

Abstract
Human health in the current medical era is facing numerous challenges, especially cancer. So, the therapeutic arsenal for cancer should be unremittingly enriched with novel small molecules that selectively target tumour cells with minimal toxicity towards normal cells. In this context, herein a new series of 3,6-disubstituted pyridazines 11a–r has been synthesised and evaluated for in vitro anticancer activity. They possessed good anti-proliferative action towards human breast cancer T-47D (IC50 range: 0.43 ± 0.01 − 35.9 ± 1.18 µM) and MDA-MB-231 (IC50 range: 0.99 ± 0.03 − 34.59 ± 1.13 µM) cell lines, whereas they displayed weak activity against the tested ovarian cancer cell line SKOV-3. Among the studied compounds, the methyltetrahydropyran-bearing pyridazine 11m emerged as the unique submicromolar growth inhibitor herein reported towards both T-47D (IC50 = 0.43 ± 0.01 µM) and MDA-MB-231 (IC50 = 0.99 ± 0.03 µM) cell lines. In addition, the biological results indicated that pyridazines 11l and 11m exerted an efficient alteration within the cell cycle progression as well as induction of apoptosis in both T-47D and MDA-MB-231 cells. Moreover, pyridazines 11l and 11m displayed good mean tumour S. I. values of 13.7 and 16.1 upon assessment of their cytotoxicity towards non-tumorigenic breast MCF-10A cells. Furthermore, an in silico study proposed CDK2 as a probable enzymatic target for pyridazines 11, and explored their binding interactions within the vicinity of CDK2 binding site. Subsequently, pyridazines 11e, 11h, 11l, and 11m were selected to be evaluated for their ability to inhibit CDK2, where they exerted good inhibitory activity (IC50 = 151, 43.8, 55.6 and 20.1 nM, respectively). Finally, the in silico study implied that target pyridazines 11 exhibited not only an efficient anticancer activity but also an acceptable ADME, physicochemical and druglikeness properties, specifically pyridazines 11l and 11m. Overall the obtained results from this study quite sustained our strategy and gave us a robust opportunity for further development and optimisation of 3,6-disubstituted pyridazine scaffold to enrich therapeutic arsenal with efficient and safe anticancer CDK inhibitors.
Keywords: Breast cancer, aminopyridazines, CDK2 inhibitors, apoptosis inducers, molecular docking, cell cycle arrest
1. Introduction
Cancer is a very complex disease, which affects diverse systems and organs within the body. Cancer develops because of the abnormal and uncontrolled cells division, initiated as a result of chemicals, viruses, smoking, or diet. Complications of cancer disease lead to death if left without treatment1. As a serious health problem, mortality due to cancer is expected to surpass that attributable to cardiovascular disorders in a short time. Around seven million cancer-related cases die per year, whereas about more than 26 million new cancer cases and 17 million cancer-related deaths are estimated to be reached per year by 20302. Accordingly, the therapeutic arsenal for cancer treatment should be urgently enriched with novel small molecules directed towards a certain signalling factor that could be implicated in tumour growth and/or tumorigenesis.
Cyclin-dependent kinases (CDKs) are a family of comparatively small proteins, from 34 to 40 kDa, which is classified as serine/threonine protein kinases3. CDKs have essential roles in the cell cycle regulation as well as in apoptosis, transcription and differentiation through binding to a regulatory protein known as “cyclin”; only the CDK-cyclin complexes are active kinases4,5.
It is well established that excessive production of CDKs, such as CDK1, CDK2, CDK4 and CDK6, or cyclins may result in a disruption of the normal regulation controls and eventually leads to cancer. For this reason, therapeutic approach based on inhibition of CDKs represents an auspicious opportunity and promising strategy for drug discovery and development of novel efficient and targeted chemical entities that can fight different types of human malignancies6. To date, three small molecule CDK inhibitors (palbociclib, abemaciclib and ribociclib) have been approved for clinical use, Figure 17, whereas there are several CDK inhibitors in the clinical trials; such as roniciclib (BAY 1000394, Figure 1)8. However, the side effects and resistance to these inhibitors somehow limits their therapeutic value and discloses the necessity of development of novel CDK inhibitors9.
Figure 1.

Chemical structure of the clinically used (palbociclib, abemaciclib and ribociclib) or currently in clinical trials (roniciclib) CDK inhibitors.
Over the last two decades, non-fused pyridazine nucleus has emerged as an important class of heterocycles that represents a promising and privileged scaffold in medicinal chemistry10. Literature survey revealed that plenty of pyridazine-based small molecules have been extensively investigated for a broad array of significant biological actions, such as anti-inflammatory11,12, anti-hypertensive13, anti-diabetic14, anti-obesity15, neuroprotective16, anti-Alzheimer’s17, anti-tubercular18, anti-HIV19 and anticancer20 activities.
While several studies and research work have explored different anticancer activities for diverse fused pyridazine derivatives, little attention has been paid to investigate the anticancer activity for the non-fused 3,6-disubstituted pyridazine derivatives over the last two decades21–30. For example; compound I (Figure 2) displayed potent in vitro and in vivo antitumor and anti-angiogenesis activities21, compound II (Figure 2) efficiently inhibited the cell proliferation in a panel of breast, colon, prostate and liver human tumours28. In addition, compound III (Figure 2) elicited excellent cytotoxic activity towards human colon cancer HT-29 cell line22, whereas compound IV (Figure 2) emerged as promising VEGFR-2 inhibitor with IC50 in the nanomolar range29.
Figure 2.

Chemical structure for some 3,6‐disubstituted pyridazine derivatives reported as efficient anticancer small molecules.
Inspired by the aforementioned findings and as part of our research work on the discovery of efficient anticancer candidates, herein we report in details the synthesis and anticancer activities assessment for a new series of non-fused 3,6-disubstituted pyridazine derivatives 11a–r (Figure 2). In this context, the anticancer actions for the 3,6-disubstituted pyridazine derivatives here reported; 11a–r will be evaluated against three human cancer cell lines, namely, T-47D and MDA-MB-231 (breast cancers), and SKOV-3 (ovarian cancer) cell lines utilising the protocol of SRB assay. Thereafter, the most potent anti-proliferative pyridazines will be selected to explore their plausible mechanism of action through cell cycle analysis as well as Anx V-FITC apoptosis assay in both breast cancer (T-47D and MDA-MB-231) cell lines. Finally, an in silico study suggested CDK2 as a probable enzymatic target for the herein reported 3,6-disubstituted pyridazines 11a–r and explored their binding interactions within the vicinity of CDK2 binding site, thereafter, target pyridazines will be explored for their potential inhibitory activity against CDK2.
2. Results and discussion
2.1. Chemistry
The target 5-(trifluoromethyl)pyridazine-3-carboxamide derivatives (11a–r) were prepared through several synthetic steps (Schemes 1–3) starting from the commercially available ethyl 3,3,3-trifluoropyruvate. With respect to Scheme 1, ethyl trifluoropyruvate (1) reacted with acetone in the existence of L-proline and DMF following a reported method31 to yield ethyl 2-hydroxy-4-oxo-2-(trifluoromethyl)pentanoate (2), the later compound was converted into 6-methyl-4-(trifluoromethyl)pyridazin-3(2H)-one (3) upon reaction with hydrazine hydrate in the existence of acetic acid32. Compound (3) was then subjected to oxidation process using potassium chromate and sulphuric acid at room temperature to give 6-oxo-5-(trifluoromethyl)-1,6-dihydropyridazine-3-carboxylic acid (4). Fischer esterification was then carried out for compound (4) through reflux with ethanol in the presence of sulphuric acid (catalytic amount) in order to afford the corresponding ester derivative (5), Scheme 1.
Scheme 1.

Preparation of ester 5; reagents and conditions: (i) Acetone, DMF, L-proline, r.t., 48 h; (ii) NH2NH2, AcOH, reflux, 6 h; (iii) K2Cr2O7, Conc. H2SO4, 0 °C to r.t. overnight; (iv) EtOH, H2SO4, reflux, 4 h.
Scheme 2.

Preparation of intermediateds 9a–e; reagents and conditions: (i) POCl3, 100 °C, 5 h; (ii) LiOH, THF, H2O, r.t., 1 h; (iii) SOCl2, DMF (catalytic), 1,2-dichloroethane, reflux, 3 h; (iv) Methylene chloride, Et3N, r.t., 4 h.
Scheme 3.

Preparation of target pyridazines 11a–r; reagents and conditions: (i) 1,4-Dioxane, Hünig’s base, reflux, 6 h; (ii) Morpholine, 1,4-dioxane, Hünig’s base, reflux, 12 h; (iii) Primary amines 8a–e, EtOH, piperdine, reflux, 6 h.
Chlorination of 6-oxo-1,6-dihydropyridazine derivative (5) was performed via its reflux with excess phosphorous oxychloride to furnish ethyl 6-chloro-5-(trifluoromethyl)pyridazine-3-carboxylate (6), which subsequently hydrolysed via treatment with the alkali lithium hydroxide in presence of THF/H2O (4/1) then chlorinated with thionyl chloride to afford the crude acid chloride derivative (7). Stirring of acid chloride (7) with the different primary amines 8a–e in dichloromethane at r.t. and in the existence of Et3N led to formation of the key amide intermediates 9a–e (Scheme 2).
The target pyridazine derivatives (11a–r) were prepared via two synthetic routes. The first route utilised the key amide intermediates 9a–e, which reacted with the amines 8a, 8e and 10a–d in refluxing 1,4-dioxane in the presence of Hünig’s base in order to furnish target pyridazines 11a–r (Scheme 3). This synthetic pathway proved successful to prepare target pyridazines 11a–r with low to good yields; 36–79%. In the second route, ethyl 6-chloro-5-(trifluoromethyl)pyridazine-3-carboxylate (6) was subjected to a nucleophilic substitution with morpholine in boiling 1,4-dioxane in the existence of Hünig’s base to furnish ethyl 6-morpholino-5-(trifluoromethyl)pyridazine-3-carboxylate (13), which subsequently underwent direct amidation that accomplished via reaction with primary amines 8a–e in absolute ethyl alcohol in the existence of piperidine to yield the corresponding target pyridazines 11d, 11g, 11i, 11m and 11r, respectively, with overall 29–54% yield for the two steps; nucleophilic substitution and direct amidation (Scheme 3)33.
It is worth emphasising that pyridazines 11d, 11g, 11i, 11m and 11r were also prepared utilising the first route, from the key amide intermediates 9a–e, with higher yields (50–73%) than those afforded from the second route (29–54%), suggesting first route as more advantageous to synthesise final target pyridazines.
2.2. Biological evaluation
2.2.1. In vitro anti-proliferative activities
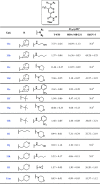
The newly herein reported pyridazines 11a–r were assessed for their anti-proliferative potential towards three human cancer cell lines, namely, T-47D (breast cancer), MDA-MB-231 (breast cancer) and SKOV-3 (ovarian cancer) cell lines by using the protocol of the SRB assay34. MDA-MB-231 cell line is classified as a triple-negative/basal-like cell line, which is hormone (estrogen and progesterone)-receptor negative and HER2 negative, whereas T-47D is considered as hormone-receptor positive and HER2 negative breast cancer cell line. The IC50 values for target pyridazines 11a–r were determined, and were represented in Table 1.
Table 1.
Anti-proliferative activities for the newly prepared pyridazines (11a–r) towards T-47D, MDA-MB-231 and SKOV-3 cell lines.
 | |||||
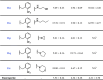 |
aIC50s are displayed as mean ± SD for three separate experiments.
bNA: Pyridazines with IC50 value more than 100 µM.
The obtained results disclosed that all the screened pyridazines possessed excellent to moderate anticancer action against the two tested breast cancer (T-47D and MDA-MB-231) cell lines, whereas they displayed weak to non-significant cytotoxic impact against the tested ovarian cancer (SKOV-3) cell line, Table 1. Nonetheless, it was noted that the screened pyridazines 11a–r exhibited differential activities against the two tested human breast cancer cell lines; 8 out of 18 compounds were more active against T-47D than MDA-MB-231, whereas 10 out of 18 compounds were more active against MDA-MB-231 than T-47D as indicated in Table 1. In details, T-47D cells were more sensitive (IC50 range 0.44 ± 0.01 − 11.44 ± 0.37 µM) to the influence of all 2-adamantyl-bearing pyridazines 11a–e (except 11d), tert-butyl-bearing pyridazines 11f–h (except 11 g) and butyl-bearing pyridazine (11i) than MDA-MB-231 cells (IC50 range 2.18 ± 0.07 − 34.59 ± 1.13 µM), Table 1. On the contrary, the methyltetrahydropyran-bearing pyridazines 11j–l and 2-(trifluoromethyl)benzyl-bearing pyridazines 11n–r displayed better cytotoxic activity against MDA-MB-231 (IC50 range 1.30 ± 0.04 − 19.71 ± 0.64 µM) than T-47D cell line (IC50 range 1.57 ± 0.05 − 35.9 ± 1.18 µM).
Concerning the anti-proliferative activity of target pyridazines 11a–r against breast cancer T-47D cell line, the results indicated that the growth of the T-47D cells was effectively inhibited by all pyridazines reported here with IC50s spanned in the range 0.43 ± 0.01 − 15.76 ± 0.51 µM, except for 11j and 11r which weakly inhibited the T-47D growth (IC50 = 35.9 ± 1.18 and 24.80 ± 0.81 µM, respectively). Outstandingly, two pyridazines displayed submicromolar potency against T-47D cells were identified, that were the butyl-bearing pyridazine (11i) and methyltetrahydropyran-bearing pyridazine (11m), whose IC50s of 0.44 ± 0.01 and 0.43 ± 0.01 µM represent the best growth inhibition here reported for T-47D cells (Table 1). Interestingly, both pyridazines 11i and 11m are decorated with morpholine moiety at C-6 position. Moreover, pyridazines 11b, 11e, 11f, 11h and 11l showed excellent cytotoxic activity against pyridazines 11m T-47D cell line with IC50 values equal 1.37 ± 0.04, 2.62 ± 0.08, 1.94 ± 0.06, 1.60 ± 0.05 and 1.57 ± 0.05 µM, respectively.
With respect to the anticancer activity against MDA-MB-231 cells, the obtained growth inhibitory data (Table 1) ascribed to most newly synthesised pyridazines 11 good efficacy in inhibiting the growth of MDA-MB-231 cells, with IC50s ranging between 0.99 ± 0.03 and 19.71 ± 0.64 µM. Uniquely, compound 11a induced weak growth inhibition towards MDA-MB-231 cell line with IC50 equals 34.59 ± 1.13 µM (Table 1). It is worth stressing that methyltetrahydropyran-bearing pyridazine (11m) superiorly displayed submicromolar potency towards MDA-MB-231 cell line (IC50 = 0.99 ± 0.03 µM), and thus 11m emerged as the unique submicromolar growth inhibitor herein reported towards both breast cancer T-47D and MDA-MB-231 cell lines. In addition, pyridazines 11d, 11h, 11l and 11n elicited excellent growth inhibitory action towards MDA-MB-231 cell lines with IC50 values of 2.18 ± 0.07, 2.44 ± 0.08, 1.30 ± 0.04 and 2.94 ± 0.09 µM, respectively (Table 1).
On the other hand, the cytotoxic action for pyridazines 11l and 11m, the most potent anti-proliferative agents with dual growth inhibitory activities towards T-47D and MDA-MB-231, against non-tumorigenic human breast (MCF-10A) cells has been assessed by the use of the MTT assay in order to explore the selectivity and safety for target pyridazines reported here towards the non-tumorigenic normal cells. The obtained IC50 values were displayed in Table 2, in addition to the calculated mean tumour selectivity index (S. I.); IC50 for MCF-10A/ IC50 average for (T-47D and MDA-MB-231). As results indicated, examined pyridazines 11l and 11m displayed good mean tumour S. I. values of 13.7 and 16.1, respectively, which discloses good safety profile for target pyridazines as antitumor agents.
Table 2.
Cytotoxic impact for pyridazines 11l and 11m against non-tumorigenic MCF-10A human breast cell line, as well as mean tumour selectivity index (S. I.) (MCF-10A/T-47D and MDA-MB-231).
| Comp. | IC50 (µM) |
Mean tumour selectivity | ||
|---|---|---|---|---|
| MCF-10A | T-47D | MDA-MB-231 | ||
| 11l | 19.63 ± 0.38 | 1.57 ± 0.05 | 1.30 ± 0.04 | 13.7 |
| 11m | 11.44 ± 0.22 | 0.43 ± 0.01 | 0.99 ± 0.03 | 16.1 |
2.2.2. Cell cycle analysis
In this study, the efficient anti-proliferative activities of target pyridazines 11a-r towards breast cancer cell lines promoted us to explore and get insights into the plausible cellular mechanisms for these small molecules. To this end, we decided to determine if the target pyridazines exerted their cytotoxic activity through alteration in cell cycle progression and/or induction of apoptosis in both breast cancer T-47D and MDA-MB-231 cells. As a reliable tool, the flow cytometric analysis was utilised to inspect the cell cycle distribution after treatment of T-47D and MDA-MB-231 cells with IC50s of pyridazines 11l and 11m; the most potent cytotoxic agents towards both examined breast cancer cell lines (Table 1).
As results indicated, pyridazines 11l and 11m were able to provoke a significant rise in the cellular population of the G2/M phase in T-47D cells by 2.7- and 2.5-fold, respectively, with concurrent significant increase in the sub-G1 phase by 13.8- and 9.5-fold, respectively, Figure 3(A). In addition, treatment of MDA-MB-231 cells by pyridazines 11l and 11m elicited a significant increase in the cell population within both G2/M phase (by 4.1 and 3.8-fold, respectively) and sub-G1 phase (by 18.9- and 15.2-fold, respectively) with respect to untreated control cells (Figure 3(B)). Conclusively, both alteration of the Sub-G1 phase as well as arrest of G2-M phase are featured as significant remarks for pyridazines 11l and 11m to trigger apoptosis in T-47D and MDA-MB-231 cancer cells.
Figure 3.

Influence of pyridazines 11l and 11 m on the distribution of cell cycle phases in breast cancer T-47D (A) and MDA-MB-231 (B) cells. Asterisk indicates a significant difference from control at p < 0.05.
2.2.3. Annexin V-FITC apoptosis assay
In this study, the apoptotic potential for pyridazines 11l and 11m, the most potent anti-proliferative agents herein reported, has been assessed in both breast cancer T-47D and MDA-MB-231 cell lines using Annexin V FITC/PI dual staining assay. Both T-47D and MDA-MB-231 cells were treated with pyridazines 11l and 11m at their IC50s for 48 h. The obtained results have been depicted in Figures 4–6.
Figure 4.

Percentage of apoptotic and necrotic cells in T-47D (A) and MDA-MB-231 (B), with respect to untreated control cells, upon treatment with pyridazines 11l and 11m.
Figure 5.

Influence of pyridazines 11l and 11m on the percentage of Annexin V+ apoptotic cells in T-47D cells.
Figure 6.

Influence of pyridazines 11l and 11m on the percentage of Annexin V+ apoptotic cells in MDA-MB-231 cells.
The outcomes from the flow cytometric analysis revealed that T-47D cells treated with pyridazines 11l and 11m exhibited a significant rise in the percent of Anx V-FITC positive (Annexin V+) apoptotic cells within both early (from 0.81% to 6.39% and 11.36%, respectively) and late (from 0.19% to 27.2% and 10.34%, respectively) apoptotic phases (UR + LR), which involves about 33- and 21-fold increase with respect to untreated control cells (Figures 4(A) and 5).
On the other hand, the flow cytometric analysis disclosed that the apoptotic rates (Annexin V+) of MDA-MB-231 cells were significantly increased from 0.56% for untreated control cells to 3.38% and 8.19%, respectively, in the early apoptotic phase, as well as from 0.24% for untreated control cells to 23.60% and 15.56%, respectively, in the late apoptotic phase; after treatment of MDA-MB-231 cells with the IC50s of pyridazines 11l and 11m (Figures 4(B) and 6).
2.3. In silico target prediction
Swiss Target Prediction is an online tool that created in 2014 in order to predict the potential targets for any small molecule35. This tool can deduce the best potential macromolecular targets for the small molecules, assumed as bioactives. The predictions are based on 2D and 3D similarity combination with a library includes more than 370’000 known actives towards more than three thousands proteins from 3 different species36. In this study we utilised the online SwissTargetPrediction tool to explore the potential enzymatic targets for the herein reported pyridazine derivatives.
The generated prediction results, for two representative compounds 11a and 11j, suggested the protein kinases as the most probable targets (Supporting materials). In particular, CDK2 is listed as one of the top suggested kinases. It is interesting to note that the suggestion of CDK2 as a potential target is perfectly tuned with its known expression in breast cancer37, and with the obtained results from the cell cycle analysis, as the role of CDK2 in the cell cycle is well-established3. Accordingly, a molecular docking work was conducted in order to examine the plausible binding modes and interactions of the target pyridazines 11a–r within the active site of CDK2.
2.4. Cdk2 inhibitory activity
2.4.1. Molecular docking
Molecular docking is among the most utilised drug design techniques that provide insights on how ligands could bind to their targets and the efficiency of theses ligands by scoring them on basis of energy and interactions. Based on suggestion of SwissTargetPrediction tool that CDK2 kinase could be a potential target for the synthesised pyridazines, docking was used to validate this assumption and also to select the most promising analogues for a further biological screening.
Initially, the protocol of the molecular docking has been validated via redocking of the co-crystalized ligand Roniciclib within the vicinity of CDK2 binding site. The performed redocking procedure re-produced the original binding manner for the co-crystallized Roniciclib quite adequately implying the convenience of the utilised steps for the desired docking analysis. This was evidenced by the small RMSD of 0.53 Å between the co-crystallized Roniciclib and the docked pose (energy score (S) = −9.2 kcal/mol), as well as by the reproducing of all interactions accomplished by the co-crystallized Roniciclib within the CDK2 active site.
Generally, all the target pyridazines 11a–r achieved acceptable binding interactions and energy scores. In particular, compounds 11m, 11e, 11h and 11l achieved the highest scores (−10.2, −10.1, −9.5 and −9.0 Kcal\mole, respectively, Table 3). The four compounds (11e, 11h, 11l, and 11m) have successfully bound strongly to the CDK2 active site; forming different types of interaction including hydrophobic interactions with non-polar residues (such as Ala31, Val18, Val64, Phe80, Ala144, Phe82 and Leu134), as well as hydrogen bonding with other residues (such as Asp86, Leu83 and Lys89), Figures 7–10. All the bonding interactions within CDK2 active site for pyridazines 11e, 11h, 11l, and 11m were outlined in Table 4. The obtained results from the docking study supported the assumption generated from the SwissTargetPrediction server and directed us to conduct a biological evaluation for the best energy scoring pyridazines 11e, 11h, 11l, and 11m against CDK2 enzyme.
Table 3.
Docking energy scores (S) for pyridazines 11a–r and the co-crystallized ligand Roniciclib (in in kcal/mol).
| Cpd. | Energy score (S) Kcal\mole | Cpd. | Energy score (S) Kcal\mole |
|---|---|---|---|
| 11a | −8.1 | 11j | −8.3 |
| 11b | −8.0 | 11k | −8.7 |
| 11c | −8.2 | 11l | −9.5 |
| 11d | −7.9 | 11m | −10.2 |
| 11e | −9.0 | 11n | −8.6 |
| 11f | −8.3 | 11o | −8.0 |
| 11g | −7.9 | 11p | −8.7 |
| 11h | −10.1 | 11q | −8.2 |
| 11i | −8.5 | 11r | −8.4 |
| Roniciclib | −9.2 |
Bold values are for pyridazines with best docking energy scores.
Figure 7.

(A) 2D, and (B) 3D diagram for pyridazine 11e demonstrating its interactions within the CDK2 active site.
Figure 8.

(A) 2D, and (B) 3D diagram for pyridazine 11h demonstrating its interactions within the CDK2 active site.
Figure 9.

(A) 2D, and (B) 3D diagram for pyridazine 11l demonstrating its interactions within the CDK2 active site.
Figure 10.

(A) 2D, and (B) 3D diagram for pyridazine 11m demonstrating its interactions within the CDK2 active site.
Table 4.
The different bonding types and their distances (in Å) for pyridazines 11e, 11h, 11l, and 11m within the CDK2 active site.
| Cpd | Bond type (Involved A. A.) | Distance | Cpd | Bond type (Involved A. A.) | Distance |
|---|---|---|---|---|---|
| 11e | Hydrogen bond (Lys33) | 2.69 | 11l | Hydrogen bond (Leu83) | 2.61 |
| Hydrogen bond (Lys33) | 2.81 | Halogen interaction (Leu83) | 3.39 | ||
| Halogen interaction (Asp145) | 3.31 | Halogen interaction (His84) | 3.14 | ||
| Halogen interaction (Asp145) | 3.35 | Halogen interaction (His84) | 3.02 | ||
| Halogen interaction (Gly13) | 2.39 | Pi-Alkyl (Ile10) | 4.35 | ||
| Halogen interaction (Gly16) | 3.62 | Non-classical hydrogen bond (Ile10) | 3.60 | ||
| Halogen interaction (Glu12) | 3.26 | Ionic bond (Asp86) | 3.37 | ||
| Pi-Anion (Asp145) | 3.65 | Pi-Alkyl (Leu134) | 5.13 | ||
| Pi-Alkyl (Leu134) | 4.57 | Pi-Alkyl (Phe80) | 4.53 | ||
| Alkyl-Alkyl (Leu134) | 4.64 | Alkyl-Alkyl (Val18) | 4.92 | ||
| Alkyl-Alkyl (Val18) | 4.99 | Alkyl-Alkyl (Val64) | 5.26 | ||
| Alkyl-Alkyl (Val18) | 4.59 | Alkyl-Alkyl (Ala144) | 4.08 | ||
| Pi-Alkyl (Ile10) | 5.02 | Alkyl-Alkyl (Lys33) | 5.37 | ||
| Alkyl-Alkyl (Lys89) | 5.37 | Non-classical hydrogen bond (Gln85) | 2.25 | ||
| Alkyl-Alkyl (Lys89) | 5.48 | ||||
| Alkyl-Alkyl (Lys89) | 5.49 | ||||
| 11h | Hydrogen bond (Asp86) | 2.13 | 11m | Hydrogen bond (Leu83) | 2.02 |
| Hydrogen bond (Leu83) | 2.59 | Alkyl-Alkyl (Lys89) | 5.34 | ||
| Halogen interaction (Asp145) | 3.15 | Halogen interaction (Ile10) | 2.94 | ||
| Halogen interaction (Asp145) | 3.19 | Halogen interaction (Ile10) | 3.10 | ||
| Halogen interaction (Asn132) | 3.14 | Halogen interaction (Asp86) | 3.00 | ||
| Halogen interaction (Leu83) | 2.86 | Halogen interaction (Asp86) | 3.27 | ||
| Halogen interaction (Leu83) | 3.17 | Halogen interaction (Gln131) | 2.80 | ||
| Pi-Alkyl (Ile10) | 4.30 | Pi-Alkyl (Ile10) | 3.94 | ||
| Alkyl-Alkyl (Ile10) | 4.71 | Alkyl-Alkyl (Ile10) | 5.42 | ||
| Pi-Alkyl (Leu134) | 5.04 | Non-classical hydrogen bond (Ile10) | 3.48 | ||
| Alkyl-Alkyl (Leu134) | 4.37 | Alkyl-Alkyl (Lys33) | 5.28 | ||
| Pi-Alkyl (Phe82) | 5.02 | Non-classical hydrogen bond (Phe82) | 2.66 | ||
| Pi-Alkyl (Val18) | 4.76 | Non-classical hydrogen bond (Glu81) | 3.74 | ||
| Alkyl-Alkyl (Val18) | 4.43 | Pi-Sigma interaction (Phe80) | 3.50 | ||
| Pi-Alkyl (Ala144) | 3.97 | Alkyl-Alkyl (Val18) | 5.02 | ||
| Pi-Pi interaction (Phe80) | 5.14 | Alkyl-Alkyl (Val64) | 5.03 | ||
| Non-classical hydrogen bond (Lys33) | 2.79 | Alkyl-Alkyl (Ala144) | 4.29 | ||
| Alkyl-Alkyl (Ala31) | 5.00 | ||||
| Pi-Alkyl interaction (Leu134) | 4.96 |
2.4.2. In vitro CDK2 kinase assay
On account of their best scores achieved in the docking study (Table 3) as well as their efficient anti-proliferative actions towards both the examined breast cancer cell lines (Table 1), pyridazines 11e, 11h, 11l, and 11m were selected to be examined for their ability to inhibit CDK2. The results have been obtained as IC50 values which listed in Table 5.
Table 5.
IC50 values for the CDK2 inhibitory action of pyridazines 11e, 11h, 11l, and 11m.
| Comp. | IC50 (nM) |
|---|---|
| CDK2 | |
| 11e | 151 ± 6.16 |
| 11h | 43.8 ± 1.79 |
| 11l | 55.6 ± 2.27 |
| 11m | 20.1 ± 0.82 |
| Staurosporine | 12.4 ± 0.51 |
The results revealed that the examined pyridazines 11e, 11h, 11l, and 11m possessed good inhibitory action towards CDK2 kinase with IC50 values equal 151 ± 6.16, 43.8 ± 1.79, 55.6 ± 2.27 and 20.1 ± 0.82 nM, respectively (Table 5). Remarkably pyridazine derivative 11m, bearing two morpholine moieties, elicited the best CDK2 inhibitory activity in this study (IC50 = 20.1 ± 0.82 nM), alongside to its superior anti-proliferative activity that has been reported above (Table 1). It’s worth stressing that this is the first study, to the best of our knowledge, that reports on 3,6-disubstituted pyridazines as anticancer CDK inhibitors.
2.5. In silico ADME calculation
As a rule, novel small molecules considered as potential drug candidates when they possess acceptable pharmacokinetic and pharmacodynamic profiles. Thus, assessment of the efficiency for the synthesised compounds should rely not only on basis of their biological activities but also with taking in consideration their pharmacokinetics, druglikness and physicochemical properties.
The SwissADME online tool has been adopted to calculate the ADME profiles for target pyridazines 11e, 11h, 11l and 11m38. Target pyridazines 11h, 11l and 11m were predicted to have high gastrointestinal tract (GIT) absorption, as they were present within the region of human intestinal absorption (HIA) in the BOILED-Egg chart39, whereas compound 11e was located outside the region of HIA in the BOILED-Egg chart (Figure 11) and thus predicted to possess low GIT absorption that may hinder its oral bioavailability. The low GIT absorption predicted for compound 11e could be attributed to the pronounced lipophilicity of the rigid polycyclic cage hydrocarbon adamantine, as well as incorporation of ortho-trifluoromethyl phenyl moiety. At the same time, the BOILED-Egg graph (Figure 11) pointed out that target pyridazines 11e and 11h have no BBB permeability and thus could be used as antitumor agents with no predicted CNS concerns, whereas, compounds 11l and 11m have the ability to penetrate through the BBB and so they could be of great value for brain malignancies (Figure 11).
Figure 11.

Boiled-Egg chart for pyridazines 11e, 11h, 11l, and 11m.
The bioavailability radar plot (Figure 12) explains the extent of GIT absorption for the four examined pyridazines 11e, 11h, 11l and 11m. The radar chart comprises six critical parameters for oral bioavailability; LIPO (Lipophilicity), INSOLU (Solubility), INSATU (saturation), FLEX (Flexibility), SIZE (SIZE), and POLAR (polarity). The pink area of the radar chart represents the optimal range for each property value within the six properties, while the red lines represents the predicted physicochemical features for the examined pyridazines 11e, 11h, 11l and 11m, Figure 12.
Figure 12.

The oral bioavailability radar chart for pyridazines 11e (A), 11h (B), 11l (C) and 11m (D), produced by swissADME online web tool.
All the predicted physicochemical properties for pyridazines 11e, 11h, 11l and 11m were located in the desired pink area for all the six parameters, with an exception for compound 11e which barely violated the LIPO (Lipophilicity) and INSOLU (Solubility) parameters (Figure 12), which clarifies its location outside the region of HIA in the BOILED-Egg graph. The other three compounds (11h, 11l and 11m) have the optimal combination of physicochemical properties that guaranteed a high GIT absorption with no single violation to any component of the bioavailability radar chart.
The metabolism for the four pyridazines 11e, 11h, 11l and 11m are predicted to take place partially in the liver by one or more of the five major Cytochrome P (CYP) isoforms (CYP2C9, CYP1A2, CYP2D6, CYP2C19, CYP3A4), and thus target pyridazines are suggested to be administered alone to minimise the possible drug-drug interactions. In details, pyridazines 11e, 11h and 11l are predicted to exert inhibition for two of CYP isoforms. Compound 11e was predicted to inhibit CYP2D6 and CYP3A4, compound 11h was predicted to inhibit CYP2C19 and CYP3A4, and compound 11l was predicted to inhibit CYP1A2 and CYP2D6. Furthermore, compound 11m was predicted to inhibit CYP1A2 only (Table 6).
Table 6.
The in silico predicted ADME for pyridazines 11e, 11h, 11l, and 11m.
| Cpd. | BBB | GIA | P-gP substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor |
|---|---|---|---|---|---|---|---|---|
| 11e | No | Low | Yes | No | No | No | Yes | Yes |
| 11h | No | High | No | No | Yes | No | No | Yes |
| 11l | Yes | High | Yes | Yes | No | No | Yes | No |
| 11m | Yes | High | Yes | Yes | No | No | No | No |
GIA: gastrointestinal absorption; BBB: Blood Brain Barrier; PgP: P-glyco protein transporter.
On another note, SwissADME online web tool revealed that all the examined pyridazines were found to comply with all druglikeness rules defined by the pioneer pharmaceutical companies; Veber’s (GSK)40, Lipinski’s (Pfizer)41, Egan’s (Pharmacia)42, Ghose’s (Amgen)43 and Muegge’s (Bayer)44 filters, with an exception for compound 11e (towards Egan, Ghose and Muegge rules) and compound 11h (towards Egan and Ghose rules) (Table 7). Interestingly enough, from the medicinal chemistry point of view the four examined pyridazines have neither PAINS (Pan Assay Interference Structures)45 alerts nor Brenks (Structural) alerts46, which emphasises that their chemical structures lack any interfering framework that could result in an artefact in any protein assay implying that the obtained results from the in vitro biological assays are to be robust.
Table 7.
Compliance with Druglikeness rules (Lipinski, Veber, Egan, Ghose and Muegge), as well as PAINS and Brenk filters for target pyridazines 11e, 11h, 11l, and 11m.
| Cpd. | Lipinski violations | Veber violations | Muegge violations | Egan violations | Ghose violations | PAINS alerts | Brenk alerts |
|---|---|---|---|---|---|---|---|
| 11e | Yes | Yes | No | No | No | 0 | 0 |
| 11h | Yes | Yes | Yes | No | No | 0 | 0 |
| 11l | Yes | Yes | Yes | Yes | Yes | 0 | 0 |
| 11m | Yes | Yes | Yes | Yes | Yes | 0 | 0 |
PAINS: Pan Assay Interference Structures.
In conclusion, the four lead pyridazines (11e, 11h, 11l and 11m) proved not only efficient biological actions but also an acceptable ADME and physicochemical properties, in particular pyridazines 11l and 11m.
3. Materials and methods
3.1. Chemistry
3.1.1. General
Melting points have been obtained on a Büchi Melting PointB-540 apparatus which uncorrected and determined. 1H NMR spectra have been recorded on a Bruker Advance III instrument at 400 MHz. Chemical shifts (δ) of NMR have been reported in parts per million (ppm) units relative to internal standard (TMS). All reactions have been monitored by thin-layer chromatography (TLC), using silica gel plates (GF254) and UV light visualisation. Flash chromatography separations have been obtained on silica gel (200–300 mesh).
3.1.2. Synthesis of ethyl 2-hydroxy-4-oxo-2-(trifluoromethyl)pentanoate (2)
Compound 2 was reported previously31.
3.1.3. Synthesis of 6-methyl-4-(trifluoromethyl)pyridazin-3(2H)-one (3)
A solution of ethyl 2-hydroxy-4-oxo-2-(trifluoromethyl)pentanoate 2 (0.45 g, 2 mmol) and 99% hydrazine monohydrate (0.53 ml, 11 mmol) in acetic acid (7 ml), was refluxed with stirring for 6 h. The solution was then poured into H2O and extracted with EtOAc, dried by NaSO4 and purified by column flash chromatography (petroleum ether/EtOAc 70:30) to produce pyridazinone 3 as white crystals (0.27 g, 69%); m.p. 184–185 °C (reported m.p. 187–189 °C32).
3.1.4. Synthesis of 6-oxo-5-(trifluoromethyl)-1,6-dihydropyridazine-3-carboxylic acid (4)
Pyridazinone 3 (0.1 g, 0.561 mmol) was dissolved in conc. H2SO4 (5 ml) and stirred at 0 °C, then K2Cr2O7 (0.24 g, 0.846 mmol) was added portionwise. The reaction mixture was left to be stir at r.t. overnight, then added to ice-water and extracted to be extracted with ethyl acetate. The organic phases were dried with NaSO4 and concentrated under reduced pressure and the residue was purified by flash chromatography (DCM/MeOH: 90/10) to obtain pyridazine-3-carboxylic acid derivative 4 as white crystals (0.06 g, 54%); m.p. 239–240 °C.
3.1.5. Synthesis of ethyl 6-oxo-5-(trifluoromethyl)-1,6-dihydropyridazine-3-carboxylate (5)
Dihydropyridazine-3-carboxylic acid derivative 4 (2.4 g, 11.5 mmol) was dissolved in ethanol (18 ml) and H2SO4 (0.5 ml), then the reaction mixture was heated under reflux for 4 h. The solution was extracted with ethyl acetate and purified by column chromatography (petroleum ether / EtOAc 70:30) to produce white solid (3.0 g, 90%); m.p. 147–148 °C.
3.1.6. Synthesis of ethyl 6-chloro-5-(trifluoromethyl) pyridazine-3-carboxylate (6)
Ethyl 6-oxo dihydropyridazine-3-carboxylate derivative 5 (2 g, 8.47 mmol) was added portionwise to a cooled stirred phosphorus oxychloride (POCl3) (10 ml) which subsequently heated at 100 °C for 5 h. The reaction mixture, after cooling, was poured into iced-water, neutralised by aqueous solution of sodium hydroxide, extracted with ethyl acetate and purified by column chromatography (petroleum ether/EtOAc 85:15) to yield ester 6 as yellow oil (1.1 g, 51%).
3.1.7. General procedures for preparation of 6-chloro-5-(trifluoromethyl)pyridazine-3-carboxamide derivatives (9a–e)
Ethyl 6-chloro-5-(trifluoromethyl)pyridazine-3-carboxylate 6 (0.04 g, 0.16 mmol) was added to a mixture of THF/H2O (4:1) and LiOH hydrate (0.80 mmol) at 0 °C, then the reaction mixture was stirred for 1 h at room temperature. The reaction mixture was then evaporated to dryness and the residue was dissolved in H2O, neutralised carefully with a 1 N HCl, and extracted with ethyl acetate. Organic layers were collected, dried over anhydrous sodium sulphate, filtered and the solvent was removed under reduced pressure to yield the corresponding carboxylic acid. The later acid derivative was dissolved in dry 1,2-dichloroethane (5 ml), then thionyl chloride (0.9 mmol) and DMF (2–3 drops) were added to the solution. The mixture was refluxed for 3 h then evaporated to dryness to produce the crude product of acyl chloride 7. This compound was used immediately for the next step to prepare key amide intermediates 9a–e via stirring with primary amines 8a–e (0.32 mmol) in methylene chloride (4 ml) and in the presence of TEA (0.80 mmol) for 4 h at room temperature. The obtained precipitate was filtered off and washed with petroleum ether, then used in the next step without further purification.
3.1.8. Synthesis of ethyl 6-morpholino-5-(trifluoromethyl)pyridazine-3-carboxylate (13)
A mixture of ethyl 6-chloro-5-(trifluoromethyl)pyridazine-3-carboxylate 6 (0.06 g, 0.23 mmol), morpholine 12 (0.1 g, 1.12 mmol) and Hünig’s base (0.29 g, 2.24 mmol) were dissolved in 1,4-dioxane (10 ml), and the reaction mixture was heated under reflux for 12 h. After cooling to room temperature, the reaction mixture was evaporated under reduced pressure and the residue was dissolved in ethyl acetate and washed with H2O. The organic layer was then dried, evaporated in vacuo and purified with flash chromatography on silica gel (petroleum ether/EtOAc 70/30) to obtain compound 13 as yellow oil (0.0476 g, 70%).
3.1.9. General procedures for the preparation of target pyridazine derivatives (11a-r)
3.1.9.1. Route (A)
The appropriate aliphatic amine 8a, 8e and 10a–d (5 mmol) and Hünig’s base (1.3 g, 10 mmol) were added to a solution of intermediates 9a–e (1 mmol) in dioxane (5 ml), and then the reaction mixture was refluxed for 6 h. After the reaction completed, the excess dioxane was evaporated under reduced pressure, the residue was dissolved in ethyl acetate and washed with water and the organic layer was dried over anhydrous Na2SO4, and evaporated in vacuo. The crude product was purified by flash chromatography (petroleum ether: ethyl acetate 70:30) to yield target pyridazine 11a–r.
3.1.9.2. Route (B)
Ethyl 6-morpholino-5-(trifluoromethyl)pyridazine-3-carboxylate (13) (2.54 g, 10 mmol) was dissolved in ethyl alcohol (8 ml), then primary amines (8a–e) (15 mmol) and piperidine (2–3 drops) were added. The reaction mixture was refluxed for 6 h and then the solvent was evaporated under reduce pressure. The crude product was purified by using flash column chromatography (petroleum ether: ethyl acetate 70:30) to afford the corresponding target pyridazines 11d, 11g, 11i, 11m and 11r, respectively33.
For the full characterisation details for the intermediates and the target pyridazines 11a-r have been provided in the Supplementary Materials.
3.2. Biological evaluations
The utilised procedures in the biological assays were performed as described earlier; cytotoxicity47, cell cycle48, Annexin V-FITC Apoptosis49 and CDK250 assays, whereas all detailed procedures were mentioned in the Supplementary Materials.
3.3. In silico studies
3.3.1. In silico target prediction
To deduce a potential mechanism of action for herein reported target pyridazines 11, the SwissTargetPrediction online tool was utilised35. Two representative compounds 11a and 11j were drawn and submitted to the server for target prediction.
3.3.2. Docking studies
The crystal structure of CDK2 in complex with roniciclib was collected from the protein data bank PDB ID (5iev) 51. The molecular docking procedures reliability was ensured through re-docking of the co-crystalized roniciclib in the vicinity of the CDK2 binding site, as mentioned above in the discussion section. Molecular docking of the target pyridazines into CDK2 active site was conducted by Vina Autodock, a more accurate and twice speed higher than Autodock 4 software52. Vina Autodock requires both ligands and receptors in pdbqt format, thus, MGL tools 1.5.7 were used to prepare all the necessary files to carry out the docking53. Also the MGL tools were used to generate a grid box around the binding site of Roniciclib. The docking results were visualised by Biovia discovery studio 2020 free visualiser54, and then the best scoring candidates were selected for further investigation.
3.3.3. In silico ADME calculation and Drug-Likeness properties prediction
The SwissADME online tool was utilised to calculate the ADME profiles for target pyridazines 11e, 11h, 11l and 11m, in addition to prediction of their medicinal chemistry friendliness and drug-likeness properties38.
4. Conclusions
To the best of our knowledge, this is the first study that reports on 3,6-disubstituted pyridazines as anticancer CDK inhibitors. Herein, a new series of 3,6-disubstituted pyridazines 11a–r has been synthesised, characterised and evaluated for in vitro anticancer activity against three human cancer cell lines, namely, T-47D (breast cancer), MDA-MB-231 (breast cancer) and SKOV-3 (ovarian cancer) cell lines by the SRB assay. While, the examined pyridazines elicited good activity against T-47D cells (IC50 range: 0.43 ± 0.01 − 35.9 ± 1.18 µM) and MDA-MB-231 cells (IC50 range: 0.99 ± 0.03 − 34.59 ± 1.13 µM), they exerted weak activity against SKOV-3 cells. Uniquely, the methyltetrahydropyran-bearing pyridazine 11m showed a submicromolar growth inhibitory potency towards both breast T-47D and MDA-MB-231 (IC50 = 0.43 ± 0.01 and 0.99 ± 0.03 µM, respectively) cell lines. In addition, the biological results indicated that pyridazines 11l and 11m exerted an efficient alteration in cell cycle progression and induction of apoptosis in both T-47D and MDA-MB-231 cells, alongside, with their good mean tumour selectivity indexes (13.7 and 16.1, respectively) upon assessment of their cytotoxicity towards non-tumorigenic breast MCF-10A cells. Based on a suggestion from a conducted in silico study, pyridazines 11e, 11h, 11l, and 11m were selected to be evaluated for their ability to inhibit CDK2, where they exerted good inhibitory activity (IC50 = 151, 43.8, 55.6 and 20.1 nM, respectively). Finally, the in silico study implied that target pyridazines 11 exhibited not only an efficient anticancer activity but also an acceptable ADME, physicochemical and druglikeness properties, specifically pyridazines 11l and 11m. Overall the obtained results from this study quite sustained our strategy and gave us a robust opportunity for further development and optimisation of 3,6-disubstituted pyridazine scaffold to enrich therapeutic arsenal with efficient and safe anticancer CDK inhibitors.
Supplementary Material
Funding Statement
This work was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University, through the Research Groups Program (Grant no. RGP-1440-0025).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Padma VV. An overview of targeted cancer therapy. Biomedicine 2015;5:19–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A.. Cancer statistics 2016. CA: Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 3.Lim S, Kaldis P.. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013;140:3079–93. [DOI] [PubMed] [Google Scholar]
- 4.Ingham M, Schwartz GK.. Cell-cycle therapeutics come of age. J. Clin. Oncol 2017;35:2949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng W, Yang Z, Wang S, et al. Q. Recent development of CDK inhibitors: an overview of CDK/inhibitor co-crystal structures. Eur J Med Chem 2019;164:615–39. [DOI] [PubMed] [Google Scholar]
- 6.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES.. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015;14:130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spring LM, Wander SA, Zangardi M, Bardia A.. CDK 4/6 inhibitors in breast cancer: current controversies and future directions. Curr Oncol Rep 2019;21:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reck M, Horn L, Novello S, et al. Phase II study of roniciclib in combination with cisplatin/etoposide or carboplatin/etoposide as first-line therapy in patients with extensive-disease small cell lung cancer. J Thorac Oncol 2019;14:701–11. [DOI] [PubMed] [Google Scholar]
- 9.Niu Y, Xu J, Sun T.. Cyclin-dependent kinases 4/6 inhibitors in breast cancer: current status, resistance, and combination strategies. J Cancer 2019;10:5504–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ritchie TJ, Macdonald SJ, Peace S, et al. The developability of heteroaromatic and heteroaliphatic rings–do some have a better pedigree as potential drug. Med Chem Comm 2012;3:1062–9. [Google Scholar]
- 11.Ahmed EM, Hassan MS, El-Malah AA, Kassab AE.. New pyridazine derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents; design, synthesis and biological evaluation. Bioorg Chem 2020;95:103497. [DOI] [PubMed] [Google Scholar]
- 12.Barberot C, Moniot A, Allart-Simon I, et al. Synthesis and biological evaluation of pyridazinone derivatives as potential anti-inflammatory agents. Eur J Med Chem 2018;146:139–46. [DOI] [PubMed] [Google Scholar]
- 13.Costas T, Besada P, Piras A, et al. New pyridazinone derivatives with vasorelaxant and platelet antiaggregatory activities. Bioorg Med Chem Lett 2010;20:6624–7. [DOI] [PubMed] [Google Scholar]
- 14.Rathish IG, Javed K, Bano S, et al. Synthesis and blood glucose lowering effect of novel pyridazinone substituted benzenesulfonylurea derivatives. Eur J Med Chem 2009;44:2673–8. [DOI] [PubMed] [Google Scholar]
- 15.Roth GJ, Heckel A, Kley JT, et al. Design, synthesis and evaluation of MCH receptor 1 antagonists-Part II: optimization of pyridazines toward reduced phospholipidosis and hERG inhibition. Bioorg. Med. Chem. Lett 2015;25:3270–4. [DOI] [PubMed] [Google Scholar]
- 16.Costas-Lago MC, Besada P, Rodríguez-Enríquez F, et al. Synthesis and structure-activity relationship study of novel 3-heteroarylcoumarins based on pyridazine scaffold as selective MAO-B inhibitors. Eur J Med Chem 2017;139:1–11. [DOI] [PubMed] [Google Scholar]
- 17.Kilic B, Gulcan HO, Aksakal F, et al. Design and synthesis of some new carboxamide and propanamide derivatives bearing phenylpyridazine as a core ring and the investigation of their inhibitory potential on in-vitro acetylcholinesterase and butyrylcholinesterase. Bioorg Chem 2018;79:235–49. [DOI] [PubMed] [Google Scholar]
- 18.Tan OU, Ozadali K, Yogeeswari P, et al. Synthesis and antimycobacterial activities of some new N-acylhydrazone and thiosemicarbazide derivatives of 6-methyl-4,5-dihydropyridazin-3(2H)-one. Med Chem Res 2012;21:2388–94. [Google Scholar]
- 19.Li D, Zhan P, Liu H, et al. Synthesis and biological evaluation of pyridazine derivatives as novel HIV-1 NNRTIs. Bioorg Med Chem 2013;21:2128–34. [DOI] [PubMed] [Google Scholar]
- 20.Jaballah MY, Serya RT, Abouzid K.. Pyridazine based scaffolds as privileged structures in anti-cancer therapy. Drug Res (Stuttg) 2017;67:138–48. [DOI] [PubMed] [Google Scholar]
- 21.Bongartz JP, Stokbroekx R, Van der Aa M, et al. Synthesis and anti-angiogenic activity of 6-(1,2,4-thiadiazol-5-yl)-3-amino pyridazine derivatives. Bioorg Med ChemLett 2002;12:589–91. [DOI] [PubMed] [Google Scholar]
- 22.Abouzid KA, Khalil NA, Ahmed EM, Mohamed KO.. [(3-[(6-Arylamino)pyridazinylamino]benzoic acids: design, synthesis and in vitro evaluation of anticancer activity. Arch Pharm Res 2013;36:41–50. [DOI] [PubMed] [Google Scholar]
- 23.Kim C, Park EH, Park MS.. Novel alkylaminopyridazine derivatives: synthesis and their anti-proliferative effects against MCF-7 cells. Bull Korean Chem Soc 2013;34:3317–21. [Google Scholar]
- 24.Kim C, Kim SB, Park MS.. Synthesis of novel 3-allylseleno-6-alkylthiopyridazines: their anticancer activity against MCF-7 cells. Arch Pharm Res 2014;37:452–8. [DOI] [PubMed] [Google Scholar]
- 25.Park HS, Kim C, Park MS.. Discovery and synthesis of novel allylthioaralkylthiopyridazines: their antiproliferative activity against MCF-7 and Hep3B cells. Arch Pharm Res 2015;38:791–800. [DOI] [PubMed] [Google Scholar]
- 26.George RF, Fouad MA, Gomaa IEO.. Synthesis and cytotoxic activities of some pyrazoline derivatives bearing phenyl pyridazine core as new apoptosis inducers. Eur J Med Chem 2016;112:48–59. [DOI] [PubMed] [Google Scholar]
- 27.Elmeligie S, Ahmed EM, Abuel-Maaty SM, et al. Design and synthesis of pyridazine containing compounds with promising anticancer activity. Chem Pharm Bull 2017;65:236–47. [DOI] [PubMed] [Google Scholar]
- 28.Sengmany S, Sitter M, Léone E, et al. Synthesis and biological evaluation of 3-amino-, 3-alkoxy- and 3-aryloxy-6-(hetero)arylpyridazines as potent antitumor agents. Bioorg Med Chem Lett 2019;29:755–60. [DOI] [PubMed] [Google Scholar]
- 29.Jaballah MY, Serya RAT, Saad N, et al. Towards discovery of novel scaffold with potent antiangiogenic activity; design, synthesis of pyridazine based compounds, impact of hinge interaction, and accessibility of their bioactive conformation on VEGFR-2 activities. J Enzyme Inhib Med Chem 2019;34:1573–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ali YM, Ismail MF, Abu El‐Azm FS, Marzouk MI.. Design, synthesis, and pharmacological assay of novel compounds based on pyridazine moiety as potential antitumor agents. J Heterocycl Chem 2019;56:2580–91. [Google Scholar]
- 31.Landge SM, Török B.. Highly enantioselective organocatalytic addition of ethyl trifluoropyruvate to ketones with subzero temperature microwave activation. Catal Lett 2009;131:432–9. [Google Scholar]
- 32.Sibgatulin DA, Volochnyuk DM, Kostyuk AN.. A convenient synthesis of 4-trifluoromethyl-(2H)-pyridazin-3-ones from methyl 3,3,3-trifluoropyruvate. Synlett 2005;2005:1907–11. [Google Scholar]
- 33.Siddiqui FA, Alam C, Rosenqvist P, et al. PDE6D inhibitors with a new design principle selectively block K-ras activity. ACS Omega 2020;5:832–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cyto-toxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–12. [DOI] [PubMed] [Google Scholar]
- 35.Daina A, Michielin O, Zoete V.. Swiss TargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucl Acids Res 2019;47:357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gfeller D, Michielin O, Zoete V.. Shaping the interaction landscape of bioactive molecules. Bioinformatics 2013;29:3073–9. [DOI] [PubMed] [Google Scholar]
- 37.Ding L, Cao J, Lin W, et al. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci 2020;21:1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daina A, Michielin O, Zoete V.. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daina A, Zoete V.. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016;11:1117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23. [DOI] [PubMed] [Google Scholar]
- 41.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ.. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 2001;46:3–26. [DOI] [PubMed] [Google Scholar]
- 42.Egan WJ, Merz KM, Baldwin JJ.. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem 2000;43:3867–77. [DOI] [PubMed] [Google Scholar]
- 43.Ghose AK, Viswanadhan VN, Wendoloski JJ.. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem 1999;1:55–68. [DOI] [PubMed] [Google Scholar]
- 44.Muegge I, Heald SL, Brittelli D.. Simple selection criteria for drug-like chemical matter. J Med Chem 2001;44:1841–6. [DOI] [PubMed] [Google Scholar]
- 45.Baell JB, Holloway GA.. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53:2719–40. [DOI] [PubMed] [Google Scholar]
- 46.Ruth B, Schipani A, James D, et al. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008;3:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sabt A, Abdelhafez OM, El-Haggar RS, et al. Novel coumarin-6-sulfonamides as apoptotic anti-proliferative agents: Synthesis, in vitro biological evaluation, and QSAR studies. J Enzym Inhib Med Chem 2018;33:1095–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eldehna WM, Hassan GS, Al-Rashood ST, et al. Synthesis and in vitro anticancer activity of certain novel 1-(2-methyl-6-arylpyridin-3-yl)-3-phenylureas as apoptosis-inducing agents. J Enzym Inhib Med Chem 2019;34:322–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eldehna WM, Hassan GS, Al-Rashood ST, et al. Marine-inspired bis-indoles possessing antiproliferative activity against breast cancer; design, synthesis, and biological evaluation. Mar Drugs 2020;18:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Said MA, Eldehna WM, Nocentini A, et al. Sulfonamide-based ring-fused analogues for CAN508 as novel carbonic anhydrase inhibitors endowed with antitumor activity: design, synthesis, and in vitro biological evaluation. Eur J Med Chem 2020;189:112019. [DOI] [PubMed] [Google Scholar]
- 51.Ayaz P, Andres D, Kwiatkowski DA, et al. Conformational adaption may explain the slow dissociation kinetics of roniciclib (BAY 1000394), a type I CDK inhibitor with kinetic selectivity for CDK2 and CDK9. ACS Chem Biol 2016;11:1710–9. [DOI] [PubMed] [Google Scholar]
- 52.Trott O, Olson AJ.. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comp Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris GM, Huey R, Lindstrom W, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comp Chem 2009;16:2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Available online : https://3dsbiovia.com/resource-center/downloads/ [last accessed 10 April 2020].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.