

Abstract
Three decades of research in hematopoietic stem cell transplantation and HIV/AIDS fields have shaped a picture of immune restoration disorders. This manuscript overviews the molecular biology of interferon networks, the molecular pathogenesis of immune reconstitution inflammatory syndrome, and post-hematopoietic stem cell transplantation immune restoration disorders (IRD). It also summarizes the effects of thymic involution on T cell diversity, and the results of the assessment of diagnostic biomarkers of IRD, and tested targeted immunomodulatory treatments
Keywords: Immune reconstitution, Type I/II interferons, allo-HSCT, IFNG, T cells, AIDS, HIV, IRIS
Introduction
Alterations in the balanced recovery of innate and adaptive immunity represent the immune restoration disorders. The spectrum of them have been grown throughout the past decade, and their pathology somewhat clarified. The common theme among them is that they occurred in lymphopenic individuals, who can be divided into two large groups: 1. In patients with primary immunodeficiency state due to advanced HIV infection, who initiated antiretroviral therapies (ART), and 2. In transplant patients after high-dose chemo-radiation-therapy who underwent hematopoietic stem cell transplantation. These poorly defined conditions require prolonged infection surveillance, careful choice of anti-inflammatory medications, as well as expensive laboratory monitoring throughout the immune reconstitution phases. Because these patients experience either prolonged and severe immune deficiency due to HIV, or acute myeloablation due to treatment, the immune reconstitution kinetics differ, but the T cell immunity is particularly affected in both.
High occurrence of inflammatory immune restoration is exclusively reported in patients who initiated antiretroviral therapy after the onset of severe immunodeficiency. This disorder is reported as immune reconstitution inflammatory syndrome (IRIS), or immune reconstitution/restoration disease (IRD). IRIS is a common complication in AIDS patients who initiated ART. It can be described as a paradoxical inflammatory reaction that occurs in response to persistent opportunistic pathogens: mycobacterial, viral, fungal, or parasitic [1,2]. Among diverged clinical symptomatology, one common denominator is that the patient’s immune system begins improving due to ART and subsequently deteriorate [3]. Since the antigen-specific T cell-mediated immunity is nearly wiped out by HIV, the restoration of it after therapy initiation trigger a dysfunctional response against persisting antigens, whether present as whole microorganisms, or in the form of pathogen-associated molecules, which trigger a highly inflammatory immune activation [4].
The allogeneic hematopoietic stem cell transplantation (allo-HSCT) has proven to be lifesaving for many patients recovering from immuno-hematological malignancies [5]. Outcomes however are dependent on the eradication of malignant cells, donor compatibility, and on how successful is the reconstitution of recipients’ immuno-hematopoiesis [6]. The phases of immune reconstitution are usually assessed at several milestones, such as neutrophil engraftment, the innate immune recovery, and eventually an adaptive immune recovery, which may finalize by 24 months post-transplantation [7]. Delayed or incomplete immune reconstitution has been associated with significant morbidity and mortality, especially in adults due to infections, transplant rejection, or malignant disease relapse [8]. The restoration of T cell immune responses is increasingly recognized as the main determinant which sets apart favorable immune reconstitution from pathological one, which is accompanied by immune exhaustion, pathogenic autoimmune activation, or graft versus host reactions (GVHD) [9].
Three decades of research in transplantation and HIV fields have shaped a picture of immune restoration disorders. In the absence or poor recovery of T cells, other immune cells synergize their effort to control infections while preventing excessive inflammation or autoimmunity. Particularly, monocytes, natural killer cells (NK) or B cells are able to undergo faster homeostatic expansion and produce and overlapping repertoire of immune mediators (e.g. cytokines of interferon family). It remains unclear whether complete T cell immune reconstitution ever occurs in older individuals who are most susceptible to immune reconstitution disorders.
Thus, cumulative knowledge about innate and adaptive immune cells and the secretions of cytokines during immune recovery has an important clinical implication for assessment of favorable or the diagnosis of pathological immune reconstitution. One of the cytokine family we are going to highlight in this review is type I/II interferons, which exhibit defined kinetics during immune reconstitution phases. Type I/II interferon networks play a unique role in the connection between innate and adaptive immunity, therefore essential in normal and pathological immune reconstitution.
Interferon Signaling
Interferons (IFNs) are a group of signaling proteins that are being made and released by immune and non-immune cells to heighten their defenses against intracellular pathogens [10]. The release of interferons causes a range of flu-like symptoms from subfebrile fever, mild muscle or body aches, to cytokine storm-like symptoms when IFNs are overproduced along with other cytokines and pro-inflammatory molecules resulting in multiple organ failure. IFNs play a dual role in inflammation: pro- and anti-inflammatory [11]. IFNs enhance antigen presentation by virtue of increasing the expression of human leukocyte antigens (HLA) on antigen-presenting cells and enhancing lysis potentials of natural killer (NK) cells [12]. Interferons display protective anti-inflammatory functions via direct inhibition of pro-inflammatory cytokines, inductions of cytokine antagonists or re-directing the signaling through negative feedback loops [13]. All types of IFNs bind to a specific heteromeric cell surface receptor complexes known as the IFN receptors (IFNR) that play a crucial role in direction of many cellular processes toward pro- or anti-inflammatory outcomes [14]. Based on the type of receptor through which they signal, human interferons have been classified into three major types [15,16].
Interferon Type I, III
IFNs are long known to inhibit viral replication in virus-infected cells, thus represent one of the most important anti-viral innate immunity defenses [17,18]. Additionally, type I IFNs play a significant role in the response to bacterial infections [19]. Type I IFNs are composed of IFNB, IFNA that can further be classified into 13 different subtypes, and the extended family of IFN E/K/W [18]. A number of cells produce IFNA and IFNB, including macrophages/monocytes, fibroblasts, and endothelial cells also called natural interferon-producing cells. Additionally, plasmacytoid dendritic cells (pDCs) are able to secrete up to one thousand times more interferons type I than the others above [20].
Type I IFNs and more recently discovered type III IFNs (IFN-lambda, IFNL1–4) signaling rely on a group of intracellular transcription factors STAT 1–4 (signal transducer and activator of transcription 1–4). Activated through IFN receptors, these cells quickly secrete type I IFNs to enhance the cytotoxic function of NK, B, and T cells, which links innate and adaptive immune responses. Type I IFN binds to IFNAR receptor and activate a robust transcriptional pathway through a JAK-STAT signaling, interferon response factors (IRF 1–9), or partially overlapping but distinct interferon stimulated genes (ISGs), to action [21,22]. The transcriptional complexes activated by type I IFN signaling bind to specific interferon stimulating responsive element (ISRE) and gamma interferon activation sites (GAS) sequences within promoter regions of ISGs and lead to the expression of numerous genes important for cell death, cell proliferation and immune responses [23].
Although the specifics of IFN immune activation are complex, it appears to begin with activation of innate phagocytic leukocytes by antigens. Type I IFN synthesis is induced by two groups of antigenic challenges: the pathogen-associated molecular patterns (PAMPs) and danger-associated molecular pattern (DAMPs) through the pattern recognition receptors (PRRs). These antigen-sensor receptors can be found in the cytosol or in the endosome of cells. The signal is transmitted through four smaller networks of PRRs:
Toll-like receptors, TLRs1–11.
Nucleotide-binding oligomerization domain (NOD)-like receptors, NLRs.
Retinoic acid-inducible gene-1 (RIG1)-like receptors, RLR.
C-type lectin receptors, CLRs.
All these receptors utilize the common downstream serine/threonine protein kinases (e.g. TBK1 and IKKE) to transfer signals to IFN regulatory factors (IRFs) and nuclear factor kappa B (NFkB), and thus activating type I IFN transcription.
TLRs are cytoplasmic and endosomal receptors that are specialized in detecting specific PAMP molecules. For example, expression of type I and III IFNs can be induced upon recognition of viral components by TLR3, 7, or 9. TLR signaling is transmitted through TRIF (TRL3 and TLR4) or Myd88 (TLR4, 7, 8, and 9), and multiple IRFs to activate transcriptions of IFNA and IFNB in infected cell [24]. NLRs family is also activated by the same PAMPs (e.g. pathogen derived lipopolysaccharides, peptidoglycans, glycoproteins, RNA and DNA). The NOD2, for example, activates the downstream TBK1 and IRF5 signaling pathway, leading to the production of type I IFN [25].
However, NLR signaling frequently leads to the synthesis of absent in melanoma 2 (AIM2), interleukin 1 beta (IL1B) and IL18, the protein products of NLRP3-inflammasome activation. NLRP3 (NOD-like receptor family pyrin domain containing 3) pathway has been reported to negatively regulate the type I IFN production and set a pro-inflammatory state accompanied by necrosis [26]. Thus, NLR-inflammasome represents an alternative (to interferon type II) and very damaging route of responses to PAMPs [27]. The RLR activation by RNA viruses in numerous non-immune cells also leads to type I and III interferon production [28,29]. The upregulation of interferon and interferon response pathways helps limit viral replication, but since these defense pathways are accompanied by apoptosis it may contribute to slow immune reconstitution and immune complication, such as IRIS [30].
Recent information demonstrates the importance of type III IFNs in not only viral [31–33], but also in some fungal infections [34]. IFNLs are potent antiviral agents, with very mild pro-inflammatory effects, since they are not expressed in macrophages [35]. This fact may suggest important, yet unknown, role of IFNL in AIDS immune reconstitution. The production of IFNA and IFNB can be induced after recognition of fungal antigens through CLR signaling and IRF5. Sensing of some bacterial and fungal pathogens by C-type lectin receptors has also been reported to induce type I IFN production by innate immune cells, which may represent a clinically relevant path to understand the pathophysiology of immune reconstitution [36].
Interferon Type II
Type II IFN has only one representative, IFN gamma (IFNG). IFNG, originally known as the immune interferon, plays a key role in host defense against cellular and intracellular pathogens, including fungal, viral, bacterial and parasitic [37]. This cytokine plays a major role in mammalian adaptive immune responses, as it is secreted by activated CD8+ cytotoxic T cells and CD4+ T helper cells type 1 (Th1) [38]. IFNG possesses diverse biological properties, including immunomodulatory activities on innate immune cells, such as macrophages, monocytes, NK cells, and neutrophils [39,40]. The B and NK cells are also able to produce this cytokine, yet to a lesser extent [41,42].
Macrophages, activated by IFNG, exhibit improved microbial killing ability via heighten pinocytosis and phagocytosis. IFNG can also act as a cell growth inhibitor, via autocrine activation loop, directly triggering apoptosis in target cells (infected or transformed) by activating cytotoxic CD8+ T cells producing granzymes [43]. IFNG signals are transmitted via IFNG receptors (IFNGR1,2) which dictate the strength of interferon signaling [12]. Upon engagement with IFNG, IFNG receptors activate Janus-activated kinase 1 (JAK1), JAK2, and STAT1 signaling to regulate the transcription of many IFNG-inducible genes through activation of interferon-regulatory factors IRF1 and IRF2 [44,45]. The IRFs translocate to the nucleus members where they interact with interferon-stimulated response element (ISRE) to regulate expression of numerous interferon-stimulated genes (ISG) resulting in various physiological responses ranging from cell apoptosis, cell senescence to cell proliferation [46]. Involvement of IFNG in autophagy, inflammasome formation in target cells, or antibody-mediated complement activation is just beginning to be reported [47,48].
Interferon response comprises a series of reactions that alter the expression of a variety of human genes [49]. Since interferons shared signaling molecules downstream the receptors and common transcription factors, the overall effect on target cells depends on the density of different receptors, how well intracellular signaling is transmitted, and the level of soluble IFNs produced [50,51]. Interferons are of high importance for proper communication between innate and adaptive immunity (Figure 1). IFNG, for example, induces transcription of IL15 in monocytes which in turn promote the proliferation of memory CD8+ T cells, NK, and natural killer T (NKT) cells re-direct immune responses toward pro- and anti-inflammatory depending upon cellular milieu [52]. IFNG is activated by IL12 and IL18 which are secreted by dendritic cells, monocytes, macrophages, neutrophils and epithelial cells [53].
Figure 1:
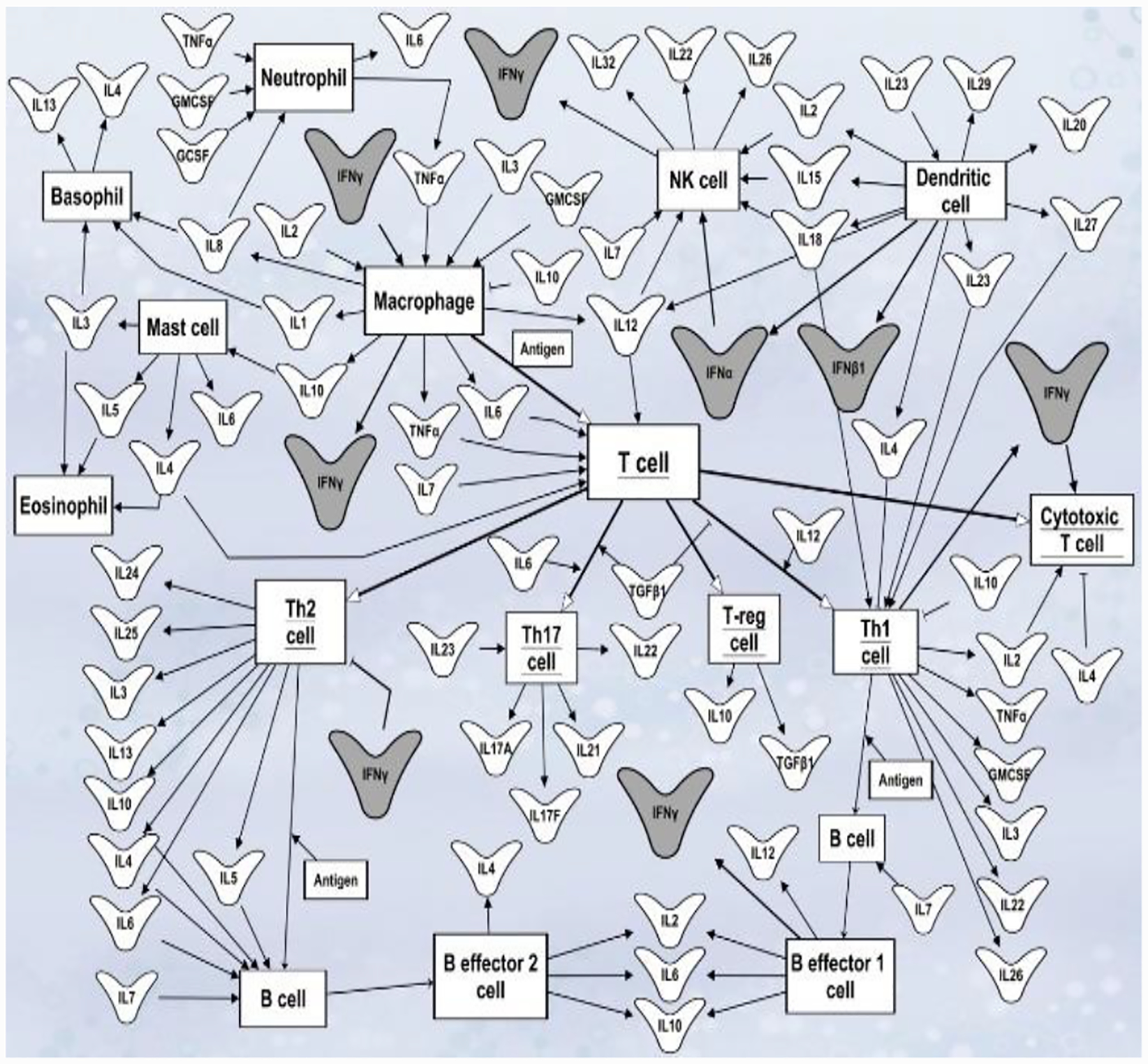
Role of cytokines in mediating communication between immune cells. Cytokines mediate cross-regulation of numerous signaling pathways between innate and adaptive immune cells. Outward arrows represent cytokine release. Point arrows represent cytokine activation function. The dull arrows represent cytokine inhibitory function. Cell’s names are shown in boxes. Interferons are highlighted in dark grey. This network diagram was built using Qiagen Pathway Assistant software.
IL27 plays an important role in naïve T cells clonal proliferation and differentiation into the Th1 lineage [54]. Cooperatively with IL12, it increases IFNG production by naïve T cells [55]. Human peripheral blood cells treated with type I IFN can increase dendritic cell maturation and IL12 production which increases priming and production of IFNG by T cells [56]. IL4 and IL10 are examples of negative regulators of IFNG production [57]. Addition of IFNB at the time of infection has been shown to negatively affect IFNG production via IL10-dependent- and independent mechanisms [58]. Conversely, IFNA acts synergistically with IFNG in development of T effector cells [59].
Interferons are required for communication between lymphocytes and macrophages, and play a unique role in macrophage M1-M2 polarization [60]. M1-IFNG type predominantly occurs during acute infection when proinflammatory M1 macrophages are stimulated by IFNG (along with PAMPs and TLRs). M1 type macrophages express CD86 and secrete inflammatory mediators like tumor necrosis factor alpha (TNFA), IL1B, IL6, IL8, IL12 and IL23 [61,62].
Activated M1 macrophages travel to the site of infection, induce inflammation via nitric oxide (NO) and reactive oxygen intermediates (ROI), and damage infected cells. Subsequently, polarized into M2 type by IL4, IL10, IL13, and transforming growth factor beta (TGFB), macrophages phagocytose cellular debris in order to resolve inflammation and to facilitate wound healing [63,64]. M2 macrophages abundantly express mannose receptor, dectin-1, CD163, CD209, scavenger receptor A and B1, CCR2, CXCR1, and CXCR2.
Additionally, M2 exhibit different metabolic profile: high production of ornithine and polyamines through the arginase pathway. The proper switch between M1/M2 phenotype is important to pro-resolution of inflammation, restoring Th1/Th2 balance and immune homeostasis during immune reconstitution [58,65–67]. However, inappropriate polarization drives disease pathology. In the absence of IFNG signaling (IFNR knockout), macrophage activation by type I IFNs is likely to take over, and this justify the beneficial effects of IFNA treatment in patients with compromised IFNG responses [68].
Thymic Involution and Effect on Interferon-Producing Cells
Thymus is a vital organ of the adaptive immune system. Thymopoiesis is a fundamental route for generation and maturation of naive T cells into CD4+ helper, CD8+ cytotoxic effector and CD4+CD25+ and Forkhead Box protein 3 (FOXP3+) regulatory T cells, among others [69,70]. The bone marrow-derived T cell progenitors traverse to the thymus, become committed to the T cell lineage, and undergo proliferative expansion and maturation [69]. During thymopoiesis, the T cell receptor (TCR) diversity is generated through recombinant rearrangement of variable, diversity and joiner genes, resulting in a broad antigen-specific repertoire of T cells [71].
At every stage of T cell development and maturation, T cells are sensitive to signals from cytokines. Age-related regression of thymus is a well-known phenomenon associated with a decline in naive T cell output and changes cytokine profiles [72]. Regression escalates during chronic HIV infection, or by treatments with chemo- or radio-therapeutic agents. When peripheral T cell populations are severely depleted, a renewal of thymic activity is essential to T cell reconstitution. Thymic involution is presented as a decrease in total thymic cellularity, increase in perivascular space, and disruption of the thymic architecture, which thought to contribute to the reduction in naïve T cell diversity and restriction in the peripheral TCR repertoire [73,74]. As seen in older individuals, thymic involution is linked with increased susceptibility to infections, autoimmune diseases, and cancer. Since T lymphocytes are major producers of IFNG, the health of thymus is the utmost importance for the development of IFNG-producing cells.
The immunosuppressive nature of pre-allo-HSCT conditioning therapies is toxic to pre-thymic, thymic, and post-thymic stages of T-cell development. Following the resolution of the acute insult, the thymus is somewhat capable of intrinsic recovery in younger patients, however the restoration of thymopoiesis in adults is highly questionable [75,76]. Myeloablative conditioning (MAC) consists of high doses of radiation or chemotherapy and aims to eradicate resistant cancer cells. MAC regimens result in the destruction of bone marrow cellularity and are lethal in one hundred percent of patients without immediate hematopoietic stem cell transplantation. However, many patients are unable to tolerate MAC, even with allo-HSCT. The reduced-intensity conditioning (RIC) was offered to older patients and to those with increased comorbidities [77]. Subsequent clinical studies comparing MAC and RIC showed no significant difference in overall survival (OS) between MAC and RIC groups [78,79].
The leukemia-free survival and non-relapse mortality also did not differ significantly between the compared groups. Moreover, the cumulative incidence of chronic GVHD and all types of infectious complications were less frequent with RIC than with MAC [78,79]. Avetisyan et. al. showed that allo-HSCT recipients receiving MAC had a higher CMV viral load and weakened T cell reconstitution as detected by low IFNG production than those receiving RIC [80]. Thus, RIC regimens are more favorable toward the thymic recovery and shorten the duration of post-transplant immunodeficiency, thereby reducing susceptibility to viral infections [81].
The homeostatic expansion of the peripheral T cell pool can facilitate T cell recovery within several months post-transplantation, although clearly the TCR repertoire diversity never recovers [69,82–84]. Immunosuppressive therapies to prevent graft versus host diseases (GVHD) during the post-transplant period also impairs thymic function and thus increase the risk of infections. Anti-thymocyte globulin (ATG) regimens used to prevent GVHD after allo-HSCT showed a significantly prolonged thymic dysfunction and delayed recovery of total CD4+ T cells in ATG-treated patients [85].
The post-HSCT thymic activity has conventionally been monitored radiologically and through immunophenotyping [86,87]. Peripheral naive and memory T cells can be distinguished by their expression patterns of cell surface markers, including CD62L, CCR7, CD27, CD45RO, CD45RA, CD28, CD103, or alpha E beta 7 integrin [88]. The quantification of recent thymic emigrants (RTEs) can be detected as TCR rearrangement DNA excision circles (TRECs) [89]. TRECs are indicators of recovery of naïve and memory T cell that significantly correlate with virologic suppression and improved long-term clinical outcome in adult allo-HSCT recipients [66,90].
Thymus regression is a feature of AIDS pathogenesis. Chronic HIV infection induces a substantial suppression of thymocyte proliferation. Resulted loss of generations of naïve T cells contributes to dysbalanced immune restoration in patients commencing ART [91]. For example, the disproportionate ratio of Th17/Treg (T helper 17/FOXP3+ T regulatory) yield T cell immune responses ineffective towards opportunistic infections, enhancing pro-inflammatory state and predisposing to IRIS [92]. An abnormal overrepresentation of cells with CD127 low FOXP3+ CD25+ Tregs phenotype, due to significant expansion, and a higher ratio of Tregs to effector/memory T cells was found in IRD patients as the main contributor to dysregulated of T cell repopulation [93]. The lower absolute number of Tregs pre-ART commencement was noted in patients who later develop IRIS [94]. The defects in IL7/IL7R(CD127) pathway are maybe behind poor reconstitution of thymic cell lineages, as IL7 cytokine is essential in primary T cells development [95].
Akilimali et al. reported that dysregulated production of IL7 and expression of IL-7R lead to aberrant T cell responses to cryptococcal antigens as an underlying factor in the immunopathogenesis of IRIS [96,97]. There have been attempts to restore thymic function and improve thymopoiesis with growth hormone (GH) in HIV-infected immunodeficient adults. GH treatment is associated with the significant increase in thymic density, the number of circulating TRECs and naive CD4+ T cells within peripheral blood monocyte population (PBMC) when co-administered at the initiation of antiretroviral therapy [98]. The use of GH is limited due to arthralgia, alteration in glucose metabolism and other harmful side effects in a significant number of patients [99].
Immune Reconstitution Inflammatory Syndrome in AIDS Patients
Normal and pathological kinetics of immune reconstitution have been observed in AIDS patients after initiation of antiretroviral therapy (ART). Unlike a normal immune system, which clears infection and returns to quiescence after being activated, the ART-related restoration of the immune function is often associated with a spike of pro-inflammatory responses to opportunistic pathogens. Immune reconstitution inflammatory syndrome (IRIS) is a prevalent complication in AIDS patients in sub- Saharan Africa and a significant cause of morbidity and mortality [100,101]. It can be described as a severe inflammatory reaction that occurs in response to numerous subclinical, latent, undiagnosed, or previously treated opportunistic infections. IRIS manifestations have multifaceted symptomatology, from meningitis or focal neurological signs to development of lymphadenopathy, pneumonitis, enlargements of Kaposi sarcoma lesions, etc. [102]. Paradoxically, IRIS symptoms develop in spite of the longitudinal decrease of HIV viral load, increased CD4+ T cell counts, and microbiologic treatment success as evidenced by improved antigen clearance [103,104]. Thus, one common denominator is that the patient’s immune system begins improving due to ART and subsequently deteriorates. The predominant morbidity occurs in patients’ populations that are co-infected with Mycobacterium tuberculosis and Cryptococcus species [105]. Therefore, AIDS clinicians and researchers are searching for better approaches to diagnose and treat these forms of IRIS.
Two common features put patients at risk for IRIS development are: 1. profound baseline immunosuppression with a median CD4+ T cell count of around 25 cells/uL; 2. high antigen burden in blood or cerebrospinal fluid (CSF) [106–108]. Few associations between levels of pro-inflammatory mediators at baseline followed by IRIS onset have been established in blood and CSF [109–111]. At presentation, IRIS is highly inflammatory, with the involvement of multiple mediators that appear systemically in blood as well as locally in organs such as lungs or CSF [110].
However, specific laboratory tests that can be used for IRIS detection have still not moved into routine patient care. The mechanisms that underlie the development of IRIS is still poorly understood. Patients with IRIS seem unable to control pathological inflammatory reactions, or properly regulate the immune activation pathways during reconstitution. Several immune pathways, including interferon regulatory pathways, are implicated in IRIS pathogenesis, suggesting a pathological switch from severe immunodepression to pronounced inflammatory state that occurs during IRIS event [112].
Interferon Signaling in the Immunopathogenesis of AIDS and IRIS
There is a wide array of evidence for the importance of interferons in the clearance of opportunistic infections, such as cryptococcosis or tuberculosis [113–115]. In immunocompetent patients, the pathogens activate the pathogen recognition receptors, which elicit intracellular signaling cascades in immune cells that rapidly lead to the activation of transcription factors, such as nuclear factor kappa B (NFkB) and IRFs, and secretion of interferons. This ultimately shapes the adaptive immune response and coordinate the elimination of pathogens and infected cells [116]. Primary immunodeficiency state, driven by CD4+ T cell loss, results in severe impairment of all branches of the immune system. In immunocompromised patients with CD4+ T cell count <50 cells/uL, low T cell responses, as evidenced by low production of IFNG, IL8, IL6, and TNFA, leads to inefficient cytotoxic response, macrophage activation and pathogen clearance [117].
Several immune abnormalities were identified before ART commencement in patients who developed IRIS in comparison with those who did not [118]. The predominant abnormality was the down-regulation of interferon-response genes in individuals who went on to develop IRIS. Poor cellular interferon responses in CSF and paucity of anti-viral gene expression in the blood at the time of ART initiation have been previously suggested to predispose patients to IRIS [119,120] and was also associated with higher mortality from IRIS [121].
Several studies reported very low interferon levels secretion by PBMC in response to antigenic stimuli prior to and after ART initiation as predictors of subsequent IRIS events [92,122]. Low concentration of IFNG at baseline may be explained by severe depletion of interferon-producing cells. Patients who exhibit such signature may require a longer course of antimicrobial therapy before initiation of ART to achieve clearance or supplemental immunotherapy [123]. Thus, an ineffective baseline immune response characterized by low production of interferons and interferon-response genes leads to poor antigen clearance and worsen outcomes.
In advanced AIDS the innate immune defense becomes the primary branch that is able to combat opportunistic infections. However, in the absence of T cell regulation, the antigen presentation is compromised by opportunistic diseases and skewed toward alternative M2 macrophage activation pathway [124,125]. Such macrophages/monocytes are unable to secrete protective concentrations of proinflammatory chemokines and cytokines (e.g. TNFA, CCR2 or IL6) at the site of infection in an attempt to attract lymphocytes during immune restoration, thus become more permissive to infection relapse [126]. After initiation of antimicrobial therapy and ART, the reduction of microbial load may result in re-polarization of monocyte/macrophage population toward M1, however, they become hyper-reactive toward regulatory stimuli, such as IFNG [127].
Kinetics of Immune Reconstitution on ART
The role for IFNs in AIDS immune reconstitution has been described in two studies. One was conducted in peripheral blood from advanced stage HIV-infected patients without opportunistic infections who were commenced on ART and did not exhibit IRIS. The other was conducted in the South African cohort of patients with opportunistic cryptococcal infections [118]. Both cohorts exhibited an upregulation of IFN I/II-STAT and NFkB pathways at baseline, followed by a subsequent decline as early as 4 weeks after ART initiation [128].
Similar gene expression is noticed in patients with active tuberculosis: they have initial upregulation of IFN genes that down-regulated following successful treatment [129,130]. Evidence of macrophage activation and protective levels of IL6 and IFNG, IL4, IL10, and IL17 in CSF or plasma associated with favorable baseline signature for subsequent recovery on ART [109,131]. The most plausible mechanism for this is maintaining innate host defenses in an active state through the direct stimulation of the immune system by the virus itself [132]. During favorable immune reconstitution, the longitudinal upregulation of cytokines such as IL7, IL2, HLA molecules during 12 weeks on ART reflect the effective immune recovery in lymphocyte populations and improvement of the antigen-presentation.
On the contrary, IFN pathway along with IL6 was upregulated within first few weeks on ART, and at the time of IRIS events, perhaps due to activation of cytotoxic NK cells and monocytes [103]. PBMCs from tuberculosis-IRIS patients exhibit high expression of cytotoxic mediators (perforin and granzyme B), which also suggest the involvement of cytotoxic natural killer T cells [133]. Longitudinal increase of plasma levels of IL2, IFNG, TNFA, IL17, and IL8 preceded IRIS and remained elevated at the time of IRIS [134]. It is unclear which immune cells are primarily responsible for the rise of cytokine levels. It is suggested that unbalanced restoration of T cell subpopulations and miscommunication with innate immune responses may play a role.
Majority of conducted studies described IRIS events that occurred during the first 3 weeks on ART, and thus called early IRIS. High levels of IL1, IL6, IL7, IL8, granulocyte colony stimulation factor (GCSF), or IL18 cytokines can be detected in patient’s plasma or serum at the time of early IRIS events [50,119,135]. During the first 2–3 months on ART, the gain in absolute numbers of CD4+ T cells rarely occurred more than 30 cells per microliter per month [136,137]. Thus, it is not surprising that immune activation occurs via NLR-inflammasome pathways, representing exaggerated innate cells response toward ongoing viral replication and microbial antigens.
It had been recently shown that the inflammasome pathway drives CD4+ T cell depletion in HIV-1 infection and delayed immune reconstitution [138]. Thus, inflammasome pathology seems to be behind the IRIS symptoms [139]. In IRIS patients involving the deterioration of the central nervous system, the inflammasome activation may represent a peripheral biomarker of brain inflammation that crosses the blood-brain barrier [137,140,141].
The similarity of transcriptomic biomarkers between cryptococcal IRIS and tuberculosis IRIS suggests that the symptoms of deterioration may involve activation of patrolling monocytes and neutrophils [142,143]. Triggered by PAMPs, these cells secrete pro-inflammatory mediators that can be observed in blood at the time of IRIS events [144]. Thus, during poor restoration of adaptive T cell immunity, the innate immune system activation is redirected via NLR-inflammasome route, resulting in more damage to target organs and accumulation of cellular debris.
The clinical manifestations of IRIS usually begin within the first four weeks of ART initiation; however, late presentation beyond 8 weeks has also been reported [105,145]. We recently described peripheral blood changes during cryptococcal late IRIS events that occurred after 10 weeks of immune reconstitution on ART [118]. The screening analysis revealed biomarker genes that encode a variety of molecules in T, B, NK cells and neutrophils. Significant differences in gene expression between early IRIS and late IRIS events suggest that late IRIS has distinct molecular phenotypes. High level of expression of IFNG and IL27 were discovered as biomarkers of late IRIS. In our study both Th1 and Th2 response genes were upregulated in late IRIS, other studies showed that Th2 responses were predominant [146].
The upregulation of numerous chemokines, chemokine receptors, and adhesion molecules preceded late IRIS events. The CSF chemokines expression was predictive of IRIS in other studies as well [109]. Thus, late IRIS showed a signature of heightened T cell proliferation, cytokine, and chemokine production, but delayed T cell maturation, leading to inability to resolve inflammation. The innate immune system activation is still present in late IRIS, due to impaired communication between players of activation/suppression in innate and adaptive immunity, and unresolved inflammation. The clinical implication of these findings is that the onset of late IRIS can be detected in peripheral blood through monitoring aberrant kinetics of immune reconstitution. This may provide an opportunity for intervention before clinical deterioration occurs.
Potential Treatment Approaches for Immune Reconstitution in AIDS
During immune reconstitution in AIDS patients the chronic inflammation which has not been resolved for years, derails the protective immune responses toward damaging. The clearance of opportunistic pathogens requires robust mobilization of Th1 type immunity and sufficient production of IFNG at the site of pathology. Thus, the addition of short-course IFNG to standard treatment may be beneficial to restore impaired communication between innate and adaptive immune branches. The assessment of the efficacy of adjunctive IFNG for the treatment of HIV-associated opportunistic infections has been performed by several groups of clinical investigators.
Adjunctive IFNG (given in addition to antimicrobial treatment) has been shown to be safe, with no adverse effect on CD4+ T cell count or viral load [147,148]. This is especially important since AIDS patients showed impaired type I IFN signaling [149]. A recent pre-clinical study provided evidence for the development of macrophage innate memory through IFNG priming which may lead the way to pathogen-specific vaccine development [150]. Adjunctive IFNA treatment in chronic HIV patients resulted in modest reductions of viral load, but poor recovery of CD4+T cells, prompting the majority of IFNA clinical trials to stop for futility [151].
Anti-TNF agents such as antibodies, chloroquine, pentoxifylline, or thalidomide can be useful if administered with ART to prevent or treat certain forms of IRIS [152–154]. The use of prednisone has successfully utilized for the treatment of TB-IRIS to reduce acute symptoms in the short term [152,155]. However, a combination of corticosteroids with intermittent regimens of IL2 increased T lymphocyte deaths [156,157]. Immunotherapy with IL2 alone was insufficient to improve immune restoration in AIDS patients, thus, proved to be low efficacy [158]. A deeper understanding of the pathophysiology of immune reconstitution and the immunopathogenesis of IRIS would perhaps shed light not only on the choice of immunomodulators [159] but also on the timing of ART initiation [160,161].
Kinetics of Immune Reconstitution in Allo-HSCT Recipients
Slow and dysbalanced immune reconstitution is a deprecatory issue for patients who undergo allo-HSCT, as it is associated with the increased risk of infection-associated mortality [162–170]. Allo-HSCT from HLA-matched sibling donors (MSD) generally provides the best clinical outcomes and thus is regarded as the gold standard for transplantation [171]. However, because only one-third of patients have an MSD, many patients receive allo-HSCT from banked umbilical cord blood (UCB) [172]. In adults, the UCB transplantation is associated with lower rates of GVHD [172], but with a significantly higher frequency of viral infections and delayed immune cell reconstitution. UCB immune cells are considered more immature and antigen-inexperienced, which may explain the poor recovery of T cell immunity and the higher risk of viral infections caused by human Epstein-Barr virus, adenovirus, baculovirus, herpes viruses or cytomegalovirus (CMV) [173–176]. To evaluate quantitative immune recoveries several studies have been conducted [167,177–181].
Recent comparisons of immune reconstitution rates in 157 adult recipients who received MSD or UCB revealed that natural killer (NK) cells and B cells exhibited higher quantitative rates of recovery in UCB recipients during the first 6 months to 1 year after transplantation [178]. However, UCB recipients had slower T cell subset recovery, with lower numbers of CD3+CD8+ (naïve and effector), CD4+ (naive and memory), and regulatory T cells than MSD recipients from day 60 to one year of observation. Delayed quantitative recoveries of T cell most likely explain the increased rate of reactivation of latent viral infections in UCB recipients. The observation of rapid quantitative recovery of NK and B cells in UCB patients support the hypothesis that other immune cells synergize their effort to control latent infections, but in the absence of thymic function and the full recovery of T cells, the immune reconstitution is inefficient.
It is hypothesized that the increased rate of viral infections could be due to delayed quantitative or functional recovery of immune cells, or both. Cytomegalovirus (CMV) specific response can be used to evaluate the functional role of T cells derived from the homeostatic proliferation of the graft and the T cells generated by thymic T cell neogenesis in the adult recipient.
Several studies used enzyme-linked immune absorbent spot assay (ELISPOT) to evaluate the IFNG production by CD4+ and CD8+T cells toward CMV antigens [182,183]. Results showed that patients with high IFNG production were protected from developing CMV infection, whereas patients with low IFNG production were significantly more prone to CMV disease progression and in need of antiviral therapy. A study [90] showed that although naive cord blood T cells transferred to adult UCB recipients could initiate immune responses to CMV and become CMV specific effectors as early as 8 weeks after transplantation. Yet, they failed to clear CMV viremia. Thus, assessment of IFNG responses may be clinically relevant to differentiate the uneventful immune recovery from the pathological immune reconstitution that leads to viral reactivation.
Treatment Approaches for Improvement of Allo-HSCT Immune Recovery
A number of immunotherapeutic approaches using T cell transfer to combat viral reactivation, improve the rates of immune reconstitution, as well as to prevent GVHD or disease relapse after allo-HSCT. There is clear evidence that infections can be treated by the adoptive transfer of T cells specifically targeting viral antigens [184]. Heslop et al. [185] showed that adoptively transferred EBV-specific cytotoxic CD4+ and CD8+ T lymphocytes can reconstitute the patient’s immune responses against EBV. Similarly, donor-derived adenovirus-specific T cells have been used for the treatment of patients with adenovirus infections after allo-HSCT [186]. Also, drug-refractory CMV infections after allo-HSCT have been successfully treated with CMV-specific T cells [187,188]. Invasive fungal infections, in particular, aspergillosis, represent another opportunistic life-threatening infection during immune reconstitution [189]. A clinical trial using aspergillus-specific T cell therapy showed suppression of antigenemia and prevention of invasive aspergillosis in a considerable number of patients [190]. Thus, the adoptive cellular immune therapy demonstrates high efficacy in restoring the anti-infectious T cell immunity after allo-HSCT [191].
Considering the importance of cytokine receptors in IFNG signaling cascade, several early phase clinical studies test cytokine agonist-receptor complexes. For example, IL15/IL15R complexes enhance immune activation in patients who relapsed within 60 days after allo-HSCT. The agonist complex was well-tolerated and did not increase the rate of adverse events [192]. Preclinical and clinical studies demonstrated the effectiveness of IL15 analogs to stimulate cytotoxic functions of CD8+ T cells and NK cells toward tumor antigens [193]. However, the limitation to its use is due to NK cell-mediated hyper-cytotoxicity though CD95, granzymes, and perforins, which is driven by abnormal IFNG production as shown in the settings of contact-dependent cardiac allograft rejection [194]. A phase 1 clinical trial of recombinant human IL7 (hIL7) in recipients of T cell-depleted allogeneic HSCT showed that CD3+, CD4+ and CD8+ counts are increased in hIL7 treated patients [195]. Exogenous administration of IL7 was found to enhance antigen-specific T cell responses to viral infections [196]. Thus, carries a promising potential for new treatment approaches for immune reconstitution disorders.
The efficacy of mesenchymal stem cells (MSC) transfer had been assessed to combat GVHD in ongoing clinical trials [197,198]. MSC are spindle-shaped multipotent progenitor cells with immunomodulatory capacities that reside in the bone marrow [199]. A pre-clinical study in mice showed that IFNG was required to initiate MSC efficacy. Recipients of T cells with poor IFNG secretion did not respond to MSC treatment and succumbed to GVHD. MSC, pre-treated with IFNG, became immediately active and suppress GVHD more efficiently [200]. Therefore, MSC may require activating signals from proliferating T cells to induce their suppressive effects. Gao et al. showed that repeated infusions of MSC might inhibit chronic GVHD symptoms in allo-HSCT patients, due to improved quantitative and functional recovery of T, B, and NK cell subtypes, leading to the acquisition of immune tolerance [198].
Adoptive transfer of Tregs has been additionally shown to be effective in the prevention and treatment of GVHD in preclinical models [201,202]. Tregs are able to inhibit immune responses without proinflammatory side effects and regulate immune cells from the adaptive and innate compartment including NK cells and antigen presenting cells (APCs) to prevent inflammation [203]. Clinical studies already revealed the potential of treatment with in vitro expanded Tregs [204–206]. Phase one clinical study presented that after Tregs transfusion, two of five patients showed clinical response with an improvement of chronic GVHD symptoms [205].
Conclusion and Future Prospective
Deciphering the pathogenesis of immune reconstitution disorders remains a challenge [1]. Lower levels of interferons, but higher levels of other cytokines have been suggested as a risk factor. Thus, it becomes rational to consider the possibility of simultaneous supplementation of some cytokines and neutralization of the others to provide long-term control of inflammation [207].
Adjunctive IFNG treatment has been investigated with various outcomes, depending upon regimens. Interferons are potent inducers of other cytokines and numerous interferon-response genes, many of which are key hematopoietic transcription factors. However, the chronic and excessive production of IFNG or repetitive supplementation with downstream cytokines (e.g. IL15) induces cell exhaustion, bone marrow failure, accompanied by anemia [208]. The definitive data supporting the beneficial effect of interferons in host protective immunity during homeostatic repopulation are still lacking.
There still several unanswered questions concerning the role of interferon networks during immune recovery [209]. Which phase of immune reconstitution would benefit from interferon therapy the most? Which cell type should be targeted early during immune reconstitution, and which one is later? Are systemic interferon biomarkers as informative as those measured at the site of inflammation? In cells, the secretion of endogenous interferons and expression of interferon-response genes can be regulated by other molecules through transcriptional, posttranscriptional, and posttranslational mechanisms [132,210–214]. Thus, the investigations to search for novel drugs to timely alter the kinetics of immune reconstitution will have significant implications to optimize outcomes [215].
The evidence suggests that immunoregulatory success will depend not only on suppression of inflammation but also on re-directing the immune response toward resolution. The assessment of metabolic signaling in the maintenance of immune homeostasis and the settings of immune reconstitution is also under intense investigation [216]. Assessment of host IFN or IFNR genes polymorphism as factors that influence thymic recovery and resistance to latent infections may lead to the development of novel therapeutic strategies to combat immune reconstitution disorders [217].
Footnotes
Conflict of Interests
None declared.
References
- 1.Delliere S, Guery R, Candon S, et al. Understanding pathogenesis and care challenges of immune reconstitution inflammatory syndrome in fungal infections. J Fungi (Basel) 2018; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boulougoura A, Sereti I. HIV infection and immune activation: The role of coinfections. Curr Opin HIV AIDS 2016; 11: 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kestens L, Seddiki N, Bohjanen PR. Immunopathogenesis of immune reconstitution disease in HIV patients responding to antiretroviral therapy. Curr Opin HIV AIDS 2008; 3: 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saharia KK, Koup RA. T cell susceptibility to HIV influences outcome of opportunistic infections. Cell 2013; 155: 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chabannon C, Kuball J, Bondanza A, et al. Hematopoietic stem cell transplantation in its 60s: A platform for cellular therapies. Sci Transl Med 2018; 10. [DOI] [PubMed] [Google Scholar]
- 6.Kolb HJ. Hematopoietic stem cell transplantation and cellular therapy. Hla 2017; 89: 267–277. [DOI] [PubMed] [Google Scholar]
- 7.Ogonek J, Kralj Juric M, Ghimire S, et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front Immunol 2016; 7: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norkin M, Wingard JR. Recent advances in hematopoietic stem cell transplantation. F1000Res 2017; 6: 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Koning C, Nierkens S, Boelens JJ. Strategies before, during, and after hematopoietic cell transplantation to improve T-cell immune reconstitution. Blood 2016; 128: 2607–2615. [DOI] [PubMed] [Google Scholar]
- 10.Stanifer ML, Pervolaraki K, Boulant S. Differential regulation of Type I and Type III interferon signaling. Int J Mol Sci 2019; 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Utay NS, Douek DC. Interferons and HIV infection: The good, the bad, and the ugly. Pathog Immun 2016; 1: 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hotter D, Kirchhoff F. Interferons and beyond: Induction of antiretroviral restriction factors. J Leukoc Biol 2018; 103: 465–477. [DOI] [PubMed] [Google Scholar]
- 13.Bolivar S, Anfossi R, Humeres C, et al. IFN-beta plays both pro- and anti-inflammatory roles in the rat cardiac fibroblast through differential STAT protein activation. Front Pharmacol 2018; 9: 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green DS, Young HA, Valencia JC. Current prospects of type II interferon gamma signaling and autoimmunity. J Biol Chem 2017; 292: 13925–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piehler J, Thomas C, Garcia KC, et al. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev 2012; 250: 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Weerd NA, Nguyen T. The interferons and their receptors--distribution and regulation. Immunol Cell Biol 2012; 90: 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haller O, Kochs G, Weber F. The interferon response circuit: Induction and suppression by pathogenic viruses. Virology 2006; 344: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McNab F, Mayer-Barber K, Sher A, et al. Type I interferons in infectious disease. Nat Rev Immunol 2015; 15: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder DT, Hedges JF, Jutila MA. Getting “inside” type I IFNs: Type I IFNs in intracellular bacterial infections. J Immunol Res 2017; 2017: 9361802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fitzgerald-Bocarsly P, Feng D. The role of type I interferon production by dendritic cells in host defense. Biochimie 2007; 89: 843–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Batra S, Sassano A, et al. Activation of mitogen-activated protein kinase kinase (MKK) 3 and MKK6 by type I interferons. J Biol Chem 2005; 280: 10001–10010. [DOI] [PubMed] [Google Scholar]
- 22.Rauch I, Muller M, Decker T. The regulation of inflammation by interferons and their STATs. Jakstat 2013; 2: e23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy DE, Marie IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol 2011; 1: 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah M, Anwar MA, Kim JH, et al. Advances in antiviral therapies targeting toll-like receptors. Expert Opin Investig Drugs 2016; 25: 437–453. [DOI] [PubMed] [Google Scholar]
- 25.Pandey AK, Yang Y, Jiang Z, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 2009; 5: e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Groslambert M, Py BF. Spotlight on the NLRP3 inflammasome pathway. J Inflamm Res 2018; 11: 359–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kopitar-Jerala N The role of interferons in inflammation and inflammasome activation. Front Immunol 2017; 8: 873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onoguchi K, Yoneyama M, Fujita T. Retinoic acid-inducible gene-I-like receptors. J Interferon Cytokine Res 2011; 31: 27–31. [DOI] [PubMed] [Google Scholar]
- 29.Raicevic G, Najar M, Busser H, et al. Comparison and immunobiological characterization of retinoic acid inducible gene-I-like receptor expression in mesenchymal stromal cells. Sci Rep 2017; 7: 2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singhania A, Wilkinson RJ, Rodrigue M, et al. The value of transcriptomics in advancing knowledge of the immune response and diagnosis in tuberculosis. Nat Immunol 2018; 19: 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pervolaraki K, Rastgou Talemi S, Albrecht D, et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog 2018; 14: e1007420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ank N, West H, Bartholdy C, et al. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol 2006; 80: 4501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zanoni I, Granucci F, Broggi A. Interferon (IFN)-lambda takes the helm: Immunomodulatory roles of Type III IFNs. Front Immunol 2017; 8: 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Espinosa V, Dutta O, McElrath C, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol 2017; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou JH, Wang YN, Chang QY, et al. Type III interferons in viral infection and antiviral immunity. Cell Physiol Biochem 2018; 51: 173–185. [DOI] [PubMed] [Google Scholar]
- 36.Troegeler A, Mercier I, Cougoule C, et al. C-type lectin receptor DCIR modulates immunity to tuberculosis by sustaining type I interferon signaling in dendritic cells. Proc Natl Acad Sci U S A 2017; 114: E540–e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schroder K, Hertzog PJ, Ravasi T, et al. Interferon-gamma: An overview of signals, mechanisms and functions. J Leukoc Biol 2004; 75: 163–189. [DOI] [PubMed] [Google Scholar]
- 38.Razaghi A, Owens L, Heimann K. Review of the recombinant human interferon gamma as an immunotherapeutic: Impacts of production platforms and glycosylation. J Biotechnol 2016; 240: 48–60. [DOI] [PubMed] [Google Scholar]
- 39.Kak G, Raza M, Tiwari BK. Interferon-gamma (IFN-gamma): Exploring its implications in infectious diseases. Biomol Concepts 2018; 9: 64–79. [DOI] [PubMed] [Google Scholar]
- 40.Kalyan S, Kabelitz D. When neutrophils meet T cells: Beginnings of a tumultuous relationship with underappreciated potential. Eur J Immunol 2014; 44: 627–633. [DOI] [PubMed] [Google Scholar]
- 41.Zhang S, Saha B, Kodys K, et al. IFN-gamma production by human natural killer cells in response to HCV-infected hepatoma cells is dependent on accessory cells. J Hepatol 2013; 59: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olalekan SA, Cao Y, Hamel KM, et al. B cells expressing IFN-gamma suppress Treg-cell differentiation and promote autoimmune experimental arthritis. Eur J Immunol 2015; 45: 988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhat P, Leggatt G, Waterhouse N, et al. Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis 2017; 8: e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFkappaB in Inflammation. Front Immunol 2018; 9: 2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol 2012; 32: 23–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alsamman K, El-Masry OS. Interferon regulatory factor 1 inactivation in human cancer. Biosci Rep 2018; 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie CB, Qin L, Li G, et al. Complement membrane attack complexes assemble NLRP3 inflammasomes triggering IL-1 activation of IFN-gamma-primed human endothelium. Circ Res 2019; 124: 1747–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang YP, Chen CL, Chen SO, et al. Autophagy facilitates an IFN-gamma response and signal transduction. Microbes Infect 2011; 13: 888–894. [DOI] [PubMed] [Google Scholar]
- 49.Michalska A, Blaszczyk K, Wesoly J, et al. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol 2018; 9: 1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goovaerts O, Jennes W, Massinga-Loembe M, et al. LPS-binding protein and IL-6 mark paradoxical tuberculosis immune reconstitution inflammatory syndrome in HIV patients. PLoS One 2013; 8: e81856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szelag M, Piaszyk-Borychowska A, Plens-Galaska M, et al. Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease. Oncotarget 2016; 7: 48788–48812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang X, Sun S, Hwang I, et al. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 1998; 8: 591–599. [DOI] [PubMed] [Google Scholar]
- 53.Giacomini E, Remoli ME, Gafa V, et al. IFN-beta improves BCG immunogenicity by acting on DC maturation. J Leukoc Biol 2009; 85: 462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Owaki T, Asakawa M, Morishima N, et al. A role for IL-27 in early regulation of Th1 differentiation. J Immunol 2005; 175: 2191–2200. [DOI] [PubMed] [Google Scholar]
- 55.Yu S, Jia L, Zhang Y, et al. IL-12 induced the generation of IL-21- and IFN-gamma-co-expressing poly-functional CD4+ T cells from human naive CD4+ T cells. Cell Cycle 2015; 14: 3362–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ismaili J, Olislagers V, Poupot R, et al. Human gamma delta T cells induce dendritic cell maturation. Clin Immunol 2002; 103: 296–302. [DOI] [PubMed] [Google Scholar]
- 57.Varano B, Fantuzzi L, Puddu P, et al. Inhibition of the constitutive and induced IFN-beta production by IL-4 and IL-10 in murine peritoneal macrophages. Virology 2000; 277: 270–277. [DOI] [PubMed] [Google Scholar]
- 58.McNab FW, Ewbank J, Howes A, et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol 2014; 193: 3600–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Curtsinger JM, Agarwal P, Lins DC, et al. Autocrine IFN-gamma promotes naive CD8 T cell differentiation and synergizes with IFN-alpha to stimulate strong function. J Immunol 2012; 189: 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol 2014; 5: 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee S, Huen S, Nishio H, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 2011; 22: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams H, Cassorla G, Pertsoulis N, et al. Human classical monocytes display unbalanced M1/M2 phenotype with increased atherosclerotic risk and presence of disease. Int Angiol 2017; 36: 145–155. [DOI] [PubMed] [Google Scholar]
- 63.Wolfs IM, Donners MM, de Winther MP. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb Haemost 2011; 106: 763–771. [DOI] [PubMed] [Google Scholar]
- 64.Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 2015; 30: 183–194. [DOI] [PubMed] [Google Scholar]
- 65.Chistiakov DA, Myasoedova VA, Revin VV, et al. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 2018; 223: 101–111. [DOI] [PubMed] [Google Scholar]
- 66.Slavcev A, Rybakova K, Svobodova E, et al. Pre-transplant donor-specific Interferon-gamma-producing cells and acute rejection of the kidney allograft. Transpl Immunol 2015; 33: 63–68. [DOI] [PubMed] [Google Scholar]
- 67.Boyette LB, Macedo C, Hadi K, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PLoS One 2017; 12: e0176460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreira-Teixeira L, Mayer-Barber K, Sher A, et al. Type I interferons in tuberculosis: Foe and occasionally friend. J Exp Med 2018; 215: 1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mackall CL, Fleisher TA, Brown MR, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med 1995; 332: 143–149. [DOI] [PubMed] [Google Scholar]
- 70.Shen Z, Chen L, Hao F, et al. Transcriptional regulation of Foxp3 gene: Multiple signal pathways on the road. Med Res Rev 2009; 29: 742–766. [DOI] [PubMed] [Google Scholar]
- 71.Hakim FT, Memon SA, Cepeda R, et al. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest 2005; 115: 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee N, Shin MS, Kang KS, et al. Human monocytes have increased IFN-gamma-mediated IL-15 production with age alongside altered IFN-gamma receptor signaling. Clin Immunol 2014; 152: 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Surh CD, Sprent J. Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J Exp Med 2000; 192: F9–F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murray JM, Kaufmann GR, Hodgkin PD, et al. Naive T cells are maintained by thymic output in early ages but by proliferation without phenotypic change after age twenty. Immunol Cell Biol 2003; 81: 487–495. [DOI] [PubMed] [Google Scholar]
- 75.Alpdogan O, Hubbard VM, Smith OM, et al. Keratinocyte growth factor (KGF) is required for postnatal thymic regeneration. Blood 2006; 107: 2453–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dudakov JA, Hanash AM, Jenq RR, et al. Interleukin-22 drives endogenous thymic regeneration in mice. Science 2012; 336: 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Slavin S, Nagler A, Naparstek E, et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood 1998; 91: 756–763. [PubMed] [Google Scholar]
- 78.Alatrash G, Kidwell KM, Thall PF, et al. Reduced intensity vs. myeloablative conditioning with fludarabine and PK-guided busulfan in allogeneic stem cell transplantation for patients with AML/MDS. Bone Marrow Transplant 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim SH, Kee SY, Lee DG, et al. Infectious complications following allogeneic stem cell transplantation: Reduced intensity vs. myeloablative conditioning regimens. Transpl Infect Dis 2013; 15: 49–59. [DOI] [PubMed] [Google Scholar]
- 80.Ljungman P, Perez-Bercoff L, Jonsson J, et al. Risk factors for the development of cytomegalovirus disease after allogeneic stem cell transplantation. Haematologica 2006; 91: 78–83. [PubMed] [Google Scholar]
- 81.Krenger W, Blazar BR, Holländer GA. Thymic T-cell development in allogeneic stem cell transplantation. Blood 2011; 117: 6768–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature 1999; 402: 255–262. [DOI] [PubMed] [Google Scholar]
- 83.Weinberg K, Annett G, Kashyap A, et al. The effect of thymic function on immunocompetence following bone marrow transplantation. Biol Blood Marrow Transplant 1995; 1: 18–23. [PubMed] [Google Scholar]
- 84.Storek J, Witherspoon RP, Storb R. T cell reconstitution after bone marrow transplantation into adult patients does not resemble T cell development in early life. Bone Marrow Transplant 1995; 16: 413–425. [PubMed] [Google Scholar]
- 85.Na IK, Wittenbecher F, Dziubianau M, et al. Rabbit antithymocyte globulin (thymoglobulin) impairs the thymic output of both conventional and regulatory CD4+ T cells after allogeneic hematopoietic stem cell transplantation in adult patients. Haematologica 2013; 98: 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roux E, Dumont-Girard F, Starobinski M, et al. Recovery of immune reactivity after T-cell-depleted bone marrow transplantation depends on thymic activity. Blood 2000; 96: 2299–2303. [PubMed] [Google Scholar]
- 87.Nasseri F, Eftekhari F. Clinical and radiologic review of the normal and abnormal thymus: Pearls and pitfalls. Radiographics 2010; 30: 413–428. [DOI] [PubMed] [Google Scholar]
- 88.Ribeiro RM, Perelson AS. Determining thymic output quantitatively: Using models to interpret experimental T-cell receptor excision circle (TREC) data. Immunol Rev 2007; 216: 21–34. [DOI] [PubMed] [Google Scholar]
- 89.Douek DC, McFarland RD, Keiser PH, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature 1998; 396: 690–695. [DOI] [PubMed] [Google Scholar]
- 90.Brown JA, Stevenson K, Kim HT, et al. Clearance of CMV viremia and survival after double umbilical cord blood transplantation in adults depends on reconstitution of thymopoiesis. Blood 2010; 115: 4111–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ho Tsong Fang R, Colantonio AD, Uittenbogaart CH. The role of the thymus in HIV infection: A 10 year perspective. Aids 2008; 22: 171–184. [DOI] [PubMed] [Google Scholar]
- 92.Meintjes G, Wilkinson KA, Rangaka MX, et al. Type 1 helper T cells and FoxP3-positive T cells in HIV-tuberculosis-associated immune reconstitution inflammatory syndrome. Am J Respir Crit Care Med 2008; 178: 1083–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seddiki N, Sasson SC, Santner-Nanan B, et al. Proliferation of weakly suppressive regulatory CD4+ T cells is associated with over-active CD4+ T-cell responses in HIV-positive patients with mycobacterial immune restoration disease. Eur J Immunol 2009; 39: 391–403. [DOI] [PubMed] [Google Scholar]
- 94.Zaidi I, Peterson K, Jeffries D, et al. Immune reconstitution inflammatory syndrome and the influence of T regulatory cells: A cohort study in The Gambia. PLoS One 2012; 7: e39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol 2011; 11: 330–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Akilimali NA, Chang CC, Muema DM, et al. Plasma but not cerebrospinal fluid interleukin 7 and interleukin 5 levels pre-antiretroviral therapy commencement predict cryptococcosis-associated immune reconstitution inflammatory syndrome. Clin Infect Dis 2017; 65: 1551–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Akilimali NA, Muema DM, Specht C, et al. Cryptococcosis-associated immune reconstitution inflammatory syndrome is associated with dysregulation of IL-7/IL-7 receptor signaling pathway in T cells and monocyte activation. J Acquir Immune Defic Syndr 2019; 80: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ye P, Kirschner DE, Kourtis AP. The thymus during HIV disease: Role in pathogenesis and in immune recovery. Curr HIV Res 2004; 2: 177–183. [DOI] [PubMed] [Google Scholar]
- 99.Lindboe JB, Langkilde A, Eugen-Olsen J, et al. Low-dose growth hormone therapy reduces inflammation in HIV-infected patients: A randomized placebo-controlled study. Infect Dis (Lond) 2016; 48: 829–837. [DOI] [PubMed] [Google Scholar]
- 100.Chang CC, Sheikh V, Sereti I, et al. Immune reconstitution disorders in patients with HIV infection: From pathogenesis to prevention and treatment. Curr HIV/AIDS Rep 2014; 11: 223–232. [DOI] [PubMed] [Google Scholar]
- 101.Rajasingham R, Smith RM, Park BJ, et al. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect Dis 2017; 17: 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walker NF, Scriven J, Meintjes G, et al. Immune reconstitution inflammatory syndrome in HIV-infected patients. HIV AIDS (Auckl) 2015; 7: 49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meya DB, Okurut S, Zziwa G, et al. Cellular immune activation in cerebrospinal fluid from ugandans with cryptococcal meningitis and immune reconstitution inflammatory syndrome. J Infect Dis 2015; 211: 1597–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Musubire AK, Meya BD, Mayanja-Kizza H, et al. Challenges in diagnosis and management of Cryptococcal immune reconstitution inflammatory syndrome (IRIS) in resource limited settings. Afr Health Sci 2012; 12: 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Meya DB, Manabe YC, Boulware DR, et al. The immunopathogenesis of cryptococcal immune reconstitution inflammatory syndrome: Understanding a conundrum. Current opinion in infectious diseases 2016; 29: 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chang CC, Dorasamy AA, Gosnell BI, et al. Clinical and mycological predictors of cryptococcosis-associated immune reconstitution inflammatory syndrome. AIDS 2013; 27: 2089–2099. [DOI] [PubMed] [Google Scholar]
- 107.Narendran G, Andrade BB, Porter BO, et al. Paradoxical tuberculosis immune reconstitution inflammatory syndrome (TB-IRIS) in HIV patients with culture confirmed pulmonary tuberculosis in India and the potential role of IL-6 in prediction. PLoS One 2013; 8: e63541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Siedner MJ, Ng CK, Bassett IV, et al. Trends in CD4 count at presentation to care and treatment initiation in sub-Saharan Africa, 2002–2013: A meta-analysis. Clinical infectious diseases: An Official Publication of the Infectious Diseases Society of America 2015; 60: 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jarvis JN, Meintjes G, Bicanic T, et al. Cerebrospinal fluid cytokine profiles predict risk of early mortality and immune reconstitution inflammatory syndrome in HIV-associated cryptococcal meningitis. PLoS Pathog 2015; 11: e1004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boulware DR, Meya DB, Bergemann TL, et al. Clinical features and serum biomarkers in HIV immune reconstitution inflammatory syndrome after cryptococcal meningitis: a prospective cohort study. PLoS Med 2010; 7: e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chang CC, Omarjee S, Lim A, et al. Chemokine levels and chemokine receptor expression in the blood and the cerebrospinal fluid of HIV-infected patients with cryptococcal meningitis and cryptococcosis-associated immune reconstitution inflammatory syndrome. J Infect Dis 2013; 208: 1604–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gopal R, Rapaka RR, Kolls JK. Immune reconstitution inflammatory syndrome associated with pulmonary pathogens. Eur Respir Rev 2017; 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Elsegeiny W, Marr KA, Williamson PR. Immunology of Cryptococcal infections: Developing a rational approach to patient therapy. Front Immunol 2018; 9: 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Donovan ML, Schultz TE, Duke TJ, et al. Type I interferons in the pathogenesis of tuberculosis: Molecular drivers and immunological consequences. Front Immunol 2017; 8: 1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chin KL, Anis FZ, Sarmiento ME, et al. Role of interferons in the development of diagnostics, vaccines, and therapy for tuberculosis. J Immunol Res 2017; 2017: 5212910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lai RP, Meintjes G, Wilkinson KA, et al. HIV-tuberculosis-associated immune reconstitution inflammatory syndrome is characterized by Toll-like receptor and inflammasome signalling. Nat Commun 2015; 6: 8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meya DB, Okurut S, Zziwa G, et al. HIV-associated Cryptococcal immune reconstitution inflammatory syndrome is associated with aberrant t cell function and increased cytokine responses. J Fungi (Basel) 2019; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vlasova-St Louis I, Chang CC, Shahid S, et al. Transcriptomic predictors of paradoxical cryptococcosis-associated immune reconstitution inflammatory syndrome. Open Forum Infect Dis 2018; 5: ofy157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boulware DR, Bonham SC, Meya DB, et al. Paucity of initial cerebrospinal fluid inflammation in cryptococcal meningitis is associated with subsequent immune reconstitution inflammatory syndrome. J Infect Dis 2010; 202: 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Boulware DR, von Hohenberg M, Rolfes MA, et al. Human immune response varies by the degree of relative cryptococcal antigen shedding. Open forum infectious diseases 2016; 3: ofv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Scriven JE, Graham LM, Schutz C, et al. A glucuronoxylomannan-associated immune signature, characterized by monocyte deactivation and an increased interleukin 10 level, is a predictor of death in cryptococcal meningitis. J Infect Dis 2016; 213: 1725–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chang CC, Lim A, Omarjee S, et al. Cryptococcosis-IRIS is associated with lower cryptococcus-specific IFN-gamma responses before antiretroviral therapy but not higher T-cell responses during therapy. J Infect Dis 2013; 208: 898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dyal J, Akampurira A, Rhein J, et al. Reproducibility of CSF quantitative culture methods for estimating rate of clearance in cryptococcal meningitis. Medical mycology 2016; 54: 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Davis MJ, Tsang TM, Qiu Y, et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio 2013; 4: e00264–00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Panackal AA, Wuest SC, Lin YC, et al. Paradoxical immune responses in non-HIV cryptococcal meningitis. PLoS Pathog 2015; 11: e1004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Heung LJ, Hohl TM. Inflammatory monocytes are detrimental to the host immune response during acute infection with Cryptococcus neoformans. PLoS Pathog 2019; 15: e1007627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Meya DB, Okurut S, Zziwa G, et al. Monocyte phenotype and ifn-gamma-inducible cytokine responses are associated with cryptococcal immune reconstitution inflammatory syndrome. J Fungi (Basel) 2017; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boulware DR, Meya DB, Bergemann TL, et al. Antiretroviral therapy down-regulates innate antiviral response genes in patients with AIDS in sub-saharan Africa. Journal of acquired immune deficiency syndromes 2010; 55: 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bloom CI, Graham CM, Berry MP, et al. Detectable changes in the blood transcriptome are present after two weeks of antituberculosis therapy. PLoS One 2012; 7: e46191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Berry MP, Graham CM, McNab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010; 466: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Musselwhite LW, Andrade BB, Ellenberg SS, et al. Vitamin D, D-dimer, Interferon gamma, and sCD14 Levels are independently associated with immune reconstitution inflammatory syndrome: A prospective, international study. EBioMedicine 2016; 4: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Doyle T, Goujon C, Malim MH. HIV-1 and interferons: Who’s interfering with whom? Nat Rev Microbiol 2015; 13: 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wilkinson KA, Walker NF, Meintjes G, et al. Cytotoxic mediators in paradoxical HIV-tuberculosis immune reconstitution inflammatory syndrome. J Immunol 2015; 194: 1748–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Grant PM, Komarow L, Lederman MM, et al. Elevated interleukin 8 and T-helper 1 and T-helper 17 cytokine levels prior to antiretroviral therapy in participants who developed immune reconstitution inflammatory syndrome during ACTG A5164. J Infect Dis 2012; 206: 1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Barber DL, Andrade BB, McBerry C, et al. Role of IL-6 in Mycobacterium avium--associated immune reconstitution inflammatory syndrome. J Immunol 2014; 192: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Corbeau P, Reynes J. Immune reconstitution under antiretroviral therapy: The new challenge in HIV-1 infection. Blood 2011; 117: 5582–5590. [DOI] [PubMed] [Google Scholar]
- 137.Tan HY, Yong YK, Andrade BB, et al. Plasma interleukin-18 levels are a biomarker of innate immune responses that predict and characterize tuberculosis-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2015; 29: 421–431. [DOI] [PubMed] [Google Scholar]
- 138.Doitsh G, Galloway NL, Geng X, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014; 505: 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tan HY, Yong YK, Shankar EM, et al. Aberrant inflammasome activation characterizes tuberculosis-associated immune reconstitution inflammatory syndrome. J Immunol 2016; 196: 4052–4063. [DOI] [PubMed] [Google Scholar]
- 140.Marais S, Lai RP, Wilkinson KA, et al. Inflammasome activation underlies central nervous system deterioration in HIV-associated tuberculosis. J Infect Dis 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Oliver BG, Elliott JH, Price P, et al. Mediators of innate and adaptive immune responses differentially affect immune restoration disease associated with Mycobacterium tuberculosis in HIV patients beginning antiretroviral therapy. J Infect Dis 2010; 202: 1728–1737. [DOI] [PubMed] [Google Scholar]
- 142.Tran HT, Van den Bergh R, Loembe MM, et al. Modulation of the complement system in monocytes contributes to tuberculosis-associated immune reconstitution inflammatory syndrome. AIDS (London, England) 2013; 27: 1725–1734. [DOI] [PubMed] [Google Scholar]
- 143.Tran HT, Van den Bergh R, Vu TN, et al. The role of monocytes in the development of tuberculosis-associated immune reconstitution inflammatory syndrome. Immunobiology 2014; 219: 37–44. [DOI] [PubMed] [Google Scholar]
- 144.Tan DB, Lim A, Yong YK, et al. TLR2-induced cytokine responses may characterize HIV-infected patients experiencing mycobacterial immune restoration disease. Aids 2011; 25: 1455–1460. [DOI] [PubMed] [Google Scholar]
- 145.Muller M, Wandel S, Colebunders R, et al. Immune reconstitution inflammatory syndrome in patients starting antiretroviral therapy for HIV infection: A systematic review and meta-analysis. Lancet Infect Dis 2010; 10: 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Antonelli LR, Mahnke Y, Hodge JN, et al. Elevated frequencies of highly activated CD4+ T cells in HIV+ patients developing immune reconstitution inflammatory syndrome. Blood 2010; 116: 3818–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pappas PG, Bustamante B, Ticona E, et al. Recombinant interferon- gamma 1b as adjunctive therapy for AIDS-related acute cryptococcal meningitis. J Infect Dis 2004; 189: 2185–2191. [DOI] [PubMed] [Google Scholar]
- 148.Jarvis JN, Meintjes G, Rebe K, et al. Adjunctive interferon-gamma immunotherapy for the treatment of HIV-associated cryptococcal meningitis: A randomized controlled trial. Aids 2012; 26: 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.He B, Tran JT, Sanchez DJ. Manipulation of Type I interferon signaling by HIV and AIDS-associated viruses. J Immunol Res 2019; 2019: 8685312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Leopold Wager CM, Hole CR, Campuzano A, et al. IFN-gamma immune priming of macrophages in vivo induces prolonged STAT1 binding and protection against Cryptococcus neoformans. PLoS Pathog 2018; 14: e1007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bosinger SE, Utay NS. Type I interferon: Understanding its role in HIV pathogenesis and therapy. Curr HIV/AIDS Rep 2015; 12: 41–53. [DOI] [PubMed] [Google Scholar]
- 152.Stek C, Schutz C, Blumenthal L, et al. Preventing paradoxical tuberculosis-associated immune reconstitution inflammatory syndrome in high-risk patients: Protocol of a randomized placebo-controlled trial of prednisone (PredART Trial). JMIR Res Protoc 2016; 5: e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wallis RS, van Vuuren C, Potgieter S. Adalimumab treatment of life-threatening tuberculosis. Clin Infect Dis 2009; 48: 1429–1432. [DOI] [PubMed] [Google Scholar]
- 154.Lai RP, Meintjes G, Wilkinson RJ. HIV-1 tuberculosis-associated immune reconstitution inflammatory syndrome. Semin Immunopathol 2016; 38: 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Meintjes G, Stek C, Blumenthal L, et al. Prednisone for the prevention of paradoxical tuberculosis-associated IRIS. N Engl J Med 2018; 379: 1915–1925. [DOI] [PubMed] [Google Scholar]
- 156.Tavel JA, Sereti I, Walker RE, et al. A randomized, double-blinded, placebo-controlled trial of intermittent administration of interleukin-2 and prednisone in subjects infected with human immunodeficiency virus. J Infect Dis 2003; 188: 531–536. [DOI] [PubMed] [Google Scholar]
- 157.Sereti I, Herpin B, Metcalf JA, et al. CD4 T cell expansions are associated with increased apoptosis rates of T lymphocytes during IL-2 cycles in HIV infected patients. Aids 2001; 15: 1765–1775. [DOI] [PubMed] [Google Scholar]
- 158.Della Chiara G, Fortis C, Tambussi G, et al. The rise and fall of intermittent interleukin-2 therapy in HIV infection. Eur Cytokine Netw 2010; 21: 197–201. [DOI] [PubMed] [Google Scholar]
- 159.Rhein J, Morawski BM, Hullsiek KH, et al. Efficacy of adjunctive sertraline for the treatment of HIV-associated cryptococcal meningitis: An open-label dose-ranging study. Lancet Infect Dis 2016; 16: 809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Scriven JE, Rhein J, Hullsiek KH, et al. Early ART after cryptococcal meningitis is associated with cerebrospinal fluid pleocytosis and macrophage activation in a multisite randomized trial. J Infect Dis 2015; 212: 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Boulware DR, Meya DB, Muzoora C, et al. Timing of antiretroviral therapy after diagnosis of cryptococcal meningitis. N Engl J Med 2014; 370: 2487–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jacobson CA, Turki AT, McDonough SM, et al. Immune reconstitution after double umbilical cord blood stem cell transplantation: Comparison with unrelated peripheral blood stem cell transplantation. Biol Blood Marrow Transplant 2012; 18: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Komanduri KV, St John LS, de Lima M, et al. Delayed immune reconstitution after cord blood transplantation is characterized by impaired thymopoiesis and late memory T-cell skewing. Blood 2007; 110: 4543–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Majhail NS, Rizzo JD, Lee SJ, et al. Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2012; 18: 348–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Parody R, Martino R, Rovira M, et al. Severe infections after unrelated donor allogeneic hematopoietic stem cell transplantation in adults: Comparison of cord blood transplantation with peripheral blood and bone marrow transplantation. Biol Blood Marrow Transplant 2006; 12: 734–748. [DOI] [PubMed] [Google Scholar]
- 166.Ruggeri A, Peffault de Latour R, Carmagnat M, et al. Outcomes, infections, and immune reconstitution after double cord blood transplantation in patients with high-risk hematological diseases. Transpl Infect Dis 2011; 13: 456–465. [DOI] [PubMed] [Google Scholar]
- 167.Saliba RM, Rezvani K, Leen A, et al. General and virus-specific immune cell reconstitution after double cord blood transplantation. Biol Blood Marrow Transplant 2015; 21: 1284–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sauter C, Abboud M, Jia X, et al. Serious infection risk and immune recovery after double-unit cord blood transplantation without antithymocyte globulin. Biol Blood Marrow Transplant 2011; 17: 1460–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Somers JA, Brand A, van Hensbergen Y, et al. Double umbilical cord blood transplantation: A study of early engraftment kinetics in leukocyte subsets using HLA-specific monoclonal antibodies. Biol Blood Marrow Transplant 2013; 19: 266–273. [DOI] [PubMed] [Google Scholar]
- 170.Young JH, Logan BR, Wu J, et al. Infections after transplantation of bone marrow or peripheral blood stem cells from unrelated donors. Biol Blood Marrow Transplant 2016; 22: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Majhail NS, Weisdorf DJ, Wagner JE, et al. Comparable results of umbilical cord blood and HLA-matched sibling donor hematopoietic stem cell transplantation after reduced-intensity preparative regimen for advanced Hodgkin lymphoma. Blood 2006; 107: 3804–3807. [DOI] [PubMed] [Google Scholar]
- 172.Brunstein CG, Barker JN, Weisdorf DJ, et al. Umbilical cord blood transplantation after nonmyeloablative conditioning: Impact on transplantation outcomes in 110 adults with hematologic disease. Blood 2007; 110: 3064–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Szabolcs P, Niedzwiecki D. Immune reconstitution after unrelated cord blood transplantation. Cytotherapy 2007; 9: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hill JA, Koo S, Guzman Suarez BB, et al. Cord-blood hematopoietic stem cell transplant confers an increased risk for human herpesvirus-6-associated acute limbic encephalitis: A cohort analysis. Biol Blood Marrow Transplant 2012; 18: 1638–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Silva LeP Patah PA, Saliba RM, et al. Hemorrhagic cystitis after allogeneic hematopoietic stem cell transplants is the complex result of BK virus infection, preparative regimen intensity and donor type. Haematologica 2010; 95: 1183–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.El Chaer F, Shah DP, Chemaly RF. How I treat resistant cytomegalovirus infection in hematopoietic cell transplantation recipients. Blood 2016; 128: 2624–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pei XY, Zhao XY, Chang YJ, et al. Cytomegalovirus-specific T-cell transfer for refractory cytomegalovirus infection after haploidentical stem cell transplantation: The quantitative and qualitative immune recovery for cytomegalovirus. J Infect Dis 2017; 216: 945–956. [DOI] [PubMed] [Google Scholar]
- 178.Bejanyan N, Brunstein CG, Cao Q, et al. Delayed immune reconstitution after allogeneic transplantation increases the risks of mortality and chronic GVHD. Blood Adv 2018; 2: 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yew PY, Alachkar H, Yamaguchi R, et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant 2015; 50: 1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kanda J, Chiou LW, Szabolcs P, et al. Immune recovery in adult patients after myeloablative dual umbilical cord blood, matched sibling, and matched unrelated donor hematopoietic cell transplantation. Biol Blood Marrow Transplant 2012; 18: 1664–1676.e1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Schulenburg A, Fischer M, Kalhs P, et al. Immune recovery after conventional and nonmyeloablative allogeneic stem cell transplantation. Leuk Lymphoma 2005; 46: 1755–1760. [DOI] [PubMed] [Google Scholar]
- 182.Nesher L, Shah DP, Ariza-Heredia EJ, et al. Utility of the enzyme-linked immunospot interferon-γ-release assay to predict the risk of cytomegalovirus infection in hematopoietic cell transplant recipients. J Infect Dis 2016; 213: 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.El Haddad L, Ariza-Heredia E, Shah DP, et al. The ability of a cytomegalovirus ELISPOT assay to predict outcome of low-level CMV reactivation in hematopoietic cell transplant recipients. J Infect Dis 2019; 219: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Roddie C, Peggs KS. Immunotherapy for transplantation-associated viral infections. J Clin Invest 2017; 127: 2513–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Heslop HE, Ng CY, Li C, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med 1996; 2: 551–555. [DOI] [PubMed] [Google Scholar]
- 186.Qian C, Campidelli A, Wang Y, et al. Curative or pre-emptive adenovirus-specific T cell transfer from matched unrelated or third party haploidentical donors after HSCT, including UCB transplantations: A successful phase I/II multicenter clinical trial. J Hematol Oncol 2017; 10: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med 1995; 333: 1038–1044. [DOI] [PubMed] [Google Scholar]
- 188.Neuenhahn M, Albrecht J, Odendahl M, et al. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia 2017; 31: 2161–2171. [DOI] [PubMed] [Google Scholar]
- 189.Kontoyiannis DP, Marr KA, Park BJ, et al. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: Overview of the transplant-associated infection surveillance network (TRANSNET) Database. Clin Infect Dis 2010; 50: 1091–1100. [DOI] [PubMed] [Google Scholar]
- 190.Perruccio K, Tosti A, Burchielli E, et al. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood 2005; 106: 4397–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Audehm S, Krackhardt AM. Specific adoptive cellular immunotherapy in allogeneic stem cell transplantation. Oncol Res Treat 2017; 40: 691–696. [DOI] [PubMed] [Google Scholar]
- 192.Romee R, Cooley S, Berrien-Elliott MM, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 2018; 131: 2515–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Robinson TO, Schluns KS. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol Lett 2017; 190: 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lin CM, Plenter RJ, Coulombe M, et al. Interferon gamma and contact-dependent cytotoxicity are each rate limiting for natural killer cell-mediated antibody-dependent chronic rejection. Am J Transplant 2016; 16: 3121–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chaudhry MS, Velardi E, Dudakov JA, et al. Thymus: The next (re)generation. Immunol Rev 2016; 271: 56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Colombetti S, Lévy F, Chapatte L. IL-7 adjuvant treatment enhances long-term tumor-antigen-specific CD8+ T-cell responses after immunization with recombinant lentivector. Blood 2009; 113: 6629–6637. [DOI] [PubMed] [Google Scholar]
- 197.Munneke JM, Spruit MJ, Cornelissen AS, et al. The potential of mesenchymal stromal cells as treatment for severe steroid-refractory acute graft-versus-host disease: A critical review of the literature. Transplantation 2016; 100: 2309–2314. [DOI] [PubMed] [Google Scholar]
- 198.Gao L, Zhang Y, Hu B, et al. Phase II multicenter, randomized, double-blind controlled study of efficacy and safety of umbilical cord-derived mesenchymal stromal cells in the prophylaxis of chronic graft-versus-host disease after HLA-haploidentical stem-cell transplantation. J Clin Oncol 2016; 34: 2843–2850. [DOI] [PubMed] [Google Scholar]
- 199.Nowarski R, Jackson R, Flavell RA. The stromal intervention: Regulation of immunity and inflammation at the epithelial-mesenchymal barrier. Cell 2017; 168: 362–375. [DOI] [PubMed] [Google Scholar]
- 200.Polchert D, Sobinsky J, Douglas G, et al. IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. Eur J Immunol 2008; 38: 1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Joffre O, Santolaria T, Calise D, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med 2008; 14: 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hoffmann P, Ermann J, Edinger M, et al. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med 2002; 196: 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Josefowicz SZ, Niec RE, Kim HY, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012; 482: 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Trzonkowski P, Bieniaszewska M, Juścińska J, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin Immunol 2009; 133: 22–26. [DOI] [PubMed] [Google Scholar]
- 205.Theil A, Tuve S, Oelschlägel U, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy 2015; 17: 473–486. [DOI] [PubMed] [Google Scholar]
- 206.Di Ianni M, Falzetti F, Carotti A, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011; 117: 3921–3928. [DOI] [PubMed] [Google Scholar]
- 207.Behar SM, Carpenter SM, Booty MG, et al. Orchestration of pulmonary T cell immunity during Mycobacterium tuberculosis infection: immunity interruptus. Semin Immunol 2014; 26: 559–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Felices M, Lenvik AJ, McElmurry R, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Rowan CM, Paczesny S. Biomarkers for early complications after hematopoietic stem cell transplantation. Clin Lab Med 2019; 39: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ogilvie RL, Sternjohn JR, Rattenbacher B, et al. Tristetraprolin mediates interferon-gamma mRNA decay. J Biol Chem 2009; 284: 11216–11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Vlasova-St.Louis I, Bohjanen PR. Post-transcriptional regulation of cytokine signaling during inflammatory responses: Goldstrohm AC, Menon KMJ (Eds.), Post-transcriptional mechanisms in endocrine regulation, Springer, Switzerland, 2016; 55–70. [Google Scholar]
- 212.Vlasova-St Louis I, Bohjanen PR. Post-transcriptional regulation of cytokine signaling by AU-rich and GU-rich elements. J Interferon Cytokine Res 2014; 34: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proceedings of the National Academy of Sciences of the United States of America 2007; 104: 6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Vlasova-St Louis I, Bohjanen PR. Feedback regulation of kinase signaling pathways by AREs and GREs. Cells 2016; 5: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Li Z, Qian P, Shao W, et al. Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res 2018; 28: 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Silva CAM, Graham B, Webb K, et al. A pilot metabolomics study of tuberculosis immune reconstitution inflammatory syndrome. Int J Infect Dis 2019; 84: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Bax HI, Freeman AF, Ding L, et al. Interferon alpha treatment of patients with impaired interferon gamma signaling. J Clin Immunol 2013; 33: 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]