration of albumin in blood in comparison to other S-donor ligands (free Cys, etc.).
Albumin forms with Hg2+ also mixed-ligand complexes with Cys at a ratio of two Cys molecules per one Hg-HSA site. Constant stability for such complex is estimated as 9.05 [96]. This complex is suggested to play an important role in the transport of Hg(II) between different tissues and blood.
Human serum albumin (HSA) contains four metal-binding sites characterized by different structures and chemical nature of the metals bound [97].
The N-terminal site (NTS) is formed by the first three amino acid residues of aspartic acid, alanine, and histidine. Metal-binding is carried out by histidine residue, terminal amino group, and two nitrogen atoms of the polypeptide chain, resulting in the coordination of metals forming square complexes (e.g., Cu(II)) [98]. Varshney et al. (2010) noted the ability of Hg to interact with the N-terminal site, although the authors fail to provide any data on the stability or structure of the formed complex [99].
Metal-albumin coordination is achieved by the Cys34 residue and the surrounding structures. The majority of authors have postulated that Cys34 is the main Hg-binding site on the albumin molecule [97,100]. The Cys34 in the IA subdomain does not take part in disulfide bond formation, and the SH group of the Cys residue binds Hg2+ ions and other heavy metals [91]. In general, albumin contains 35 Cys residues. However, 34 of them form 17 disulfide bridges joining the α-helixes and forming protein domains and subdomains [101]. The only Cys residues not taking part in -S-S- bridge formation and containing free mercapto-group is located at position 34 (Cys34). The same -SH group is prone to dimerization, interaction with GSH, as well as metal binding. According to a number of studies, Cys34 is located in the cleft between helixes 2 and 3 of the IA subdomain [102,103]. Although Asp38 and His39 are also located in the region, these amino acid residues do not take part in metal ion coordination due to their location [97]. Therefore, this site is specific for metal ions that may effectively bind a protein through a single S bond secondary to high enthalpy. Using fluorescence and UV–VIS spectroscopy Santos et al. (2018) studied the geometry of albumin complexes with thiomersal (ethyl(2-mercaptobenzoato-(2-)-O,S) mercurate (1-) sodium) [104]. Binding thimerosal causes significant structural alterations of the polypeptide chain characterized by an increased proportion of α-helix from 43.9 to 47.8% [105]. The binding mechanism proposed by the authors includes binding of thiomersal to albumin via the free Cys34 residue, resulting in BSA-HgC2H5 adduct formation. The process is accompanied by thiosalicylic acid (C7H6O2S) release that electrostatically interacts with amino acids of the modified protein side-chain.
The third site of metal binding is the A site, also known as a multi-metal binding site (MBS). MBS plays a key role in Cd2+ and Zn2+ binding [106,107], although it may also take part in the transport of Ni(II) and Cu(II) ions. NMR studies of the structure demonstrated that metal binding is determined by the nitrogen atoms of His67 and His247, as well as Asp249 and Asn99. Metal-binding also occurs at site B, showing an affinity for Cd(II) and Mn(II), although its particular location and configuration have yet to be estimated [91]. Based on the role of nitrogen atoms as electron donors in sites A and B, it can be proposed that they do not significantly contribute to Hg binding.
Although the other roles of Hg-HSA conjugates apart from transport have yet to be clarified, studies these compounds may have a direct impact on Hg toxicity. Particularly, a U-shaped response to MeHg-albumin conjugate (MeHg-HSA) was noted in N9 microglial cells. Low MeHg-HAS treatment resulted in increased cell viability and proliferation as well as reduced expression of proinflammatory TNFα and IL1β, whereas high-concentrations caused the opposite effects. ERK/MAPKs and STAT3 pathway activation appear to underly both effects [108]. In addition, exposure to MeHg resulted in a remarkable elevation of MeHg-HSA adduct transport from plasma to CSF preceding brain damage, suggestive of efficient mechanisms for Hg transport into the brain [109].
3.4. Metallothioneins
Metallothioneins represent a family of Cys-rich proteins being capable of binding divalent metals [110]. Mammalian MTs have a molecular mass of 6–7 kDa containing 61–62 amino acids with 20 Cys residues [89], although aromatic His and Tyr are absent. One MT molecule may transport up to 7 divalent or 12 monovalent metals [111]. Four MT isoforms have been identified in mammals with MT1 and MT2 is the most widespread [112].
Leiva-Presa et al. (2004) have stated that stoichiometry, stereochemistry, and folding of Hg-binding MTs are determined by solution pH, counterion nature (chloride or perchlorate), as well as the period of time between addition of Hg(II) salts to the protein solution [113].
Structural NMR studies revealed two sites of where metal-binding occurs: N-terminal α-domain and C-terminal β-domain [114]. These sites are located in domains separated by the Cys-free part of the polypeptide chain. The first one possesses a high affinity to zinc (Zn) and binds four Zn2+ ions, whereas the second one is specific for Cd and binds three Cd2+ ions [111].
Taking into account certain similarities in the properties of Cd (II) and Hg(II), it is reasonable to propose that this site may be responsible for Hg binding. According to Muñoz and Rodríguez (1995), MT forms more stable complexes with Hg than those with Zn and Cd [115]. The stability constants for HgL (L = rabbit liver MTs) at pH = 7.5 are estimated as 30.9 and 31.1–32 for isoforms I and II, respectively. At the same time, the respective values for Cd and Zn complexes are 15.0 and 14.8, as well as 11.9 and 12.1, respectively [115].
Studies on the structural features of interaction between Hg and MTs performed by Henkel and Krebs (2004) and Lu et al. (1993), suggest that one molecule of MT binds up to 18 Hg ions [111,116]. The geometry of such complexes is supposed to be pseudo-tetrahedral with metal-ion bridges between protein molecules. Hg4-α-MT and Hg7-MT are characterized by tetrahedral geometry, whereas Hg11-MT is supposed to have trigonal geometry. Mercury binding results in the formation of isolated MS2–4 regions and Hg-thiolate clusters (RS)1–3MSRM(SR)1–3, where up to four S atoms of Cys residues bind metal ions both in terminal and bridge positions [116].
When studying the composition and structure of rabbit liver proteins at pH > 7, Lu and Stillman (1993) revealed Hg complexes with Hg:MT ratio of 4:1, 7:1, and 11–12:1 [117]. An in vivo study by Shen et al. (2005) demonstrated that oral HgCl2 exposure results in Hg binding predominantly to MT-2c MT subisoform resulting in the formation of Hg6Cu and Hg5Cu2-MT-2c forms [118]. Binding to other isoforms with the formation of the Hg6-MT-2β, Hg6Cu-MT-1γ, and Hg7-MT-2α forms were also observed [118]. At the same time, MALDI-TOF-MS analysis of rat serum has demonstrated that MeHg exposure results in the production of the MT2-MeHg compound [119]. Application of HILIC-ICP-MS for analysis of MT-bound Hg in the dolphin liver revealed the presence of MT–Zn6Hg [120].
MT response was species-specific with the most profound induction of renal and hepatic MT1 and MT2 expression by MeHg and Hg0, respectively [121]. A significant increase in MT1/2 expression was also observed in the salivary glands of MeHg-treated rats [122]. An in vitro study using the keratinocyte cell line also demonstrated a Hg-induced increase in MT expression in a dose-dependent manner [123]. Although MT3 does not have a significant impact on total cerebral Hg levels, it significantly affects the expression of certain Hg target chemokine genes [124].
It is also noteworthy that when compared to the effective role of toxic metals (As, Hg, Pb), Hg was reported to be the only metal being responsible for significant induction of both renal and hepatic MT levels [125].
MT-null mice were characterized by higher susceptibility to Hg0 neurotoxicity due to an increased accumulation of Hg in the brain [126]. Interesting data were obtained from MT-null mice, demonstrating that a lack of MT in pregnant mice exposed to Hg vapor (Hg0) provides a shift in Hg binding to high-molecular-weight proteins [127]. These findings fit well with the general hypothesis that MTs serve as detoxifying proteins for Cd and Hg.
Laboratory data generally corroborate the results of single human studies. Wang et al. (2012) have demonstrated that MT1A (rs8052394) GG and GA genotypes and MT1M (rs9936741) TT genotype are related to the reduced Hg accumulation as compared to MT1A AA or MT1M TC and CC genotypes, respectively [128]. Correspondingly, MT1M and MT2A gene polymorphisms were also associated with increased susceptibility to adverse neurobehavioral effects of Hg [129].
While Hg binding by MT is well established (see above), the pathways underlying Hg-induced up-regulation of MT expression and the effective role of Hg-SH reactivity in these processes remain unclear. MT synthesis is described to be modulated by metal-responsive transcription factor-1 (MTF-1) through Zn released from Zn-MT [130]. Based on the observation of higher stability constants for the Hg-MT complex as compared to Zn-MT or Cd-MT complexes [115], as well as observations on the ability of Hg2+ ions to replace Zn2+ in MT molecule [131], it has been proposed that this mechanism may underlie Hg-induced increase in MT production [132]. Such hypothetical pathways are also approved by the observed association between MT isoforms and MTF-1 polymorphisms and biomarkers of Hg exposure [133]. However, a study by Balamurugan et al. (2005) demonstrated that in mammalian cells, MTF-1-dependent MT-type promoters are not activated significantly upon Hg treatment, whereas a strong response was observed in Drosophila through dMTF-1 [134].
Another potential mechanism of Hg-induced up-regulation of MT synthesis may involve Nrf2 downstream signaling. Particularly, Hg exposure was shown to activate the Nrf2 pathway by binding Keap1 at Cys151 [135], or Cys319 [136], related to the form of Hg (see lower). In turn, recent findings demonstrate that MT expression may be Nrf2-dependent [137], although previous studies have demonstrated a more expressed effect of Nrf1 on MT synthesis as compared to Nrf2, were not significant [138]. This association may also involve modulation of glycogen synthase kinase 3β (GSK-3β), playing a significant role in Nrf2 regulation [139,140].
3.5. Exogenous thiol donors
3.5.1. N-acetylcysteine
N-acetylcysteine (NAC) is a potent antioxidant and potential antidote against toxic metals, including Hg and Cd [141]. Structural aspects of Hg(II) and NAC complexes are insufficiently studied to date despite multiple studies on its capability to bind organic and inorganic Hg species [53]. In neutral and alkaline medium, Hg2+ ions form a complex (L-NAC) of linear geometry at M:L ratio of 1:2. In excess of ligand, the particles and exist in equilibrium at neutral pH, whereas in alkaline medium and high concentration of free thiolate ion dominates . Complex geometry is diagonal, trigonal, and tetrahedral, respectively. In this context, NAC shares similar features with Cys [142]. Köszegi-Szalai et al. (2000) demonstrated high stability of two-ligand complexes with different protonation degree (47.83, 45.38, and 41.83 for , , and HgH2L2, respectively), being comparable or even exceeding that for Cys [98].
Generally, these data corroborate the reports of decreasing Hg toxicity by NAC. Particularly, NAC treatment significantly reduced MeHg-induced expression of proinflammatory IL-6 and MCP-1 in the U-87-MG human astrocytoma cell line [143]. The protective effect of NAC treatment in HgCl2-exposed rats was reported to be related to the improvement of GSH levels and antioxidant enzyme activity [144]. In view of the role of NAC as a precursor of GSH synthesis [145], the observed protective effects under Hg exposure may be mediated by GSH synthesis up-regulation, although direct NAC-Hg binding may also play an important role.
3.5.2. Alpha-lipoic and dihydrolipoic acids
Existing data demonstrate that alpha-lipoic acid (ALA) is widely used for Hg detoxication [23]. However, its complexation with Hg2+, , and organic mercurial have not been studied extensively. Investigation of complex formation between ALA and its reduced form DHLA with Hg2+ using differential pulse voltammetry has demonstrated the formation of 1:1 complexes [146]. At the same time, the formation of a 2:1 complex (or even higher L: M ratio) is also possible for DHLA. It is notable that 5-S-Lipoylhydroxytyrosol (LipoHT), a DHLA derivative possesses certain similarities to GSH. Particularly, binding Hg to LipoHT through SH groups induces a complex formation with a 1:2 M:L ratio, whereas –OH and –COOH do not play a role in metal coordination (Fig. 3) [147].
Fig. 3.
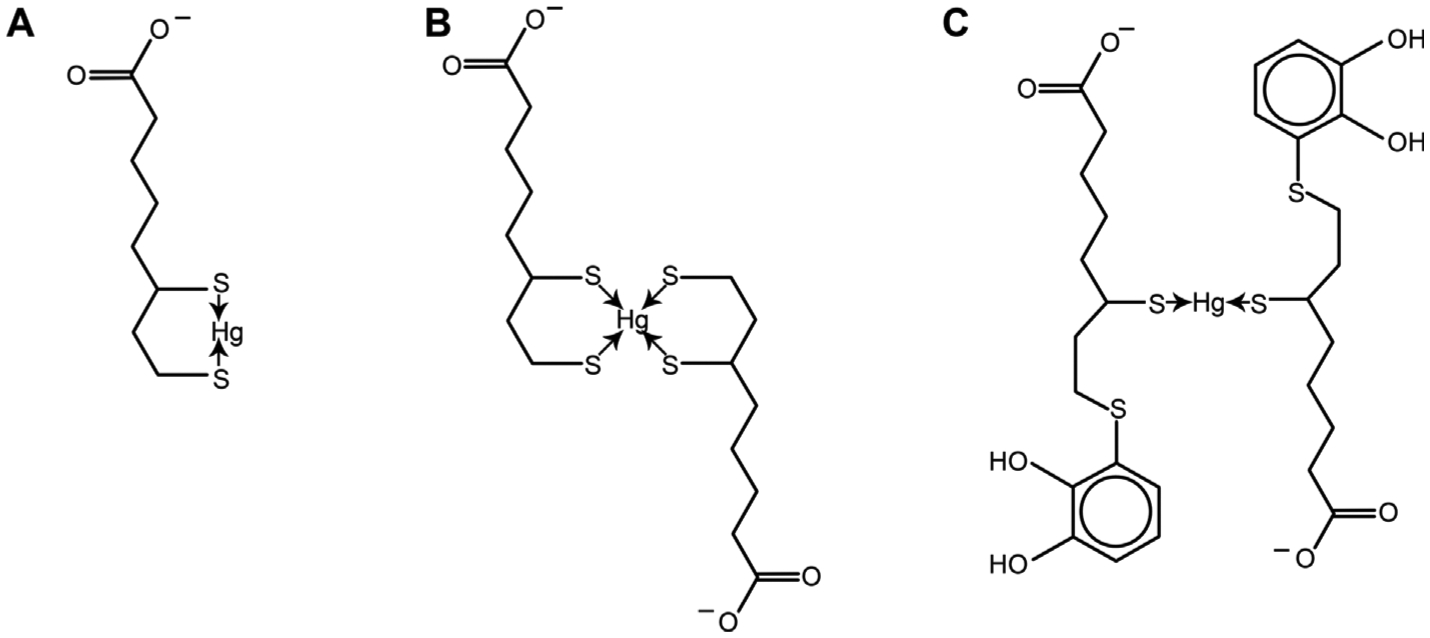
The potential mechanisms of Hg(II) binding to alpha-lipoic acid (ALA). A – Monoligand complex of Hg(II) with dihydrolipoic acid (DHLA); B – Biligand complex of mercuric Hg with dihydrolipoic acid (DHLA); C – Biligand complex of Hg2+ with 5-S-Lipoylhydroxytyrosol.
Although in vivo studies did not reveal direct interaction between Hg and ALA (or DHLA), a protective effect has been clearly demonstrated. Specifically, ALA pretreatment significantly reduced Hg-induced oxidative stress and excitotoxicity in the rat cerebral cortex [148]. Despite the scarcity of data, based on in vitro data, one can propose that direct Hg-binding activity may contribute to a greater extent to the protective effect of ALA (DHLA) vs. NAC. NAC possibly has protective effects against Hg via stimulation of GSH synthesis. Furthermore, since NAC-Hg complexes could be absorbed by the kidneys [149], NAC can potentially increase the renal toxicity of Hg2+.
4. Biological effects of Hg-thiol interaction
4.1. Mercury transport
The Hg S-conjugates contribute to Hg transport, which is in agreement with the molecular mimicry (especially at Lα region of amino acid) of transport of Hg conjugates with Cys or GSH [150]. MeHg-S-Cys complex structurally closely resembles L-methionine, being a substrate for L-type large neutral amino acid carrier transport (LAT1) system that is at least partially responsible for MeHg transport through BBB [151]. GS-CH3Hg has also been reported to be moved by LAT1 [152]. The human organic anion transporter 1 (hOAT1) was reported to transfer only Cys-S-Hg-SCys conjugate but not G-S-Hg-S-G [153]. Hypothetically, Cys-S-Hg-S-Cys may also be trafficked by the cysteine-glutamate transporter SLC7A11, playing a significant role in brain functioning [32]. It is also noteworthy that the uptake rate of GSH-S-CH3Hg is twofold higher as compared to Cys-S-CH3Hg, although both forms are effectively transported intracellularly. GSH-S-CH3Hg may be transported by amino acid transporters [128]. A detailed study by Oliveira et al. (2018) has demonstrated that MRP2 also transports Cys-S-Hg-S-Cys both in vitro and in vivo [122]. Renal GGT may also be involved in renal transport of the Hg-SG complex [154].
The transport pathways are uncertain, although the GSH-Hg complexes are the main form of Hg in the urine and the bile. It has been proposed that Cys and GSH, as Hg carriers, can modulate the Hg efflux rate into bile; therefore, the Hg secretion rate in bile appears to be independent of the actual flow of bile. The Hg content in the bile can change inversely, as the rate of bile flow is decreased or increased. Therefore, the net efflux of Hg in the liver remains constant. However, biliary secretion of MeHg in rats can increase by elevating GSH and Cys bile levels. Other investigations have reported corroborating results in numerous animal models [155–157]. In contrast, the depletion of GSH controls the biliary secretion of MeHg in animal models, and the prevention of GSH production can shut down the biliary release of Hg [157,158].
In studies with astrocytes, the use of GSH, GSH stimulators, or precursors of GSH can significantly increase Hg efflux from these cells as a complex of GSH. Fujiyama et al. reported that GSH conjugation is the main mechanism for Hg efflux from astrocytes [159]. GSH also increases the Hg elimination in the renal tissue. Investigations with mammalian renal cells reported that GSH acts as effective as the chelating agent of DMSA (2,3-dimercaptosuccinic acid) in control of the accumulation of inorganic Hg in renal cells [160].
In an in vivo study with oral administration of MeHg or MeHg-Cys conjugate, MeHg-Cys was shown to be more effectively transported to the brain and liver as compared to MeHg itself, whereas accumulation in kidneys was significantly reduced [161]. It is also noteworthy that pharmacokinetics of Hg differs significantly between MeHg-Cys and MeHg with higher circulating blood Hg levels in response to the latter, although these differences disappear after 1 h after treatment [162].
Homocysteine (Hcy) S-conjugates of MeHg (Hcy-S-CH3Hg) is considered as biologically relevant Hg species. It has been reported that Hcy-S-CH3Hg, similarly to Cys-S-Hg-S-Cys, is transported through hOAT1. However, it may be a substrate for other amino acid transporter(s) [163]. Zalups and Barfuss (1998) hypothesized that hyperhomocysteinemia might aggravate Hg transport to kidneys due to higher Hg-HCy uptake as compared to Hg itself, although this mechanism in vivo is to be further studied [149].
At the same time, NAC-Hg conjugates are efficiently absorbed in the renal cortex as compared to mercuric conjugates of Cys [149]. It has been demonstrated that retinal pigment epithelial cells effectively uptake Cys-S-Hg-S-Cys and Hcy-S-Hg-S-Hcy but no other Hg S-conjugates including NAC-S-Hg-S-NAC and GSH-S-Hg-S-GSH. In view of the Na-dependent process, the authors have proposed that Na-dependent amino acid transporters, including B0,+, and ASC-type amino acid transporter (ASC), may be responsible for the uptake [164]. Cys-S-CH3Hg, as well as Hcy-S-CH3Hg, may also be transported through the membrane by B0,+ system [165].
In addition to transmembrane transport, S-conjugates of Hg may exert toxic effects through ligand exchange and covalent binding of Hg to enzyme molecules. Particularly, the Cys-S-Hg-S-Cys conjugate is considered a physiologically relevant form of Hg which has been shown to induce cytoskeletal alterations, as well as lipid peroxidation, mitochondrial dysfunction, and induction of necroptosis in renal proximal tubule cells [12]. It has been also demonstrated that CH3Hg-S-Cys and Cys-S-Hg-S-Cys, as well as inorganic HgCl2, are irreversible inhibitors of cystathionine γ-lyase. At the same time, Cys (CH3Hg-S-Cys and Cys-S-Hg-S-Cys) and Hcy (Hcy-S-Hg-S-Hcy and CH3Hg-S-Hcy) S-conjugates are both substrates and reversible inhibitors of glutamine transaminase K [166]. The metal–ligand ratio significantly determines the biological effects of Hg2+-thiol (Cys, GSH, and albumin) complexes with the overall toxic effect of 1:1 ratio and protective effect of 1:2 M: L ratio [52]. In contrast to cysteinates, Hg-GSH complexes do not only play a role as Hg transporters (see lower) but apparently also exert prooxidant activity [167].
4.2. Enzyme inactivation
Mercury-induced inhibition of enzymatic activity may be one of the most important heading factors in Hg toxicity, as discussed in detail by Ynalvez et al. (2016) and Oliveira et al. (2018) [16,122]. Despite an abundance of enzymes inactivated by Hg, Ynalvez et al. (2016) have clearly demonstrated that only a select number of these effects result from direct covalent modification of enzyme molecules [16]. The inhibited enzymes are involved in multiple processes and systems functioning [16]. Therefore, only certain enzymes inhibited by covalent Hg-induced modification will be reviewed herein, providing specific examples.
The majority of studies have demonstrated the impact of MeHg on enzyme activity with covalent modification of thiol-containing biomolecules by MeHg being known as “S-mercuration”. Kumagai et al. (2013) considered Mn-SOD, arginase I, NOS (discussed in “Apoptosis” section), and sorbitol dehydrogenase (SDH) as the key molecular targets for MeHg and substrates for S-mercuration [135]. Specifically, the covalent binding of MeHg to rat SDH through Cys44, Cys119, Cys129, and Cys164 results in decreased enzyme activity and its aggregation. Moreover, the S-mercuration of Cys44 causes a release of Cys44-bound active site Zn2+ [168].
δ-aminolevulinate dehydratase was also reported to be controlled by Hg exposure through its reactivity with thiol groups at the enzyme active site [169]. Molecular docking studies by Oliveira et al. (2018) have demonstrated that Hg-protein adduct formation occurs due to EtHg/MeHg binding to Cys124 and/or Cys132 residues [170]. It is also interesting that the inhibitory effect of Hg on ALAD was more pronounced in vivo, whereas in vitro lead(II) was the stronger inhibitor as compared to Hg [171].
Brain creatine kinase (CKB) was also shown to be subjected to MeHg-induced inactivation, as estimated in C6 glioma cell lineage [172]. Being in accordance with the finding of the earlier study, it has been demonstrated that organic Hg species may bind to the residue of Cys283 in the active site of CKB [170,173].
It has been also demonstrated that Hg2+ exposure significantly inhibits helicase, DNA-binding, and the ATPase activity of recombinant helicase (BLM642–1290) through alteration of the enzyme conformation and formation of Cys940-S-Hg-S-Cys944 and Cys1063-S-Hg-S-Cys1064 intramolecular bridges [174].
In a proteoliposome model, Pochini et al. (2012) have demonstrated that both HgCl2 and MeHg are shown to inhibit carnitine (OCTN2) transporter activity [175] through Cys50 and to a lesser extent Cys136 Hg-binding [176].
Several studies demonstrated the involvement of Hg-SH interaction in the alteration of ion channel function. Particularly, Na+-K+-ATPase is considered as the potential target of Hg toxicity due to Hg-thiol reactivity. Hg-induced thiol-mediated inhibition of Na+-K+-ATPase activity was observed in platelets [177], brain [178], and kidneys [179]. Thiol-dependent inactivation may also be involved in the Hg-induced reduction of myosin Ca2+-ATPase activity in the ventricular myocardium of the rat [180]. Bhattacharya et al. (1997) proposed a significant role of Na+/K+-ATPase binding in nuclear transport of Hg2+ [181]. It has been proposed that Hg binding to Na+/K+ pump molecule in Xenopus laevis oocytes may occur at the Cys residue of the first transmembrane segment [182]. In a study in HeLa cells, Zichittella et al. (2000) demonstrated that HgCl2 might cause rapid inactivation of Na,K-ATPase due to binding Cys964 [183]. Several studies have also demonstrated that canonical transient receptor potential channels (TRPC), namely TRPC4 and TRPC5, may also be subjected to Hg-induced modulation through binding of extracellular Cys residues, resulting in their activation [161]. Although the inhibitory effect of both HgCl2 and MeHg on neuronal Cav3.1 (T-type) calcium channels was demonstrated [184], the particular contribution of Cys residues binding is not estimated.
In addition to Na,K-ATPase, Hg2+ was shown to reduce mitochondrial NADH–O2 oxidase activity with a concomitant increase in F1FO-ATPase (F-ATPase) activity through a thiol-dependent mechanism, thus providing an additional link between Hg exposure and mitochondrial dysfunction [185]. It is also noteworthy that the affinity is elevated in the presence of the substrate (Mg-ATP) due to conformational changes and higher availability of thiol groups for Hg2+ [16].
However, a few examples of Hg-inhibited enzymes were reviewed, of which the data clearly demonstrate that covalent modification of enzymes may impair multiple processes, including DNA replication, carbohydrate and lipid metabolism, and heme synthesis. In addition to enzyme inhibition, Hg exposure can also affect cofactor availability, thus contributing to the impairment of enzymatic reaction. Particularly, Hg is capable of binding thiol and amide groups of coenzyme A (CoA) with the formation of Hg-CoA complex and alteration of mitochondrial β-oxidation and hypothetically other CoA-dependent processes [186].
4.3. Antioxidant enzymes and oxidative stress
Mercury is considered a potent prooxidant affecting both enzymatic and non-enzymatic antioxidants, including SOD, catalase, GSH, and thioredoxin systems [11], as well as other antioxidant selenoproteins [19]. However, only a part of the observed prooxidant effects of Hg may be considered as a consequence of direct Hg-SH interaction.
4.3.1. Superoxide dismutase
It has been proposed that Hg-induced inactivation of renal but not hepatic Mn-SOD may be at least partially mediated by direct binding of Hg to protein sulfhydryls [187]. Shinyashiki et al. (1996) demonstrated that MeHg selectively inactivates Mn-SOD but not Cu, Zn-SOD [188]. The authors have proposed that the available -SH group (Cys6) in Cu, Zn-SOD, is located within β-structure of the protein, being unavailable for S-mercuration [188]. In turn, binding of MeHg at the Cys196 residue significantly decreases mitochondrial Mn-SOD activity, thus contributing to oxidative stress and mitochondrial dysfunction [189]. Another study has demonstrated that Hg exposure results in a remarkable decline in Cu-SOD peak in parallel with an elevation of Hg-SOD peak, being indicative of the potential competition between Hg and Cu for binding the SOD active site [190]. However, the role of Hg-SH in this process is non-significant due to the absence of Cys residues bound to Cu2+ ion in the active center of Cu, Zn-SOD.
4.3.2. Catalase
An inhibitory effect of Hg on catalase has been clearly demonstrated both in vivo and in vitro [11] while a detailed analysis involving molecular docking by Chen et al. (2015) revealed that none of the Hg2+ species (HgCl2, [HgCl3]−, [HgCl4]2−) were surrounded by Cys residues in the catalase molecule [191]. However, the interaction of Hg-S-complexes (for instance, Cys and GSH, and even homocysteine) with catalase has not been studied. As the free salts listed are not expected to be the true forms to be found physiologically, one can still not exclude the potential role of Hg-SH interaction in Hg-induced inhibition of catalase activity
4.3.3. Glutathione system
It has been shown that Hg has a significant inhibitory function on the GSH system [192]. Direct effects may occur due to the formation of Hg-SG conjugates (see the former section) and subsequent reduction of reduced glutathione pool. However, the contribution of Hg-SH interaction into inhibition of GSH-handling antioxidant enzymes has yet to be clearly demonstrated. It has been thought that the role of Hg on GPX might be because of the Hg interaction with the selenol group [193]. Particularly, MeHg but not Hg2+ may bind GPX target site at SeCys49 [194]. These observations are confirmed by an observation of delayed decreased in GPX activity following a MeHg-induced reduction of thiol levels [195]. It is also notable that Farina et al. (2009) demonstrated that a decrease in the expression of GPx (the level of the enzyme was detected by immunoblotting) correlated with cerebellar neuronal death [151]. Thus, in addition to the potential direct interaction of mercurial forms with GPx, they can decrease the antioxidant defenses of brain cells via a decrease in the expression of important antioxidant enzymes such as GPx1 [151].
In vitro studies also showed significant inhibition of GR by Hg [196]. Picaud and Desbois demonstrated that GR-Hg interaction involves Hg binding both to the thiol group and catalytic histidine of the reduced form of the enzyme (GR-H2) [197]. Although no direct data have been obtained, Kanda et al. (2012) also propose that binding Cys residues in GR may play a significant role [168].
Notably, Hg-thiol interaction may play a significant role in the inhibition of another GSH-dependent [198] selenoprotein, namely Selenoprotein W (SelW), through Hg interaction with SeCys and GSH-binding Cys residues [199].
4.3.4. Thioredoxin system
Thioredoxin/thioredoxin reductase system is evaluated as one of the key targets for Hg toxicity, although the particular contribution of Hg binding to thiol groups is unclear [200]. It has been demonstrated that S-mercuration of Trx may occur at Cys69, Cys62, Cys32, Cys35 and Cys73 either with the formation of dimers through Cys73 (Cys73-S-Hg-S-Cys73) and intramolecular S-Hg-S bridges (Cys32-Cys35 and Cys62-Cys69) in the case of HgCl2 exposure or binding of Hg-CH3 to each of five residues of Cys in the case of MeHg exposure [201]. Mercury-thiol interaction may also be at least partially involved in the inactivation of TrxR even despite higher reactivity of selenol group in TrxR molecule with Hg due to its ionization at physiological pH [202]. Specifically, both thiol (–SH) and selenol (-SeH) groups of Cys497 and Cys498, respectively, contribute to the Hg interaction with the formation of -S-Hg-Se-bridge [203]. Therefore, the existing data demonstrate that Hg-thiol interaction in the Trx/TrxR system may possess an inhibitory effect not only through Trx but also TrxR interaction. It has also been proposed that MeHg-induced inhibition of glutaredoxin 1 (Grx1) in cultured astrocytoma cells may also involve covalent modification at Cys22 [204].
Generally, the existing data clearly demonstrate that direct Hg-SH interaction induces oxidative stress through inhibition of SOD, TrxR, and SelW activity, as well as decreasing Trx and GSH levels and availability (Fig. 4). The particular effect of Hg-thiol reactivity on catalase and GPX activity is still questionable. However, the proposed scheme may be considered as rather mechanistic in view of the potential up-regulation of GSH synthesis resulting from Hg-induced Nrf2 activation (see below).
Fig. 4.
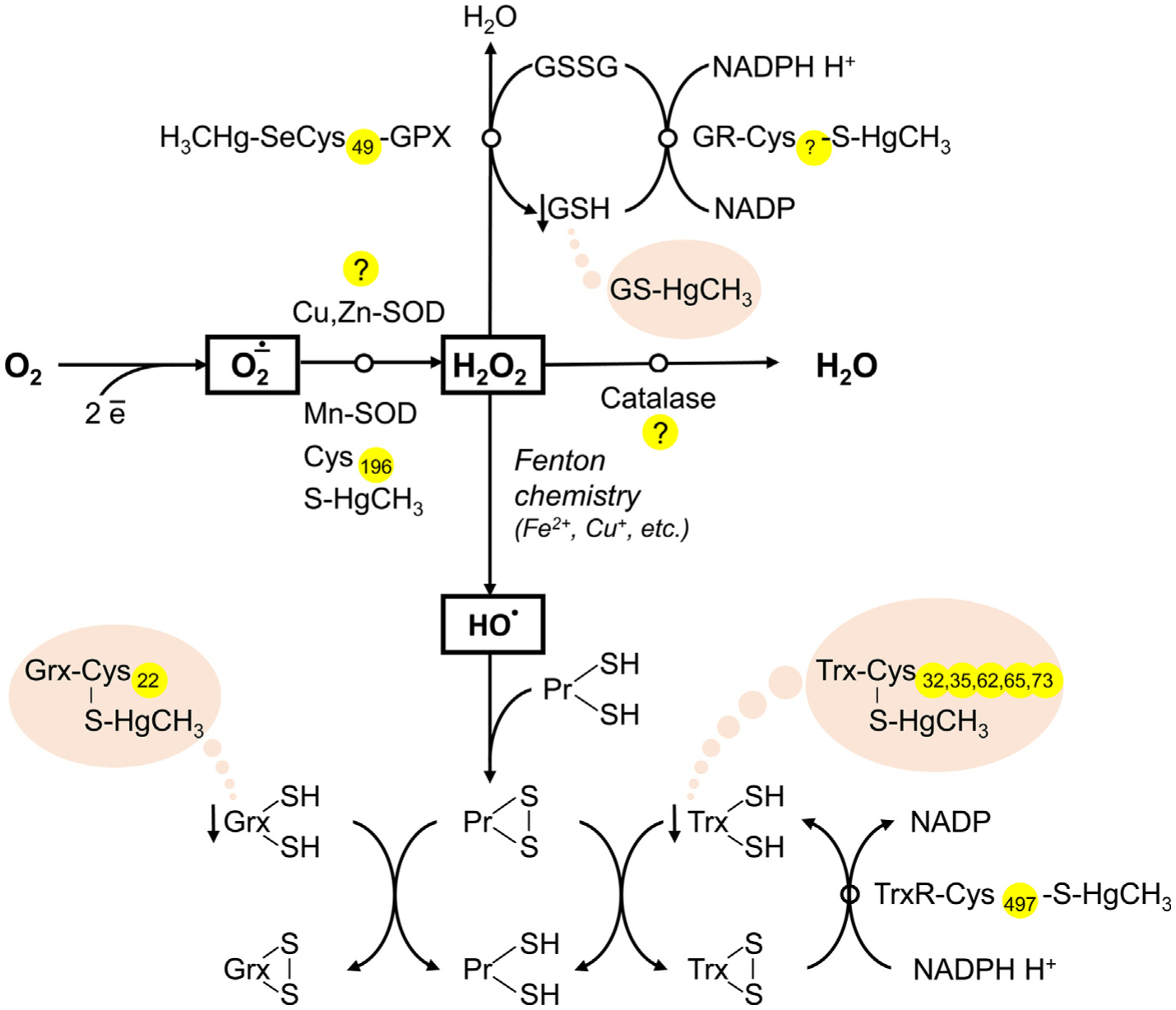
The role of Hg-SH binding in Hg-induced decrease in antioxidant system activity. Mercury was shown to inhibit antioxidant enzymes, including Cu,Zn-SOD, Mn-SOD, catalase, GPX, GR, and TrxR, although the particular role of Hg-SH interaction in this process is unclear. Inhibition of Mn-SOD was shown to be associated with Hg-SH binding at Cys196, whereas the particular location of Hg-binding cysteine residue in GR is not estimated. Inactivation of GPX may occur due to Hg binding to SeCys49 residue. In turn, recent data demonstrate that direct Hg-SH interaction is unlikely to be involved in inhibition of Cu,Zn-SOD, and catalase. In addition to Hg-induced antioxidant enzyme inactivation, Hg may also bind thiol groups of proton-donor cofactors (GSH, Trx, Grx), thus limiting their availability for reduction.
4.4. Modulation of apoptosis
Induction of apoptosis by Hg is considered as one of the key mechanisms of Hg toxicity [205], although the effect was shown to be species-dependent [206]. Despite a plethora of mechanisms linking Hg exposure and apoptosis revealed the particular contribution of Hg-SH interplay in apoptosis modulation remains unclear. A review of the existing studies in the field demonstrated that Hg-SH might contribute to apoptosis modulation through interference with Akt/CREB, Keap1/Nrf2, NF-κB, and mitochondrial pathways (Fig. 4).
4.4.1. Akt/CREB pathway
Unoki et al. (2018) have demonstrated that at low levels of MeHg S-mercuration of phosphatase and tensin homolog deleted from chromosome 10 (PTEN) results in Akt activation and Bcl-2 up-regulation and anti-apoptotic effects [207]. However, at the increasing MeHg concentrations, Hg binding to cAMP response element-binding protein (CREB) through Cys286 results in disruption of Akt/CREB/Bcl-2 signaling and apoptosis [207].
4.4.2. Keap1/Nrf2 pathway
Mercury exposure is known to modulate intracellular signaling and, in particular, Nrf2 through a number of mechanisms, including S-mercuration of different targets [207]. Methylmercury binding to Keap1 occurs at Cys151, Cys368, and Cys489, with the leading role of Cys151 S-mercuration in structural alteration and subsequent Nrf2 activation and up-regulation of antioxidant synthesis [135]. It is also notable that GSH adduct of MeHg (MeHg-SG) also covalently modifies Keap1 causing Nrf2 activation in human neuroblastoma SH-SY5Y cells, although modification occurs at Cys319. It has also been proposed that it modifies Keap1 protein through the S-transmercuration process with subsequent release of reduced glutathione (GSH) [136]. Subsequent activation of Nrf2 signaling was shown to down-regulate oxidative stress through antioxidant synthesis [198] as well as Bcl2 up-regulation [208], all resulting in suppression of apoptosis. It has been also demonstrated that MeHg might increase Nrf2 activity through the down-regulation of Fyn (Src family kinase) [209], although the particular role of Hg-SH interaction in this mechanism is unclear.
4.4.3. NF-κB pathway
It has also been demonstrated that Hg2+-induced inhibition of NF-κB-p50 binding to DNA may involve direct interaction between Hg and Cys62, which may significantly affect the susceptibility of renal cells to toxicity and apoptosis. It is also notable that binding Hg2+ to the other five Cys residues on the molecule may also possess an inhibitory effect, although being less significant [210]. In view of the antiapoptotic effect of p50 binding to Bcl3 and subsequent antiapoptotic effects, Hg-induced inhibition of p50 binding activity may predispose the cell to apoptosis. These findings also corroborate earlier data by Shumilla et al. (1998), demonstrating the involvement of the thiol mechanism in Hg-induced inhibition of NF-κB binding to DNA [211].
4.4.4. Mitochondrial pathway
The earlier discussed inhibition of Mn-SOD activity through MeHg binding to Cys196 contributes to oxidative stress [189]. The latter results in mitochondrial dysfunction characterized by decreased membrane potential (Δψ), and increased membrane permeability for cytochrome c, is an activator of caspase-mediated apoptosis [212].
4.4.5. Arginase pathway
Hypothetically, the overall anti-apoptotic effect may be at least partially mediated by modulation of NO levels and arginase. It has been demonstrated that both arginase activity and NO levels may be implicated in apoptosis regulation [213] and affected by Hg through covalent modification of the proteins. Specifically, the inhibitory effect of Hg2+ on brain constitutive NOS may be at least partially mediated by direct binding and subsequent conformational changes [214]. In turn, a dose-dependent decrease in hepatic arginase I activity may occur due to Cys119, Cys168, and Cys303 S-mercuration, also resulting in leakage of Mn2+ ions from the active site. In contrast, no such effect was demonstrated for renal arginase II despite the presence of five Cys residues [215].
4.4.6. Other targets and involvement of endoplasmic reticulum stress
Caspase-3 was also considered as the target for Hg binding because of the presence of active site cysteinyl residue, although such an interaction may be observed only at high Hg concentrations [216]. S-mercuration of ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) through Cys152 results in a dose-dependent decrease in enzyme activity resulting in an alteration of cellular monoubiquitin levels that may be linked to neurotoxicity and neurodegeneration [217]. Particularly, UCH-L1 inhibition was reported to contribute to apoptosis through the stimulation of unfolded protein response [218]. Direct interaction between protein thiols and Hg compounds also plays a significant role in endoplasmic reticulum stress [219]. In human neuroblastoma SH-SY5Y cells MeHg covalently modifies Cys residue in the C-terminal catalytic domain in protein disulfide isomerase (PDI) by S-mercuration resulting in unfolded protein response and endoplasmic reticulum stress [220] involving modulation of IRE1α-XBP1 pathway [221].
Therefore, the effect of Hg on cellular apoptosis may involve a variety of pathways depending on the cell type, species, as well as dose of Hg. It seems that at low-level exposure, Hg may possess antiapoptotic effects, whereas high Hg levels are clearly associated with pro-apoptotic signaling (Fig. 5).
Fig. 5.
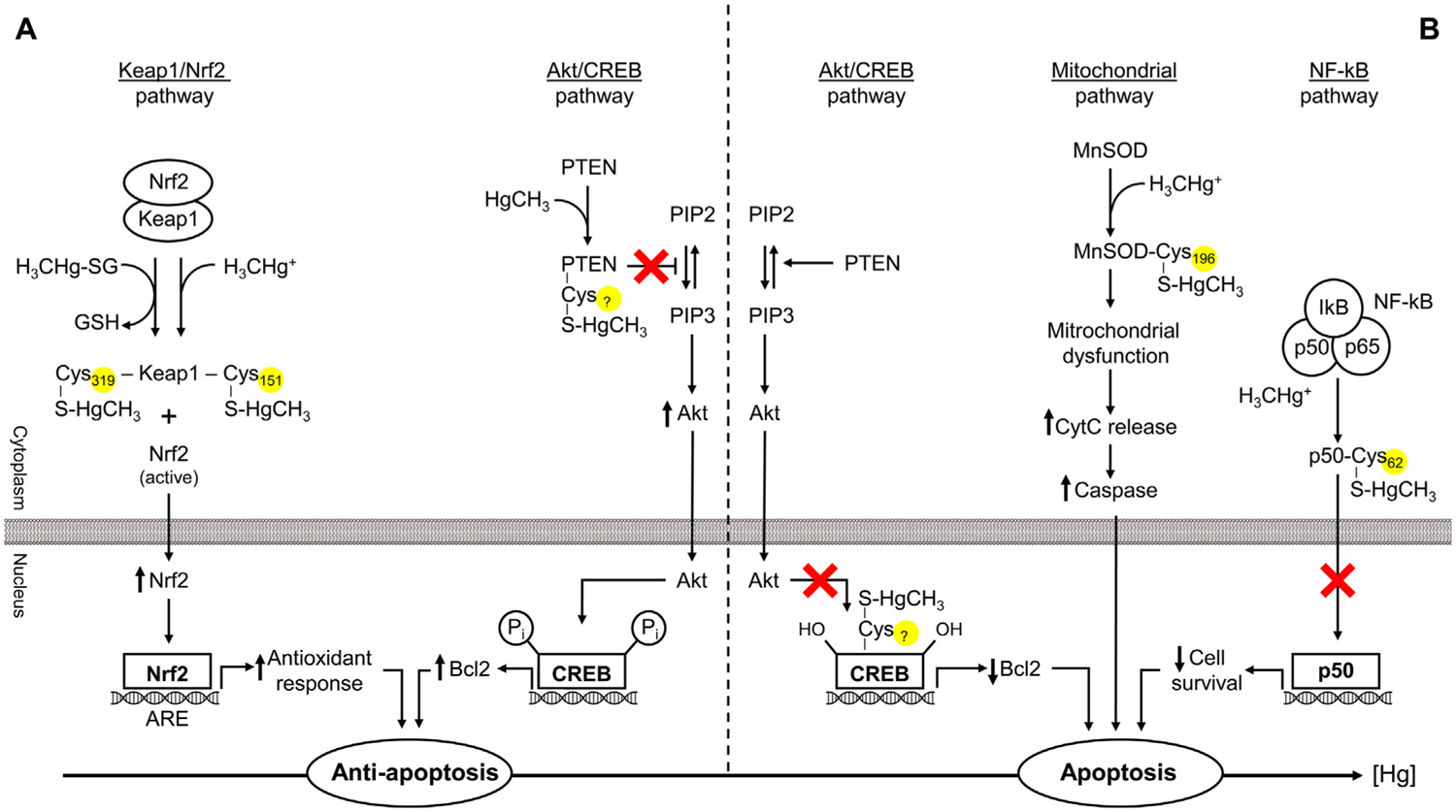
Mercury-sulfhydryl-dependent effects on apoptosis regulation. Mercury exposure may induce both anti- (low dose) and pro-apoptotic (high-dose) signaling. Covalent modification of Keap1 through Hg binding to Cys151 (MeHg) or Cys319 (MeHg-SG) results in Nrf2 activation and subsequent downstream signaling leading to up-regulation of antioxidant enzymes and anti-apoptotic signals. Akt/CREB pathway may be involved both in the anti- and pro-apoptotic effects of Hg. PTEN S-mercuration results in Akt activation and subsequent Bcl2 up-regulation. In contrast, increasing Hg levels bind Cys286 residue in CREB, thus disrupting Akt/CREB/Bcl-2 signaling and induction of proapoptotic signaling. NF-κB may also partially mediate the effects of Hg on apoptosis through S-mercuration of p50 (p50-Cys62-S-HgCH3) and alteration of its antiapoptotic effects. Finally, inhibition of mitochondrial Mn-SOD through Cys196-S-HgCH3 binding may result in mitochondrial oxidative stress and dysfunction, causing increased cytochrome c (CytC) leakage and caspase-mediated apoptosis.
5. Neurotoxicity
The brain is considered as one of the key targets for Hg toxicity by different mechanisms [33]. In turn, thiol homeostasis is critical for brain development and functioning at all steps of ontogenesis [222]. Particularly, prenatal MeHg exposure induces a remarkable elevation in cerebellar Hg levels, along with depleting thiol content, which is related to impaired motor functions [223]. In turn, impaired thiol redox balance in adults is associated with neurodegeneration [224]. Although the earlier reviewed effects on redox homeostasis and apoptosis play a significant role in Hg neurotoxicity, the specific neuronal effects of Hg-SH interaction will be reviewed in this section.
5.1. Cytoskeleton
The existing data demonstrate that the neurotoxic effect of Hg-SH binding may be mediated in close interaction with microtubules and tubulin, leading to its instability, being involved in neurodegeneration [225]. Particularly, the impact of Hg exposure on tubulin may induce a significant reduction in microtubule assembly [226]. It has been proposed that tubulin dimer contains 15 free SH groups available for Hg binding, thus affecting microtubule polymerization [227]. Correspondingly, it was with analysis of Hg-binding proteins in human neuroblastoma SK-N-SH cells shown that the majority of Hg-binding proteins are structural (including tubulin), chaperone proteins, as well as enzymes including protein disulfide isomerase (PDI) and ATP synthase [43]. It is also notable that Hg(II) is characterized by a higher affinity to cytoskeletal protein sulfhydryls as compared to EDTA [228].
Certain studies revealed a close interaction between Hg2+ and tau protein. Binding of Hg(II) to tau construct R2 [229] and R3 [230] through Cys291 and Cys322, respectively, caused changes in the conformation of unstructured R3 to β-structure and further aggregation, thus contributing to Alzheimer’s disease pathogenesis.
5.2. Neuronal receptors
Synaptic Cys residues, including those from neurotransmitter receptors, were considered as principal targets for MeHg [231].
It has been demonstrated that Hg-thiol reactivity may significantly contribute to altered glutamatergic neurotransmission [232]. Particularly, Hg2+ binding to membrane sulfhydryls has been found to play a significant role in the restriction of glutamate uptake by astrocytes [132]. In addition, Hg (both Hg2+ and MeHg) exposure was also shown to inactivate glutamine/amino acid transporter (ASCT2) in a liposome model with Cys207 and Cys210 of the CXXC metal-binding motif, with Cys233 being the most likely sites for covalent binding [233]. This mechanism may at least partially underlie MeHg-induced neuronal dysfunction secondary to metabolic disturbances in astrocytes [234].
HgCl2, and to a lesser extent MeHg, was shown to inhibit muscarinic acetylcholine (mACh) receptor subtypes (M1, M2) through thiol group binding [9,235]. These data correspond to the earlier observe differences in mACh inhibition curves [236]. It has been proposed that HgCl2 and MeHg are bound to different sites on muscarinic acetylcholine receptors, namely two thiol groups on the extracellular membrane and single thiol group in proximity to the receptor-binding domain [237]. In contrast, binding free Cys residues on extracellular α4 and β4 is unlikely to underlie the observed effects of Hg on nicotinic acetylcholine receptors [238].
Modulation of dopamine D1 [239], D2 [240], and GABA [241], receptors by Hg, may also involve Hg-thiol group interaction, although the particular target Cys residues have yet to be identified (Table 1).
Table 1.
The estimated location of mercury (Hg)-binding cysteine (Cys) residues in protein (enzyme) molecules and the respective IC50 values for Hg-induced inhibition (where available).
| Enzyme | Cys | Reference (Cys) | IC50 | Reference (IC50) |
|---|---|---|---|---|
| Thioredoxin reductase (TrxR) | Cys497 Sec498 |
[203] | MeHg: cerebral – 0.158 μM hepatic – 0.071 μM renal – 0.078 μM |
[202] |
| HgCl2: 7.2 nM MeHg: 19.7 nM (rat) |
[201] | |||
| MeHg: 1.158 μM SH-SY5Y cells |
[243] | |||
| Glutathione reductase (GR) | Cys45 Cys50 |
[197] | HgCl2: 2.2 nM MeHg: 67.3 nM (rat) |
[201] |
| Sorbitol dehydrogenase (SDH) | Cys44 Cys119 Cys129 Cys164 |
[168] | MeHg: 25.3 ± 3.2 μM (rat) |
[168] |
| δ-aminolevulinate dehydratase (ALA-D) | Cys124 Cys132 |
[170] | HgCl2: Liver 389 ± 4.2 μM Kidneys 67.59 ± 4.3 μM Brain 46.29 ± 3.7 μM |
[169] |
| HgCl2: Brain 85.57 ± 18.72 μM Kidneys 92.14 ± 11.28 μM Liver 101.64 ± 11.54 μM |
[244] | |||
| MeHg: Brain – 79.3 μM Liver – 81.8 μM Kidneys – 39.1 μM |
[171] | |||
| Brain creatine kinase (CKB) | Cys283 | [170] | MeHg preincubation: 189.6 ± 1.18 μM (15 min) 87.0 ± 1.15 μM (60 min) |
[172] |
| Na,K-ATPase | Cys964 | [183] | HgCl2: 0.90 ± 0.10 μM brain 1.42 ± 0.29 μM RBC MeHg: 55.78 ± 1.50 μM brain 66.16 ± 0.60 μM RBC |
[245] |
| HgCl2: 0.76 ± 0.2 mM rat synaptosome |
[246] | |||
| Glutamine/amino acid transporter (ASCT2) | Cys207 Cys210 Cys233 |
[233] | HgCl2: 1.4 ± 0.10 μM MeHg: 2.4 ± 0.16 μM Mersalyl: 3.1 ± 0.19 μM |
[233] |
| Organic cation (carnitine) (OCTN2) transporter | Cys50 Cys136 |
[176] | HgCl2: 2.5 μM MeHg: 7.4 μM |
[175] |
| Mn-SOD | Cys196 | [189] | – | – |
| CREB | Cys286 | [207,247] | – | – |
| Keap1 | Cys151 Cys368 Cys489 (MeHg) |
[135] | – | – |
| Cys319 (MeHg-SG) |
[136] | – | – | |
| p50 (NF-κB) | Cys62 | [210] | – | – |
| Arginase I | Cys119 Cys168 Cys303 |
[215] | – | – |
| Ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1) | Cys152 | [217] | – | – |
| Tau construct R2 | Cys291 | [229] | – | – |
| Tau construct R3 | Cys322 | [230] | – | – |
| Glucocorticoid receptor | Cys736 | [242] | – | – |
| Recombinant helicase (BLM642–1290) | Cys940 Cys944 Cys1063 Cys1064 |
[174] | – | – |
Of note, Spulber et al. (2018) demonstrated that MeHg might bind glucocorticoid receptor at Cys736, thus altering the conformation of the ligand-binding site and reducing its activation that may significantly contribute to developmental neurotoxicity [242].
Generally, the existing data demonstrate that Me-Hg interaction has a significant impact on various types of neuronal receptors, thus providing a link to a wide spectrum of diseases associated with impaired neurotransmission. Evidence for Hg-induced cytoskeletal abnormalities due to Hg-SH interaction also provides an additional link between Hg exposure and neurodegeneration characterized by microtubule instability [225].
6. Concluding remarks
Data published to date demonstrate that different species of Hg (Hg2+, HgCH3) readily interact with thiol groups of endogenous molecules, which includes tubulin, GSH, ion channels, transporters, and enzymes therefore potentially change normal biological function. The latter may underlie a significant portion of toxic effects of Hg species through modulation of oxidative stress, apoptosis, leading to the ensuing neurotoxicity. In this regard, the use of thiol-group donors (NAC, ALA) may provide a valuable tool for modulation of Hg toxicity. However, the existing data on the role of Hg-SH binding in Hg toxicity are insufficient.
Multiple other thiol groups and protein molecules responsible for binding Hg(II) in vivo are still to be estimated. Despite the illusively clear association between Hg toxicity and thiol binding, the particular data are insufficient. Separation of Hg-binding proteins with a subsequent estimation of Hg-binding amino acid residues and in silico molecular approaches are essential for the assessment of binding Hg to thiol groups of protein molecules. Particular attention should be paid to Hg(I) binding to thiol groups, as data regarding this form of metal are extremely insufficient. Also, further assessment of the behavior of Hg in complex systems containing two or more thiol-rich ligands can provide an insight into the relative affinity of Hg cations to biological thiols and reveal particular mechanisms of Hg transport (e.g., following Rabenstein’s reaction).
Although binding of Hg to critical thiol groups of proteins causes inactivation of the target thiols residues, in some cases, the interaction of Hg with reactive thiols does not lead to enzyme or protein inactivation. The human genome codifies thousands of thiol-containing proteins that can be potential targets of Hg toxicity. However, our knowledge of the actual thiol-mediated targets of Hg is still elusive.
Moreover, it is expected that certain aspects of the adverse effects of Hg may be Hg-SH-independent. Therefore, further studies are required to estimate the particular role of Hg-thiol binding in the molecular mechanisms in Hg toxicology, as well as critical thiols, to efficiently improve the strategies to counteract negative effects of Hg overload.
Acknowledgments
The study was performed with the support of the Russian Ministry of Science and Higher Education, Project № 0856-2020-0008. MA was supported by NIH grants NIEHS R01 10563, R01ES07331, and NIEHS R01ES020852.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].International Programme on Chemical Safety, in, World Health Organization, Geneva, 2010. [Google Scholar]
- [2].Evers DC, Keane SE, Basu N, Buck D, Sci. Total Environ 569 (2016) 888–903. [DOI] [PubMed] [Google Scholar]
- [3].Streets DG, Horowitz HM, Lu Z, Levin L, Thackray CP, Sunderland EM, Atmos. Environ 201 (2019) 417–427. [Google Scholar]
- [4].Pacyna EG, Pacyna J, Sundseth K, Munthe J, Kindbom K, Wilson S, Steenhuisen F, Maxson P, Atmos. Environ 44 (2010) 2487–2499. [Google Scholar]
- [5].Sundseth K, Pacyna J, Pacyna E, Pirrone N, Thorne R, Int. J. Environ. Res. Public Health 14 (2017) 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kumar A, Wu S, Huang Y, Liao H, Kaplan JO, Atmos. Environ 173 (2018) 6–15. [Google Scholar]
- [7].Rafaj P, Bertok I, Cofala J, Schoepp W, Atmos. Environ 79 (2013) 472–479. [Google Scholar]
- [8].Budnik LT, Casteleyn L, Sci. Total Environ 654 (2019) 720–734. [DOI] [PubMed] [Google Scholar]
- [9].Basu N, Horvat M, Evers D, Zastenskaya I, Weihe P, Tempowski J, in: ISEE Conference Abstracts, 2018. [DOI] [PMC free article] [PubMed]
- [10].Chen P, Miah MR, Aschner M, F1000Research, 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tinkov AA, Ajsuvakova OP, Skalnaya MG, Popova EV, Sinitskii AI, Nemereshina ON, Gatiatulina ER, Nikonorov AA, Skalny AV, Biometals 28 (2015) 231–254. [DOI] [PubMed] [Google Scholar]
- [12].Orr SE, Barnes MC, Joshee L, Uchakina O, McKallip RJ, Bridges CC, Toxicol. Lett 304 (2019) 13–20. [DOI] [PubMed] [Google Scholar]
- [13].Genchi G, Sinicropi M, Carocci A, Lauria G, Catalano A, Int. J. Environ. Res. Public Health 14 (2017) 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Park J-D, Zheng W, Preventive Med J. Public Health 45 (2012) 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nedellec V, Rabl A, Risk Anal. 36 (2016) 2096–2104. [DOI] [PubMed] [Google Scholar]
- [16].Ynalvez R, Gutierrez J, Gonzalez-Cantu H, Biometals 29 (2016) 781–788. [DOI] [PubMed] [Google Scholar]
- [17].Antunes dos Santos A, Ferrer B, Marques Gonçalves F, Tsatsakis A, Renieri E, Skalny A, Farina M, Rocha J, Aschner M, Toxics, 6 (2018) 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pollard KM, Cauvi DM, Toomey CB, Hultman P, Kono DH, Biochim. Biophys. Acta (2019). [DOI] [PMC free article] [PubMed]
- [19].Bjørklund G, Dadar M, Mutter J, Aaseth J, Environ. Res 159 (2017) 545–554. [DOI] [PubMed] [Google Scholar]
- [20].Gould NS, Evans P, Martínez-Acedo P, Marino SM, Gladyshev VN, Carroll KS, Ischiropoulos H, Chem. Biol 22 (2015) 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clarkson TW, Magos L, Crit. Rev. Toxicol 36 (2006) 609–662. [DOI] [PubMed] [Google Scholar]
- [22].Clarkson TW, Vyas JB, Ballatori N, Am. J. Ind. Med 50 (2007) 757–764. [DOI] [PubMed] [Google Scholar]
- [23].Bjørklund G, Crisponi G, Nurchi VM, Cappai R, Buha Djordjevic A, Aaseth J, Molecules 24 (2019) 3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bernhoft RA, Environ J. Public Health 2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rettig P, Health Facilities Manage. 3 (1990) 60, 62, 64. [PubMed] [Google Scholar]
- [26].Bjørklund G, J. Orthomol. Med 10 (1995) 145–146. [Google Scholar]
- [27].Goldwater LJ, Mercury: A History of Quicksilver, York Press, Baltimore, 1972. [Google Scholar]
- [28].Macrae CF, Sovago I, Cottrell SJ, Galek PT, McCabe P, Pidcock E, Platings M, Shields GP, Stevens JS, Towler M, J. Appl. Crystallogr (2020). [DOI] [PMC free article] [PubMed]
- [29].Dorea JG, Farina M, Rocha JB, J. Appl. Toxicol 33 (2013) 700–711. [DOI] [PubMed] [Google Scholar]
- [30].Bradley M, Barst B, Basu N, Int. J. Environ. Res. Public Health 14 (2017) 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rand MD, Vorojeikina D, van Wijngaarden E, Jackson BP, Scrimale T, Zareba G, Love TM, Myers GJ, Watson GE, Toxicol. Sci 149 (2015) 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bridges CC, Zalups RK, Arch. Toxicol 91 (2017) 63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Syversen T, Kaur P, Trace Elem J. Med Biol. 26 (2012) 215–226. [DOI] [PubMed] [Google Scholar]
- [34].Ward DM, Nislow KH, Folt CL, Ann. N. Y. Acad. Sci 1195 (2010) 62–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nordberg GF, Fowler BA, Nordberg M, Toxicology of metals: overview, definitions, concepts, and trends, in: Handbook on the Toxicology of Metals, Elsevier, 2015, pp. 1–12. [Google Scholar]
- [36].Brandes N, Schmitt S, Jakob U, Antioxid. Redox Signal 11 (2009) 997–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mutter J, Naumann J, Guethlin C, Crit. Rev. Toxicol 37 (2007) 537–549. [DOI] [PubMed] [Google Scholar]
- [38].Nogara PA, Oliveira CS, Schmitz GL, Piquini PC, Farina M, Aschner M, Rocha JB, Biochim. Biophys. Acta (2019). [DOI] [PubMed]
- [39].Cember H, Gallagher P, Faulkner A, Am. Ind. Hyg. Assoc. J 29 (1968) 233–237. [DOI] [PubMed] [Google Scholar]
- [40].Zalups RK, Barfuss DW, J. Toxicol. Environ. Health Part A Curr. Issues 40 (1993) 77–103. [DOI] [PubMed] [Google Scholar]
- [41].Zalups RK, Pharmacol. Rev 52 (2000) 113–144. [PubMed] [Google Scholar]
- [42].Lau SJ, Sarkar B, J. Toxicol. Environ. Health, Part A Curr. Issues 5 (1979) 907–916. [DOI] [PubMed] [Google Scholar]
- [43].Li Y, He B, Hu L, Huang X, Yun Z, Liu R, Zhou Q, Jiang G, Talanta 178 (2018) 811–817. [DOI] [PubMed] [Google Scholar]
- [44].Areti S, Verma SK, Bellare J, Rao CP, Anal. Chem 88 (2016) 7259–7267. [DOI] [PubMed] [Google Scholar]
- [45].Feroci G, Badiello R, Fini A, Trace Elem J. Med. Biol 18 (2005) 227–234. [DOI] [PubMed] [Google Scholar]
- [46].Gutknecht J, J. Membr. Biol 61 (1981) 61–66. [Google Scholar]
- [47].Cheesman BV, Arnold AP, Rabenstein DL, J. Am. Chem. Soc 110 (1988) 6359–6364. [Google Scholar]
- [48].Aaseth J, Ajsuvakova OP, Skalny AV, Skalnaya MG, Tinkov AA, Coord. Chem. Rev 358 (2018) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liem-Nguyen V, Skyllberg U, Nam K, Björn E, Environ. Chem 14 (2017) 243–253. [Google Scholar]
- [50].Guzzi G, La Porta CA, Toxicology 244 (2008) 1–12. [DOI] [PubMed] [Google Scholar]
- [51].Shindo H, Brown TL, J. Am. Chem. Soc 87 (1965) 1904–1909. [DOI] [PubMed] [Google Scholar]
- [52].Divine KK, Ayala-Fierro F, Barber DS, Carter DE, J. Toxicol. Environ. Health Part A, 57 (1999) 489–505. [DOI] [PubMed] [Google Scholar]
- [53].Jalilehvand F, Leung BO, Izadifard M, Damian E, Inorg. Chem 45 (2006) 66–73. [DOI] [PubMed] [Google Scholar]
- [54].Gruff ES, Koch SA, J. Am. Chem. Soc 112 (1990) 1245–1247. [Google Scholar]
- [55].Taylor NJ, Carty AJ, J. Am. Chem. Soc 99 (1977) 6143–6145. [DOI] [PubMed] [Google Scholar]
- [56].Starý J, Kratzer K, J. Radioanal. Nucl. Chem 126 (1988) 69–75. [Google Scholar]
- [57].Lenz GR, Martell AE, Biochemistry 3 (1964) 745–750. [DOI] [PubMed] [Google Scholar]
- [58].Berthon G, Pure Appl. Chem 67 (1995) 1117–1240. [Google Scholar]
- [59].Matlock MM, Howerton BS, Van Aelstyn M, Henke KR, Atwood DA, Water Res. 37 (2003) 579–584. [DOI] [PubMed] [Google Scholar]
- [60].Bjørklund G, Crisponi G, Nurchi VM, Cappai R, Buha Djordjevic A, J. Aaseth. Mol 24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cardiano P, Falcone G, Foti C, Giuffrè O, Sammartano S, New J Chem. 35 (2011) 800–806. [Google Scholar]
- [62].Yamamoto R, Sumino K, Nakamae K, Arch. Toxicol 69 (1994) 127–131. [DOI] [PubMed] [Google Scholar]
- [63].Taylor NJ, Wong YS, Chieh PC, Carty AJ, J. Chem. Soc. Dalton Trans (1975) 438–442. [Google Scholar]
- [64].Mah V, Jalilehvand F, J. Biol. Inorg. Chem 13 (2008) 541–553. [DOI] [PubMed] [Google Scholar]
- [65].Heyrovský M, Mader P, Vavřička S, Veselá V, Fedurco M, J. Electroanal. Chem 430 (1997) 103–117. [Google Scholar]
- [66].Sen CK, J. Nutr. Biochem 8 (1997) 660–672. [Google Scholar]
- [67].Oram PD, Fang X, Fernando Q, Letkeman P, Letkeman D, Chem. Res. Toxicol 9 (1996) 709–712. [DOI] [PubMed] [Google Scholar]
- [68].Stricks W, Kolthoff I, J. Am. Chem. Soc 75 (1953) 5673–5681. [Google Scholar]
- [69].Kapoor R, Doughty G, Gorin G, Biochim. Biophys. Acta 100 (1965) 376–383. [DOI] [PubMed] [Google Scholar]
- [70].Neville GA, Drakenberg T, Acta Chem. Scand. B 28 (1974) 473–477. [DOI] [PubMed] [Google Scholar]
- [71].Fuhr BJ, Rabenstein DL, J. Am. Chem. Soc 95 (1973) 6944–6950. [DOI] [PubMed] [Google Scholar]
- [72].Burford N, Eelman MD, Groom K, J. Inorg. Biochem 99 (2005) 1992–1997. [DOI] [PubMed] [Google Scholar]
- [73].Krężel A, Bal W, Acta Biochim. Pol 46 (1999) 567–580. [PubMed] [Google Scholar]
- [74].Rabenstein DL, J. Am. Chem. Soc 95 (1973) 2797–2803. [DOI] [PubMed] [Google Scholar]
- [75].Plumlee G, Ziegler T, Lamothe P, Meeker G, Sutley S, in: AGU Fall Meeting Abstracts, 2003
- [76].Shoukry MM, Cheesman BV, Rabenstein DL, Can. J. Chem 66 (1988) 3184–3189. [Google Scholar]
- [77].Onyido I, Norris AR, Buncel E, Chem. Rev 104 (2004) 5911–5929. [DOI] [PubMed] [Google Scholar]
- [78].Fine Z, Wood TD, Int. J. Anal Mass Spectr. Cromatogr 1 (2013) 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Rabenstein DL, Acc. Chem. Res 11 (1978) 100–107. [Google Scholar]
- [80].Carrie AM, Walker MD, Williams DR, J. Chem. Soc. Dalton Trans (1976) 1012–1015. [Google Scholar]
- [81].Miller DM, Woods JS, Chem. Biol. Interact 88 (1993) 23–35. [DOI] [PubMed] [Google Scholar]
- [82].Shoukry MM, Transition Met. Chem 15 (1990) 1–4. [Google Scholar]
- [83].Stringari J, Nunes AK, Franco JL, Bohrer D, Garcia SC, Dafre AL, Milatovic D, Souza DO, Rocha JB, Aschner M, Toxicol. Appl. Pharmacol 227 (2008) 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kaur R, Garg P, Kaur S, Singh J, Chahal HS, Int. J. Med. Sci. Clin. Invention 5 (2018) 3491–3493. [Google Scholar]
- [85].Miura K, Clarkson TW, Toxicol. Appl. Pharmacol 118 (1993) 39–45. [DOI] [PubMed] [Google Scholar]
- [86].Hansen JM, Zhang H, Jones DP, Free Radic. Biol. Med 40 (2006) 138–145. [DOI] [PubMed] [Google Scholar]
- [87].Turell L, Radi R, Alvarez B, Free Radic. Biol. Med 65 (2013) 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Giustarini D, Dalle-Donne I, Lorenzini S, Milzani A, Rossi R, Gerontol J. A Biol. Sci. Med. Sci 61 (2006) 1030–1038. [DOI] [PubMed] [Google Scholar]
- [89].Grennan AK, Plant Physiol. 155 (2011) 1750–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lavergne V, Taft RJ, Alewood PF, Curr. Top Med. Chem 12 (2012) 1514–1533. [DOI] [PubMed] [Google Scholar]
- [91].Fanali G, Di Masi A, Trezza V, Marino M, Fasano M, Ascenzi P, Mol. Aspects Med 33 (2012) 209–290. [DOI] [PubMed] [Google Scholar]
- [92].Pires de Castro C, SouzaDe J, Bloch C Jr, Protein Pept. Lett 10 (2003) 155–164. [DOI] [PubMed] [Google Scholar]
- [93].Li Y, Yan X-P, Chen C, Xia Y-L, Jiang Y, J. Proteome Res 6 (2007) 2277–2286. [DOI] [PubMed] [Google Scholar]
- [94].Simpson R, J. Am. Chem. Soc 83 (1961) 4711–4717. [Google Scholar]
- [95].Yasutake A, Hirayama K, Inoue M, Arch. Toxicol 64 (1990) 639–643. [DOI] [PubMed] [Google Scholar]
- [96].Sudmeier JL, Pesek JJ, Inorg. Chem 10 (1971) 860–863. [Google Scholar]
- [97].Bal W, Sokołowska M, Kurowska E, Faller P, Biochim. Biophys. Acta 1830 (2013) 5444–5455. [DOI] [PubMed] [Google Scholar]
- [98].Kozłowski H, Bal W, Dyba M, Kowalik-Jankowska T, Coord. Chem. Rev 184 (1999) 319–346. [Google Scholar]
- [99].Varshney A, Sen P, Ahmad E, Rehan M, Subbarao N, Khan RH, Chirality 22 (2010) 77–87. [DOI] [PubMed] [Google Scholar]
- [100].Zhang Y, Wilcox DE, J. Biol. Inorg. Chem 7 (2002) 327–337. [DOI] [PubMed] [Google Scholar]
- [101].Peters T Jr, Serum albumin, in: Advances in protein chemistry, Elsevier, 1985, pp. 161–245. [DOI] [PubMed] [Google Scholar]
- [102].Curry S, Mandelkow H, Brick P, Franks N, Nat. Struct. Mol. Biol 5 (1998) 827. [DOI] [PubMed] [Google Scholar]
- [103].Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K, Protein Eng. 12 (1999) 439–446. [DOI] [PubMed] [Google Scholar]
- [104].Santos JCN, da Silva IM, Braga TC, de Fatima A, Figueiredo IM, Santos JCC, Int. J. Biol. Macromol 113 (2018) 1032–1040. [DOI] [PubMed] [Google Scholar]
- [105].Santos JCN, da Silva IM, Braga TC, de Fátima Â, Figueiredo IM, Santos JCC, Int. J. Biol. Macromol 113 (2018) 1032–1040. [DOI] [PubMed] [Google Scholar]
- [106].Goumakos W, Laussac J-P, Sarkar B, Biochem. Cell Biol 69 (1991) 809–820. [DOI] [PubMed] [Google Scholar]
- [107].Sadler PJ, Viles JH, Inorg. Chem 35 (1996) 4490–4496. [DOI] [PubMed] [Google Scholar]
- [108].Tan Q, Zhang M, Geng L, Xia Z, Li C, Usman M, Du Y, Wei L, Bi H, Toxicol. Appl. Pharmacol 362 (2019) 59–66. [DOI] [PubMed] [Google Scholar]
- [109].Nakamura M, Yasutake A, Fujimura M, Hachiya N, Marumoto M, Arch. Toxicol 85 (2011) 911–918. [DOI] [PubMed] [Google Scholar]
- [110].Palacios Ò, Capdevila M, Encyclopedia Metalloproteins (2013) 1386–1390. [Google Scholar]
- [111].Henkel G, Krebs B, Chem. Rev 104 (2004) 801–824. [DOI] [PubMed] [Google Scholar]
- [112].Babula P, Masarik M, Adam V, Eckschlager T, Stiborova M, Trnkova L, Skutkova H, Provaznik I, Hubalek J, Kizek R, Metallomics 4 (2012) 739–750. [DOI] [PubMed] [Google Scholar]
- [113].Leiva-Presa A, Capdevila M, Gonzalez-Duarte P, Eur. J. Biochem 271 (2004) 4872–4880. [DOI] [PubMed] [Google Scholar]
- [114].Vašák M, Hasler DW, Curr. Opin. Chem. Biol 4 (2000) 177–183. [DOI] [PubMed] [Google Scholar]
- [115].Muñoz A, Rodríguez AR, Electroanalysis 7 (1995) 674–680. [Google Scholar]
- [116].Lu W, Stillman MJ, J. Am. Chem. Soc 115 (1993) 3291–3299. [Google Scholar]
- [117].Lu W, Zelazowski AJ, Stillman MJ, Inorg. Chem 32 (1993) 919–926. [Google Scholar]
- [118].Shen JC, Huang ZY, Zhuang ZX, Wang XR, Lee FS, Appl. Organomet. Chem 19 (2005) 140–146. [Google Scholar]
- [119].Li Y, Fan Y, Zhao J, Xu X, Jing H, Shang L, Gao Y, Li B, Li Y-F, Biometals 29 (2016) 893–903. [DOI] [PubMed] [Google Scholar]
- [120].Pedrero Z, Ouerdane L, Mounicou S, Lobinski R, Monperrus M, Amouroux D, Metallomics 4 (2012) 473–479. [DOI] [PubMed] [Google Scholar]
- [121].Yasutake A, Nakamura M, J. Toxicol. Sci 36 (2011) 365–372. [DOI] [PubMed] [Google Scholar]
- [122].de Oliveira Lima LA, Bittencourt LO, Puty B, Fernandes RM, Nascimento PC, Silva MCF, Alves-Junior SM, Pinheiro JDJV, Lima RR, Biol. Trace Element Res 185 (2018) 135–142. [DOI] [PubMed] [Google Scholar]
- [123].Hwang TL, Chen HY, Changchien TT, Wang CC, Wu CM, Biomed. Rep 1 (2013) 379–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lee J-Y, Tokumoto M, Hwang G-W, Kim M-S, Takahashi T, Naganuma A, Yoshida M, Satoh M, Toxics 6 (2018) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Agrawal S, Flora G, Bhatnagar P, Flora S, Cell Mol. Biol. (Noisy-le-grand) 60 (2014) 13–21. [PubMed] [Google Scholar]
- [126].Yoshida M, Watanabe C, Horie K, Satoh M, Sawada M, Shimada A, Toxicol. Lett 155 (2005) 361–368. [DOI] [PubMed] [Google Scholar]
- [127].Yoshida M, Satoh M, Shimada A, Yamamoto E, Yasutake A, Tohyama C, Toxicology 175 (2002) 215–222. [DOI] [PubMed] [Google Scholar]
- [128].Wang Y, Zalups RK, Barfuss DW, Toxicol. Lett 213 (2012) 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Woods JS, Heyer NJ, Russo JE, Martin MD, Pillai PB, Farin FM, Neurotoxicol. Teratol 39 (2013) 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Dong G, Chen H, Qi M, Dou Y, Wang Q, Mol. Med. Rep 11 (2015) 1582–1586. [DOI] [PubMed] [Google Scholar]
- [131].Coyle P, Philcox J, Carey L, Rofe A, Cell. Mol. Life Sci. CMLS 59 (2002) 627–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Aschner M, Lorscheider F, Cowan K, Conklin DR, Vimy M, Lash L, Brain Res. 778 (1997) 222–232. [DOI] [PubMed] [Google Scholar]
- [133].Basu N, Goodrich JM, Head J, Environ. Toxicol. Chem 33 (2014) 1248–1258. [DOI] [PubMed] [Google Scholar]
- [134].Balamurugan K, Egli D, Selvaraj A, Zhang B, Georgiev O, Schaffner W, Biol. Chem 385 (2004) 597–603. [DOI] [PubMed] [Google Scholar]
- [135].Kumagai Y, Kanda H, Shinkai Y, Toyama T, Oxid. Med. Cell. Longevity 2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Yoshida E, Abiko Y, Kumagai Y, Chem. Res. Toxicol 27 (2014) 1780–1786. [DOI] [PubMed] [Google Scholar]
- [137].Gu J, Cheng Y, Wu H, Kong L, Wang S, Xu Z, Zhang Z, Tan Y, Keller BB, Zhou H, Diabetes 66 (2017) 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M, J. Biol. Chem 283 (2008) 33554–33562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Culbreth M, Aschner M, F1000Research 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Wang Y, Feng W, Xue W, Tan Y, Hein DW, Li X-K, Cai L, Diabetes 58 (2009) 1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Rossignol DA, The use of N-acetylcysteine as a chelator for metal toxicity, in: Frye RE, Berk M (Eds.), The Therapeutic Use of N-Acetylcysteine (NAC) in Medicine, Adis, Singapore, 2019, pp. 169–179. [Google Scholar]
- [142].Jalilehvand F, Parmar K, Zielke S, Metallomics 5 (2013) 1368–1376. [DOI] [PubMed] [Google Scholar]
- [143].Muniroh M, Khan N, Koriyama C, Akiba S, Vogel CF, Yamamoto M, Life Sci. 134 (2015) 16–21. [DOI] [PubMed] [Google Scholar]
- [144].Ekor M, Adesanoye OA, Farombi EO, Afr. J. Med. Med. Sci 39 (Suppl) (2010) 153–160. [PubMed] [Google Scholar]
- [145].Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA, Curr. Opin. Pharmacol 7 (2007) 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Chekmeneva E, Díaz-Cruz JM, Ariño C, Esteban M, Electroanalysis 22 (2010) 177–184. [Google Scholar]
- [147].Officioso A, Panzella L, Tortora F, Alfieri ML, Napolitano A, Manna C, Oxid. Med. Cell. Longevity 2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Yang T-Y, Xu Z-F, Liu W, Xu B, Deng Y, Li Y-H, Feng S, Environ. Toxicol. Pharmacol 39 (2015) 157–166. [DOI] [PubMed] [Google Scholar]
- [149].Zalups RK, J. Toxicol. Environ. Health Part A 55 (1998) 13–29. [DOI] [PubMed] [Google Scholar]
- [150].Hoffmeyer RE, Singh SP, Doonan CJ, Ross AR, Hughes RJ, Pickering IJ, George GN, Chem. Res. Toxicol 19 (2006) 753–759. [DOI] [PubMed] [Google Scholar]
- [151].Farina M, Campos F, Vendrell I, Berenguer J, Barzi M, Pons S, Suñol C, Toxicol. Sci 112 (2009) 416–426. [DOI] [PubMed] [Google Scholar]
- [152].Yin Z, Jiang H, Syversen T, Rocha JB, Farina M, Aschner M, J. Neurochem 107 (2008) 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zalups RK, Aslamkhan AG, Ahmad S, Kidney Int. 66 (2004) 251–261. [DOI] [PubMed] [Google Scholar]
- [154].Tanaka T, Naganuma A, Imura N, Toxicology 60 (1990) 187–198. [DOI] [PubMed] [Google Scholar]
- [155].Magos L, Clarkson TW, Allen J, Biochem. Pharmacol 27 (1978) 2203–2208. [DOI] [PubMed] [Google Scholar]
- [156].Ballatori N, Clarkson TW, Fundam. Appl. Toxicol 5 (1985) 816–831. [DOI] [PubMed] [Google Scholar]
- [157].Gregus Z, Varga F, Acta Pharmacol. Toxicol 56 (1985) 398–403. [DOI] [PubMed] [Google Scholar]
- [158].Ullah H, Khan MF, Jan SU, Shaikh U, Hashmat F, Ali MZ, Khan H, Gomal J Med. Sci 13 (2015) 30. [Google Scholar]
- [159].Fujiyama J, Hirayama K, Yasutake A, Biochem. Pharmacol 47 (1994) 1525–1530. [DOI] [PubMed] [Google Scholar]
- [160].Endo T, Sakata M, Pharmacol. Toxicol 76 (1995) 190–195. [DOI] [PubMed] [Google Scholar]
- [161].Roos DH, Puntel RL, Lugokenski TH, Ineu RP, Bohrer D, Burger ME, Franco JL, Farina M, Aschner M, Rocha JBT, Basic Clin. Pharmacol. Toxicol 107 (2010) 789–792. [DOI] [PubMed] [Google Scholar]
- [162].Mori N, Yamamoto M, Tsukada E, Yokooji T, Matsumura N, Sasaki M, Murakami T, Arch. Environ. Contam. Toxicol 63 (2012) 628–636. [DOI] [PubMed] [Google Scholar]
- [163].Zalups RK, Ahmad S, J. Pharmacol. Exp. Ther 315 (2005) 896–904. [DOI] [PubMed] [Google Scholar]
- [164].Bridges CC, Battle JR, Zalups RK, Toxicol. Appl. Pharmacol 221 (2007) 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Bridges CC, Zalups RK, J. Pharmacol. Exp. Ther 319 (2006) 948–956. [DOI] [PubMed] [Google Scholar]
- [166].Bridges CC, Krasnikov BF, Joshee L, Pinto JT, Hallen A, Li J, Zalups RK, Cooper AJ, Arch. Biochem. Biophys 517 (2012) 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Aliaga ME, López-Alarcón C, Barriga G, Olea-Azar C, Speisky H, J. Inorg. Biochem 104 (2010) 1084–1090. [DOI] [PubMed] [Google Scholar]
- [168].Kanda H, Toyama T, Shinohara-Kanda A, Iwamatsu A, Shinkai Y, Kaji T, Kikushima M, Kumagai Y, Arch. Toxicol 86 (2012) 1693–1702. [DOI] [PubMed] [Google Scholar]
- [169].Farina M, Brandao R, Lara F, Soares F, Souza D, Rocha J, Toxicol. Lett 139 (2003) 55–66. [DOI] [PubMed] [Google Scholar]
- [170].Oliveira CS, Nogara PA, Ardisson-Araújo DM, Aschner M, Rocha JB, Dórea JG, Neurodevelopmental effects of mercury, in: Advances in Neurotoxicology, Elsevier, 2018, pp. 27–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Rocha J, Freitas A, Marques M, Pereira M, Emanuelli T, Souza D, Braz. J. Med. Biol. Res 26 (1993) 1077–1083. [PubMed] [Google Scholar]
- [172].Glaser V, Leipnitz G, Straliotto MR, Oliveira J, dos Santos VV, Wannmacher CMD, de Bem AF, Rocha JBT, Farina M, Latini A, Neurotoxicology 31 (2010) 454–460. [DOI] [PubMed] [Google Scholar]
- [173].Buchachenko AL, Kouznetsov DA, Shishkov AV, J. Phys. Chem. A 108 (2004) 707–710. [Google Scholar]
- [174].Chen X, Luo H, Duan L, XU Q, Zhang Y, XU H, Biomed. Environ. Sci 24 (2011) 47. [DOI] [PubMed] [Google Scholar]
- [175].Pochini L, Peta V, Indiveri C, Toxicol. Mech. Methods 23 (2013) 68–76. [DOI] [PubMed] [Google Scholar]
- [176].Galluccio M, Pochini L, Peta V, Iannì M, Scalise M, Indiveri C, Toxicol. Sci 144 (2014) 105–113. [DOI] [PubMed] [Google Scholar]
- [177].Kumar SV, Maitra S, Bhattacharya S, Biometals 15 (2002) 51–57. [DOI] [PubMed] [Google Scholar]
- [178].Omotayo T, Rocha J, Ibukun E, Kade I, Neurochem. Int 58 (2011) 776–784. [DOI] [PubMed] [Google Scholar]
- [179].Anner B, Moosmayer M, Imesch E, Biochem. Biophys. Res. Commun 167 (1990) 1115–1121. [DOI] [PubMed] [Google Scholar]
- [180].Moreira C, Oliveira E, Bonan C, Sarkis J, Vassallo D, Comp. Biochem. Physiol. C: Toxicol. Pharmacol 135 (2003) 269–275. [DOI] [PubMed] [Google Scholar]
- [181].Bhattacharya S, Bose S, Mukhopadhyay B, Sarkar D, Das D, Bandyopadhyay J, Bose R, Majumdar C, Mondal S, Sen S, Biometals 10 (1997) 157–162. [DOI] [PubMed] [Google Scholar]
- [182].Wang X, Horisberger J-D, Mol. Pharmacol 50 (1996) 687–691. [PubMed] [Google Scholar]
- [183].Zichittella A, Shi H, Argüello J, J. Membr. Biol 177 (2000) 187–197. [DOI] [PubMed] [Google Scholar]
- [184].Tarabová B, Kurejová M, Sulová Z, Drabová M, Lacinová L.u., J. Pharmacol. Exp. Therap 317 (2006) 418–427. [DOI] [PubMed] [Google Scholar]
- [185].Nesci S, Trombetti F, Pirini M, Ventrella V, Pagliarani A, Chem. Biol. Interact 260 (2016) 42–49. [DOI] [PubMed] [Google Scholar]
- [186].Gradinaru R, Ionas A, Pui A, Zbancioc G, Drochioiu G, Biometals 24 (2011) 1115–1121. [DOI] [PubMed] [Google Scholar]
- [187].Shimojo N, Kumagai Y, Nagafune J, Arch. Toxicol 76 (2002) 383–387. [DOI] [PubMed] [Google Scholar]
- [188].Shinyashiki M, Kumagai Y, Homma-Takeda S, Nagafune J, Takasawa N, Suzuki J, Matsuzaki I, Satoh S, Sagai M, Shimojo N, Environ. Toxicol. Pharmacol 2 (1996) 359–366. [DOI] [PubMed] [Google Scholar]
- [189].Kumagai Y, Homma-Takeda S, Shinyashiki M, Shimojo N, Appl. Organomet. Chem 11 (1997) 635–643. [Google Scholar]
- [190].García-Sevillano M, Jara-Biedma R, González-Fernández M, García-Barrera T, Gómez-Ariza J, Biometals 26 (2013) 651–666. [DOI] [PubMed] [Google Scholar]
- [191].Chen L, Zhang J, Zhu Y, Zhang Y, RSC Adv. 5 (2015) 79874–79881. [Google Scholar]
- [192].Farina M, Aschner M, Biochim. Biophys. Acta (2019). [DOI] [PMC free article] [PubMed]
- [193].Lubos E, Loscalzo J, Handy DE, Antioxid. Redox Signal 15 (2011) 1957–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Branco V, Canário J, Lu J, Holmgren A, Carvalho C, Free Radic. Biol. Med 52 (2012) 781–793. [DOI] [PubMed] [Google Scholar]
- [195].Usuki F, Fujimura M, Arch. Toxicol 90 (2016) 917–926. [DOI] [PubMed] [Google Scholar]
- [196].Becker A, Soliman KF, Neurochem. Res 34 (2009) 1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Picaud T, Desbois A, Biochemistry 45 (2006) 15829–15837. [DOI] [PubMed] [Google Scholar]
- [198].Jeong C-H, Joo SH, J. Cancer Prevention 21 (2016) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Kim Y-J, Chai Y-G, Ryu J-C, Biochem. Biophys. Res. Commun 330 (2005) 1095–1102. [DOI] [PubMed] [Google Scholar]
- [200].Branco V, Carvalho C, Biochim. Biophys. Acta 1863 (2019) 129255. [DOI] [PubMed] [Google Scholar]
- [201].Carvalho CM, Chew E-H, Hashemy SI, Lu J, Holmgren A, J. Biol. Chem 283 (2008) (1923) 11913–11921. [DOI] [PubMed] [Google Scholar]
- [202].Wagner C, Sudati JH, Nogueira CW, Rocha JB, Biometals 23 (2010) 1171–1177. [DOI] [PubMed] [Google Scholar]
- [203].Carvalho CM, Lu J, Zhang X, Arnér ES, Holmgren A, FASEB J. 25 (2011) 370–381. [DOI] [PubMed] [Google Scholar]
- [204].Robitaille S, Mailloux RJ, Chan HM, Toxicol. Lett 256 (2016) 1–10. [DOI] [PubMed] [Google Scholar]
- [205].Ceccatelli S, Daré E, Moors M, Chem. Biol. Interact 188 (2010) 301–308. [DOI] [PubMed] [Google Scholar]
- [206].Shenker BJ, Guo TL, Shapiro IM, Environ. Res 84 (2000) 89–99. [DOI] [PubMed] [Google Scholar]
- [207].Unoki T, Akiyama M, Kumagai Y, Gonçalves FM, Farina M, Da Rocha JBT, Aschner M, Front. Genet 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Niture SK, Jaiswal AK, J. Biol. Chem 287 (2012) 9873–9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Culbreth M, Zhang Z, Aschner M, Neurotoxicology 62 (2017) 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Dieguez-Acuña FJ, Ellis ME, Kushleika J, Woods JS, Toxicol. Appl. Pharmacol 173 (2001) 176–187. [DOI] [PubMed] [Google Scholar]
- [211].Shumilla JA, Wetterhahn KE, Barchowsky A, Arch. Biochem. Biophys 349 (1998) 356–362. [DOI] [PubMed] [Google Scholar]
- [212].Huang ML-H, Chiang S, Kalinowski DS, Bae D-H, Sahni S, Richardson DR, Oxid. Med. Cell. Longevity 2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Mori M, J. Nutr 137 (2007) 1616S–1620S. [DOI] [PubMed] [Google Scholar]
- [214].Mittal CK, Harrell WB, Mehta CS, Mol. Cell. Biochem 149 (1995) 263–265. [DOI] [PubMed] [Google Scholar]
- [215].Kanda H, Sumi D, Endo A, Toyama T, Chen C-L, Kikushima M, Kumagai Y, Arch. Toxicol 82 (2008) 803. [DOI] [PubMed] [Google Scholar]
- [216].McCabe MJ Jr, Eckles KG, Langdon M, Clarkson TW, Whitekus MJ, Rosenspire AJ, Toxicol. Lett 155 (2005) 161–170. [DOI] [PubMed] [Google Scholar]
- [217].Toyama T, Abiko Y, Katayama Y, Kaji T, Kumagai Y, Toxicol J. Sci 40 (2015) 887–893. [DOI] [PubMed] [Google Scholar]
- [218].Tan Y-Y, Zhou H-Y, Wang Z-Q, Chen S-D, Mol. Cell. Biochem 318 (2008) 109–115. [DOI] [PubMed] [Google Scholar]
- [219].Takasugi N, Hiraoka H, Nakahara K, Akiyama S, Fujikawa K, Nomura R, Furuichi M, Uehara T, Int. J. Mol. Sci 20 (2019) 1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Makino K, Okuda K, Sugino E, Nishiya T, Toyama T, Iwawaki T, Fujimura M, Kumagai Y, Uehara T, Neurotox. Res 27 (2015) 99–105. [DOI] [PubMed] [Google Scholar]
- [221].Hiraoka H, Nakahara K, Kaneko Y, Akiyama S, Okuda K, Iwawaki T, Fujimura M, Kumagai Y, Takasugi N, Uehara T, Biol. Pharm. Bull 40 (2017) 1595–1598. [DOI] [PubMed] [Google Scholar]
- [222].Calabrese V, Sultana R, Scapagnini G, Guagliano E, Sapienza M, Bella R, Kanski J, Pennisi G, Mancuso C, Stella AMG, Antioxid. Redox Signal 8 (2006) 1975–1986. [DOI] [PubMed] [Google Scholar]
- [223].Franco JL, Teixeira A, Meotti FC, Ribas CM, Stringari J, Pomblum SCG, Moro ÂM, Bohrer D, Bairros AV, Dafre AL, Environ. Res 102 (2006) 22–28. [DOI] [PubMed] [Google Scholar]
- [224].McBean GJ, Aslan M, Griffiths HR, Torrão RC, Redox Biol. 5 (2015) 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Dubey J, Ratnakaran N, Koushika SP, Front. Cell. Neurosci 9 (2015) 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Bonacker D, Stoiber T, Wang M, Böhm KJ, Prots I, Unger E, Thier R, Bolt HM, Degen GH, Arch. Toxicol 78 (2004) 575–583. [DOI] [PubMed] [Google Scholar]
- [227].Vogel DG, Margolis RL, Mottet NK, Toxicol. Appl. Pharmacol 80 (1985) 473–486. [DOI] [PubMed] [Google Scholar]
- [228].Stoiber T, Bonacker D, Böhm KJ, Bolt HM, Thier R, Degen GH, Unger E, Mutat. Res 563 (2004) 97–106. [DOI] [PubMed] [Google Scholar]
- [229].Yang DJ, Shi S, Zheng LF, Yao TM, Ji LN, Biopolymers 93 (2010) 1100–1107. [DOI] [PubMed] [Google Scholar]
- [230].Yang D-J, Shi S, Yao T-M, Ji L-N, Bull. Chem. Soc. Jpn 84 (2011) 1362–1367. [Google Scholar]
- [231].LoPachin RM, Barber DS, Toxicol. Sci 94 (2006) 240–255. [DOI] [PubMed] [Google Scholar]
- [232].Soares FA, Farina M, Santos FW, Souza D, Rocha JB, Nogueira CW, Neurochem. Res 28 (2003) 1859–1865. [DOI] [PubMed] [Google Scholar]
- [233].Oppedisano F, Galluccio M, Indiveri C, Biochem. Pharmacol 80 (2010) 1266–1273. [DOI] [PubMed] [Google Scholar]
- [234].Aschner M, Eberle N, Goderie S, Kimelberg H, Brain Res. 521 (1990) 221–228. [DOI] [PubMed] [Google Scholar]
- [235].Basu N, Scheuhammer AM, Rouvinen-Watt K, Evans RD, Grochowina N, Chan LH, Neurotoxicology 29 (2008) 328–334. [DOI] [PubMed] [Google Scholar]
- [236].Castoldi AF, Candura SM, Costa P, Manzo L, Costa LG, Neurotoxicology 17 (1996) 735–741. [PubMed] [Google Scholar]
- [237].Basu N, Stamler CJ, Loua KM, Chan HM, Toxicol. Appl. Pharmacol 205 (2005) 71–76. [DOI] [PubMed] [Google Scholar]
- [238].Mirzoian A, Luetje CW, J. Pharmacol. Exp. Ther 302 (2002) 560–567. [DOI] [PubMed] [Google Scholar]
- [239].Braestrup C, Andersen PH, J. Neurochem 48 (1987) 1667–1672. [DOI] [PubMed] [Google Scholar]
- [240].Scheuhammer AM, Cherian MG, Biochem. Pharmacol 34 (1985) 3405–3413. [DOI] [PubMed] [Google Scholar]
- [241].Huang C-S, Narahashi T, Toxicol. Appl. Pharmacol 140 (1996) 508–520. [DOI] [PubMed] [Google Scholar]
- [242].Spulber S, Raciti M, Dulko-Smith B, Lupu D, Rüegg J, Nam K, Ceccatelli S, Toxicol. Appl. Pharmacol 354 (2018) 94–100. [DOI] [PubMed] [Google Scholar]
- [243].Meinerz DF, Branco V, Aschner M, Carvalho C, Rocha JBT, J. Appl. Toxicol 37 (2017) 1073–1081. [DOI] [PubMed] [Google Scholar]
- [244].Emanuelli T, Rocha J, Pereira M, Porciuncula L, Morsch V, Martins A, Souza D, Pharmacol. Toxicol 79 (1996) 136–143. [DOI] [PubMed] [Google Scholar]
- [245].Maier WE, Costa LG, Toxicol. Lett 51 (1990) 175–188. [DOI] [PubMed] [Google Scholar]
- [246].Vasić V, Jovanović D, Horvat A, Momić T, Nikezić G, Anal. Biochem 300 (2002) 113–120. [DOI] [PubMed] [Google Scholar]
- [247].Unoki T, Abiko Y, Toyama T, Uehara T, Tsuboi K, Nishida M, Kaji T, Kumagai Y, Sci. Rep 6 (2016) 28944. [DOI] [PMC free article] [PubMed] [Google Scholar]