

ABSTRACT
In colorectal cancer, Wnt/β-catenin signaling is often aberrantly activated and associated with a T-cell-excluded phenotype which is a major obstacle for many immunotherapies. However, the effects of Wnt/β-catenin inhibition on tumor immunity and immunotherapy remain to be elucidated. In syngeneic mouse models of colorectal cancer, β-catenin/TCF inhibitor iCRT14 potently enhanced the infiltration of T and NK cells, without influencing their proliferation or the infiltration of most myeloid populations. Mechanistically, β-catenin inhibition upregulated while its overexpression suppressed the expression of T/NK cell-recruiting CXCR3 chemokines CXCL9/10/11 in both mouse and human colorectal cancer cells. Furthermore, iCRT14 treatment synergized with tumor vaccines or Treg cell ablation to achieve a complete inhibition of tumor growth in syngeneic models of CT26-OVA and MC38-S33Y.β-cat, respectively. Taken together, our work reveals that β-catenin inhibition shifts colorectal tumor microenvironment into a T-cell-inflamed phenotype and potentiates the efficacy of other immunotherapeutic strategies for colorectal cancer.
KEYWORDS: Wnt, β-catenin, tumor microenvironment, immunotherapy, colorectal cancer, CXCR3 chemokine
Introduction
Over the past few years, colorectal cancer ranks among the top three of all cancers regarding the incidence and mortality.1–3 Recent breakthroughs in immunotherapies such as CAR-T and checkpoint blockade have revolutionized the treatments for cancer. However, many tumors are resistant to immunotherapy due to their complex microenvironment. A T-cell-inflamed phenotype has been shown to be associated with better outcomes and responses to immunotherapies in patients with colorectal and other cancers.4–6 In particular, chemotaxis mediated by chemokines CXCL9/10/11 and their receptor CXCR3 is crucial for tumor infiltration of effector cells including T and NK cells, as well as responsiveness to immunotherapy.7–9
Besides their intrinsic effects on tumor cell proliferation and apoptosis, oncogenic pathways like the Wnt signaling have been found to act on tumor microenvironment and lead to immune evasion.10 In melanoma, it has been shown that Wnt/β-catenin activation results in reduced tumor infiltration of dendritic cells (DCs) and T cells via inhibition of CCL4 expression.11 Moreover, analyses of 1211 primary colorectal tumor samples12 and a panel of 31 solid tumor types13 contained within The Cancer Genome Atlas (TCGA) suggested that aberrant Wnt/β-catenin signaling might lead to a T cell-excluding phenotype. Importantly Wnt/β-catenin signaling is one of the most frequently dysregulated pathways in colorectal cancer.14,15 However, the underlying mechanism of how β-catenin signaling impacts colorectal tumor immunity is still unclear. Given that tumor T cell infiltration is crucial for immunosurveillance and immunotherapy, Wnt/β-catenin pathway potentially serves as a promising target for shaping the immune microenvironment of colorectal cancer.
To date, many Wnt/β-catenin pathway inhibitors target the upstream components of the, including but not limited to blocking the secretion of Wnt ligands, 16 upregulating the expression of GSK3β17 and stabilizing Axin.18 However, these therapies have been found to be ineffective for colorectal cancer with mutations in APC or CTNNB1 (β-catenin), which points us to target the downstream of the Wnt/β-catenin pathway. In the current study, we used a small molecule inhibitor iCRT14 to block T cell factor (TCF)/lymphoid enhancer factor (LEF) responsive transcription of canonical Wnt pathway.19 iCRT14 has been shown to interfere with β-catenin/TCF interaction and TCF binding to DNA. We aim to investigate the effects of β-catenin/TCF inhibition on the tumor immunity and immunotherapy in colorectal cancer, and to uncover the underlying mechanisms.
Results
iCRT14 inhibits β-catenin-mediated transcriptional activity and colorectal tumor cell growth in vitro
To block Wnt/β-catenin pathway, we chose a small-molecule compound iCRT14 that targets β-catenin/TCF transcriptional complex and was able to inhibit the activity of mutant β-catenin.19 Indeed, iCRT14 inhibited GSK3 inhibitor-induced canonical Wnt pathway activity in a dose-dependent manner (Supplemental. S1A). Moreover, it was able to suppress the activities of two mutant forms of β-catenin, S33Y (replacing serine 33 with tyrosine) and Δ45 (lacking serine 45), that are resistant to GSK3β/CK1α-mediated phosphorylation and destruction, while XAV939 which could stabilize Axin to downregulate β-catenin, 18 failed to exhibit any inhibitory effect (Supplemental. S1B). Accordingly, iCRT14 was able to inhibit the proliferation of human colorectal cancer cell line HCT116 which expressed the degradation-resistant mutant β-catenin while XAV939 had no effect (Supplemental. S1C). As compared to XAV939 that targets the upstream components of β-catenin destruction complex and is not able to inhibit the degradation-resistant mutant β-catenin, iCRT14 targets the downstream β-catenin/TCF transcriptional complex, and can block the aberrant activity of Wnt signaling mediated by abnormalities in any upstream pathway components or downstream transcriptional members. Therefore, iCRT14 might be applicable in a broader cohort of colorectal cancer patients including those with β-catenin mutations.
To establish a Wnt/β-catenin-active colorectal tumor model, we examined the activity of the Wnt/β-catenin in two mouse colorectal cancer cell lines CT26 and MC38. As compared to MC38, CT26 clearly showed higher β-catenin expression (Supplemental. S2A) and TCF reporter (TOP) activity (Supplemental. S2B). iCRT14 dose-dependently decreased the TCF reporter activity without affecting β-catenin protein levels in CT26 cells (Supplemental. S2C-D). Moreover, iCRT14 potently inhibited the proliferation of CT26 cells and the expression of Wnt target genes including Axin2, Ccnd1 and Myc in vitro (Supplemental. S2E-F). Together with the previous study, 19 these data indicate that iCRT14 effectively inhibits the transcriptional activity of β-catenin/TCF in colorectal cancer cells.
β-Catenin inhibition enhances T and NK cell infiltration in colorectal tumors in vivo
To study the effect of iCRT14 on tumor immunity, CT26 cell line was first used to establish the syngeneic subcutaneous mouse model. Notably, iCRT14 treatment significantly increased CD45+ leukocytes in the CT26 tumors (Figure 1a). Among the total leukocytes, the ratios of cytotoxic CD8+ T cells (Figure 1b) and Foxp3− conventional CD4+ T cells (Figure 1c) were elevated by iCRT14. NK cell ratio in total cells was also significantly raised by iCRT14, although the change in the ratio among CD45+ cells did not reach statistically significant level (Figure 1d). T regulatory (Treg) cells appeared to accumulate in the tumors received iCRT14 treatment too (Figure 1e). However, the ratio of conventional CD4+ T cells to Treg cell seemed to be upregulated in iCRT14-treated tumors (Figure 1f). In contrast, iCRT14 did not change the ratios of most myeloid populations in the tumors, including M2 macrophages and myeloid-derived suppressor cells (MDSCs) (Figure 1g), conventional DCs (cDCs) or cDC subtypes (CD103+ or CD11b+) (Supplemental. S3A). Only M1 macrophages were enriched in iCRT14-treated tumors (Figure 1g). Of note, the splenic CD4+ T, CD8+ T and NK cells were not influenced by iCRT14 (Supplemental. S3B). These data indicate that the treatment specifically modulates the tumor lymphocytes including T and NK cells.
Figure 1.
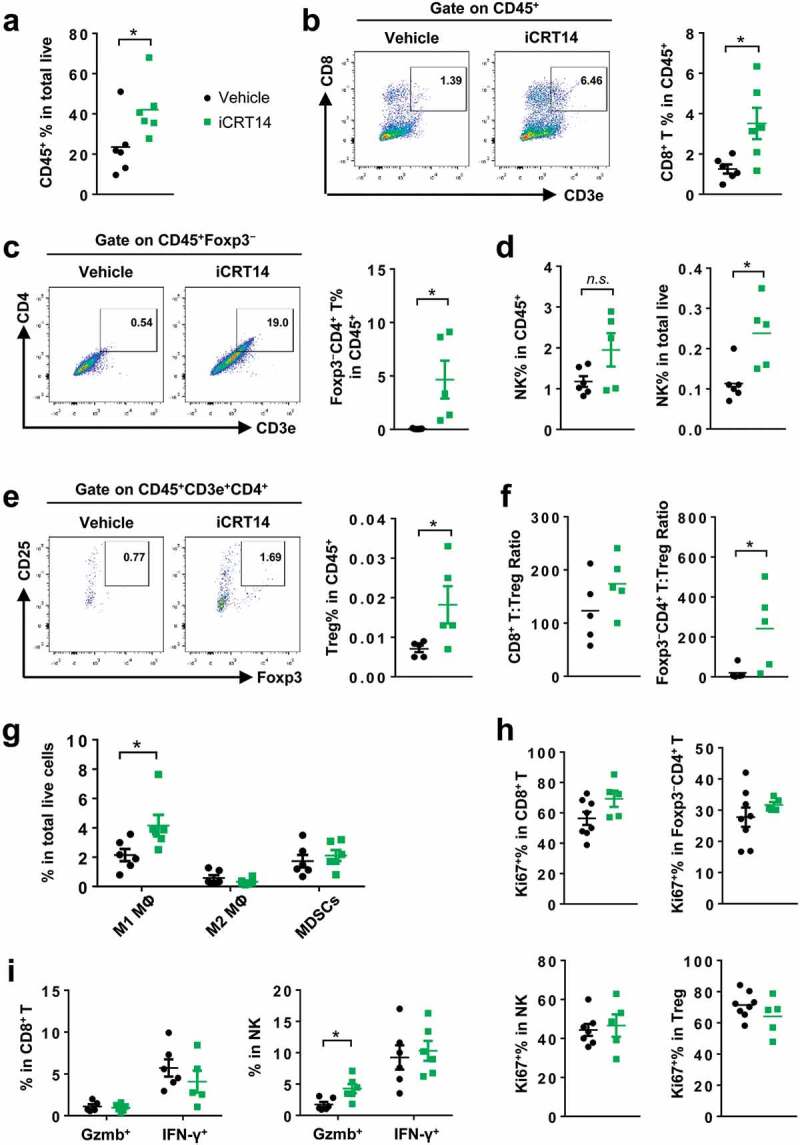
β-Catenin inhibition induces lymphocyte infiltration in CT26 tumors. Mice bearing CT26 tumors were treated with iCRT14 or vehicle every other day for 12 d. FACS was performed to analyze tumor-infiltrating immune cells. (a) Percentage of CD45+ cells in total live cells. (b) Percentage of CD8+ T cells (CD45+CD3+CD8+) in CD45+ cells. (c) Percentage of conventional CD4+ T cells (CD45+CD3+CD4+Foxp3−) in CD45+ cells. (d) Percentage of NK cells (CD45+NKp46+CD3−) in CD45+ cells and total live cells. (e) Percentage of Treg cells (CD45+CD3+CD4+CD25+Foxp3+) in CD45+ cells. (f) Ratios of CD8+ T to Treg cells, and conventional CD4+ T to Treg cells. (g) Percentages of macrophages (MΦ) (M1: CD45+CD11b+F4/80+ CD206lowMHC-IIhigh; M2: CD45+CD11b+F4/80+CD206highMHC-IIlow) and MDSCs (CD45+CD11b+Gr-1+CD11c−MHC-II−/low) in total live cells. (h) Percentages of Ki67+ cells in CD8+ T, conventional CD4+ T, NK and Treg cells. (i) Percentages of Granzyme B+ (Gzmb+) and IFN-γ+ cells in CD8+ T and NK cells. * P < .05, n.s. = not significant
FACS analysis of Ki67+ cell in T and NK cells showed that expression of this proliferation marker was not changed by iCRT14 (Figure 1h), indicating that the increased ratios of these cells in the tumors were likely due to an enhanced infiltration rather than increased proliferation. In addition, iCRT14 did not affect the expression of antitumor factors granzyme B (Gzmb) and interferon (IFN)-γ in CD8+ T or NK cells (Figure 1i). These results demonstrate that β-catenin blockade shapes tumor immune microenvironment mainly through regulation of the T infiltration but not the proliferation or function of T cells.
Next, we explored the effect of iCRT14 on tumor immune microenvironment in another syngeneic model of MC38 overexpressing the constitutively active S33Y. β-catenin (MC38-S33Y.β-cat). As shown by immunofluorescence, significantly more CD4+ T, CD8+ T and NK cells infiltrated the tumors upon iCRT14 treatment (Figure 2a-c). In accordance with observations in the CT26 model (Figure 1), β-catenin/TCF inhibition was able to turn the Wnt-signaling-active tumors into a T/NK-cell-inflamed phenotype.
Figure 2.
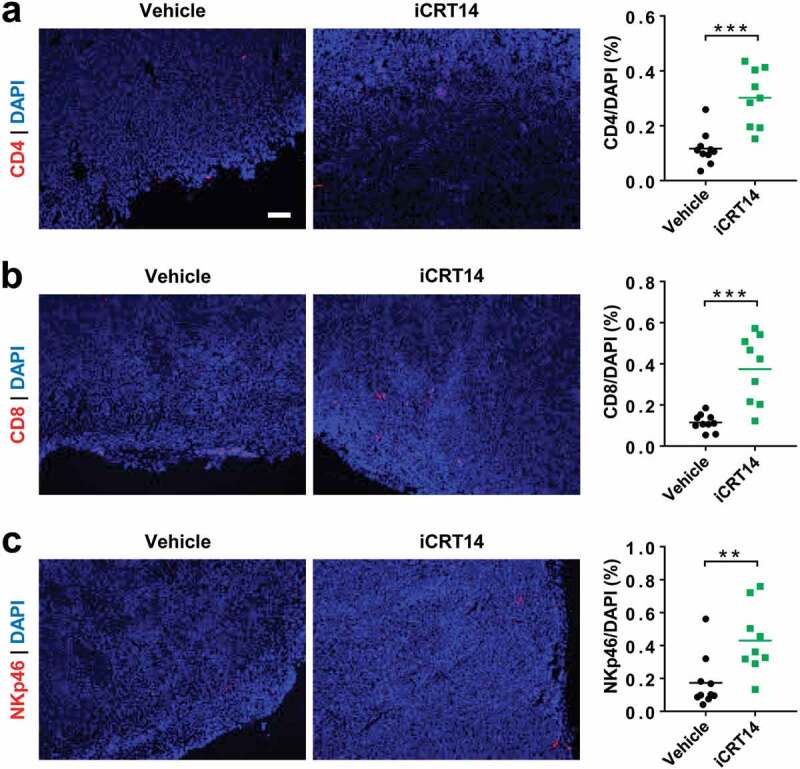
β-Catenin blockade induces lymphocyte infiltration in MC38-S33Y.β-cat tumors. Mice bearing MC38-S33Y.β-cat tumors were treated with iCRT14 or vehicle every other day for 12 d. Immunofluorescence was performed to analyze tumor-infiltrating CD4+ T (by CD4 staining) (a), CD8+ T (by CD8 staining) (b), and NK cells (by NKp46 staining) (c). Total cell number was counted by nucleus staining (DAPI). Scale bar = 100 µm. ** P < .01, *** P < .001
β-Catenin inhibition upregulates the expression of CXCR3 chemokines in colorectal tumor cells
Since it had been well established that CXCR3 chemokines CXCL9/10/11 were crucial for tumor infiltration of T and NK cells, 7–9 we then examined whether these chemokines were changed by β-catenin blockade. In iCRT14-treated CT26 tumors, there was an increase in the mRNA expression of Cxcl9, 10 and 11 (Figure 3a). To verify whether the enhanced chemokine expression was tumor cell intrinsic, CT26 cells were treated with iCRT14 in vitro and they subsequently showed an upregulation of these chemokines too (Figure 3b), suggesting that β-catenin inhibition directly regulated their expression in tumor cells. In consistence with the change in mRNA expression, secretion of CXCL10 proteins into the medium was also elevated by iCRT14 (Figure 3c). On the contrary, overexpression of the constitutively active mutant S33Y.β-catenin downregulated the expression of these CXCR3 chemokines in both CT26 and MC38 cells (Figure 3d,e). In MC38-S33Y.β-cat cells, iCRT14 restored the expression of all three chemokines (Figure 2f). In human colorectal cancer cell lines HCT116 and SW480 with mutations in APC and/or CTNNB1, 20 at least two of the CXCR3 chemokines were significantly upregulated by iCRT14 treatment (Figure 3g). These data point out that β-catenin inhibition-induced CXCR3 chemokine upregulation may contribute to the tumor infiltration of T and NK cells.
Figure 3.
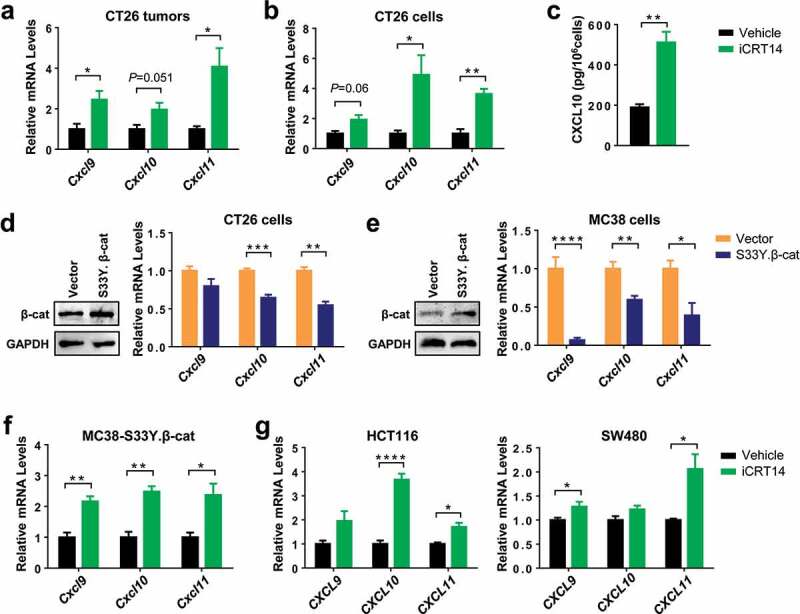
β-Catenin inhibition upregulates the expression of CXCR3 chemokines in colorectal tumor cells. (a) qPCR analysis of chemokine mRNA expression in CT26 tumors of iCRT4- or vehicle-treated mice. n = 4. (b) qPCR analysis of chemokine mRNA expression in CT26 cells treated with iCRT14 or vehicle in vitro. n = 4. (c) Quantification of CXCL10 proteins in the media of CT26 cell culture by ELISA. n = 3. (d-e) CT26 (d) or MC38 (e) were transduced with lentivirus containing S33Y.β-catenin or empty vector. Expression of β-catenin protein and chemokine mRNA expression was measured by Western blot and qPCR respectively. n = 4. (f) MC38-S33Y.β-cat cells was treated with iCRT14 or vehicle. Chemokine mRNA expression was analyzed by qPCR. n = 4. (g) qPCR analysis of chemokine mRNA expression in HCT16 and SW480 cell lines treated with iCRT14 or vehicle in vitro. n = 4. * P < .05, ** P < .01, *** P < .001, **** P < .0001
iCRT4 synergies with tumor vaccine and Treg cell-targeted immunotherapy
Given the findings that iCRT14 treatment potently enhanced tumor infiltration of T and NK cells, we hypothesized that β-catenin inhibition would potentiate the efficacy of other immunotherapies. To test this hypothesis, ovalbumin (OVA) was overexpressed in CT26 cells as tumor-specific antigens to establish the subcutaneous model of CT26-OVA that was potentially suitable for vaccine therapy using OVA protein plus poly(I:C) (Figure 4a). Monotherapy with iCRT14 or tumor vaccine alone only moderately but not significantly reduced tumor growth (Figure 4b). However, the combination of iCRT14 and vaccine completely inhibited the tumor growth (Figure 4b), demonstrating a synergistic effect of these two monotherapies.
Figure 4.
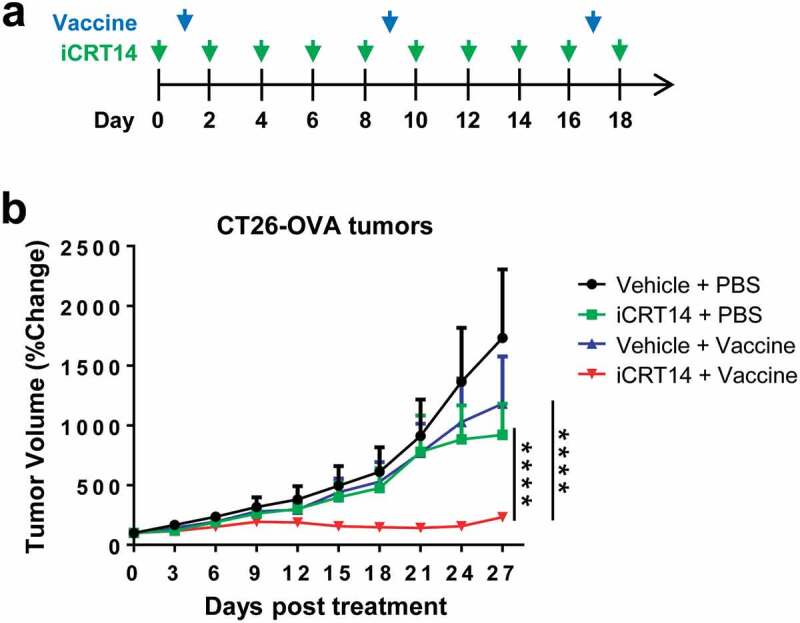
iCRT14 synergizes with vaccine therapy for tumor control. (a) Scheme of tumor vaccine and iCRT14 treatments. (b) Tumor growth of CT26-OVA tumors upon tumor vaccine, iCRT14 or combination treatments. n = 5/group. **** P < .0001
As there was an influx of Treg cells into the tumors which accompanied the infiltration of other effect T cells upon iCRT14 treatment, this provoked us to explore if β-catenin inhibition would synergize with Treg cell-targeted therapy. We established a syngeneic model of MC38-S33Y.β-cat cells in DEREG (Foxp3-DTR) mice in which Foxp3+ Treg cell could be depleted by DT administration (Figure 5a).21 Monotherapy of iCRT14 or DT-mediated Treg ablation both significantly suppressed MC38-S33Y.β-cat tumor growth while the combination therapy eventually completely eradicated the tumors (Figure 5b). In wild-type mice, DT treatment did not change MC38-S33Y.β-cat tumor growth or tumor-infiltrating T cells (Supplemental. S4), excluding any unspecific effects of DT on tumor or immune cells. These results confirm the synergistic effect of β-catenin inhibition by iCRT14 with other immunotherapy for colorectal cancer.
Figure 5.
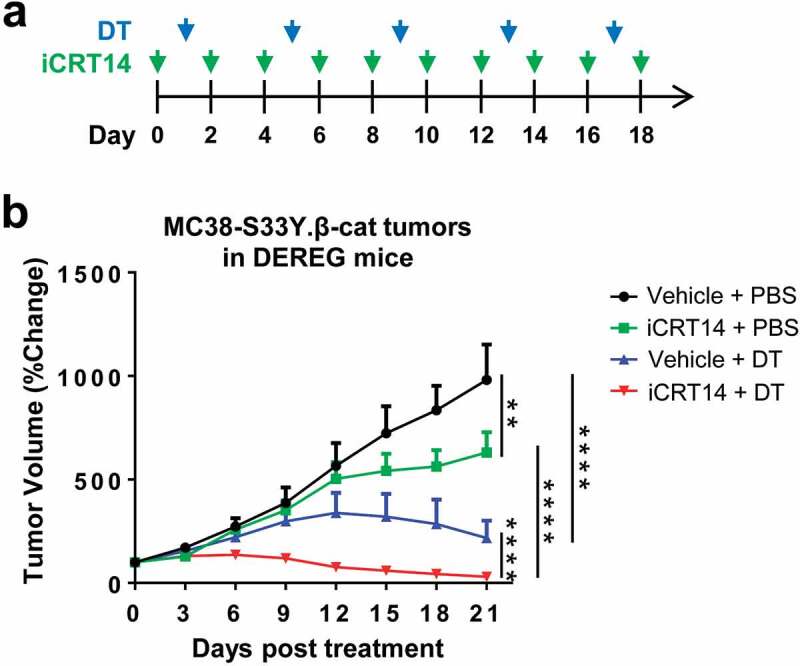
iCRT14 synergizes with Treg cell ablation to eradicate colorectal tumors. (a) Scheme of DT and iCRT14 treatments. (b) Tumor growth of MC38-S33Y.β-cat tumors in DEREG mice upon tumor vaccine, iCRT14 or combination treatments. n = 6/group. ** P < .01, **** P < .0001
Discussion
The oncogenic role of Wnt/β-catenin pathway has been extensively studied during the past two decades. However, most research focused on its effect on tumor cell proliferation and apoptosis, until recently an immunomodulatory function of Wnt/β-catenin signaling has drawn an increasing attention.11,22-24 Wnt/β-catenin pathway activation counteracts the antitumor immunity via various mechanisms such as tumor exclusion of DCs, 11 downregulation of tumor cell surface innate immune sensors and suppression of DC maturation.23 Here, by examining the tumor immune microenvironment upon treatment of β-catenin/TCF inhibitor iCRT14, we uncovered a novel mechanism that β-catenin inhibition enhanced tumor infiltration of T and NK cells via upregulation of tumor cell-intrinsic expression of CXCR3 chemokines. These findings also contribute to the explanations for the correlation of active Wnt/β-catenin signaling with T cell exclusion observed in the tumor samples of colorectal cancer patients.12
In the syngeneic model of colorectal cancer, β-catenin inhibition has only an impact on the infiltration of T and NK cells, but not the proliferation or antitumor factor expression of cytotoxic T cells. Therefore, the effects of β-catenin inhibition on colorectal tumor immune microenvironment largely rely on the regulation of lymphocyte migration, which was also supported by the upregulation of CXCR3 chemokine expression. By overexpression of S33Y.β-catenin or treatment with the inhibitor iCRT14, a clear suppressive effect of β-catenin activation on the expression of these T/NK cell-recruiting chemokines has been demonstrated, although the in-depth molecular mechanism remains to be investigated in the future. It provides a direct control of the T-cell-excluded phenotype by the Wnt/β-catenin pathway, rather than indirect regulation through other immune cells such as DCs that might further attract T cells. Of note, in colorectal tumors iCRT14 treatment did not influence the infiltration of most other myeloid populations, especially CD103+ DCs that were shown to be excluded by Wnt-active melanoma.11 Such discrepancy is presumably due to cell/tumor context-dependent mechanisms as Wnt target genes are quite variable in different cell types.
Since T cell exclusion is one of the major hurdles for immunotherapy of solid tumors, the most important clinical implication of our study is that β-catenin blockade drastically promotes T cell infiltration and the efficacy of other immunotherapeutic strategies. This finding broadens not only the applicability of various immunotherapies such as tumor vaccines and Treg cell-targeted antibodies but also the use of Wnt/β-catenin inhibitors. Although lots of inhibitors for Wnt pathway have been developed, their clinical applications have not been very successful.25 Given the fact that a substantial proportion of colorectal cancer patients show aberrantly activated canonical Wnt signaling, inhibitors targeting β-catenin and the transcriptional complex may exhibit better clinical outcomes than inhibitors for the upstream components. In addition to its effect on T/NK cell infiltration, the direct impact of β-catenin inhibitor on the differentiation, maturation and function of immune cells still needs to be clarified. Nevertheless, our study provides a rationale that the combination of β-catenin inhibitor and immunotherapy will result in a synergistic effect for the treatment of colorectal cancer.
Materials and methods
Cell lines
Mouse colorectal cancer cell line CT26 was from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). Human colon cancer cell lines HCT116 and SW480 were purchased from the American Type Culture Collection (ATCC). Mouse colorectal cancer cell line MC38 was a kind gift from the laboratory of Dr Yan Zhang (Shanghai Jiao Tong University, China). CT26 cells were cultured in RPMI. MC38, HCT116 and SW480 cells were cultured in DMEM. Basic media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Authentication of these cell lines was performed by Shanghai Biowing Applied Biotechnology Co. Ltd. using the short tandem repeat genetic analysis. CT2-OVA and MC38-S33Y.β-cat cells were generated by transduction of wild-type cell lines with lentiviruses expressing chicken ovalbumin or mouse S33Y.β-catenin, respectively. All cell lines were cultured for no more than 2–3 weeks after thawing and routinely checked for mycoplasma infection.
Reagents and antibodies
iCRT14 was purchased from BOC Sciences. LY20900314 and XAV939 were from Selleck. The antibodies used for Western blotting were anti-β-catenin and anti-GAPDH (Abcam). Antibodies for flow cytometry analysis were purchased from BioLegend, eBioscience or BD Biosciences.
Cell proliferation assay
The CT26 and HCT116 cell lines were cultured in 96-well plate and treated with iCRT14 or XAV939. Numbers of viable cells were quantified using Cell Counting Kit-8 (CCK-8) (Dojindo, Japan).
Wnt/β-catenin pathway reporter assays
Wnt/β-catenin pathway reporter construct M50 Super 8X TOPflash (Addgene plasmid #12456) or its mutant control M51 Super 8X FOPflash (Addgene plasmid #12457) were kind gifts from Randall Moon.26 Transient plasmid transfection in 293 T cells was performed using jetPRIME transfection reagent (Polyplus Transfection). Transfection efficiency was normalized by co-transfection with a pRL-CMV reporter containing Renilla luciferase gene. Luciferase activity was measured using the Dual-Glo luciferase assay system (Promega).
To analyze Wnt/β-catenin pathway activity in CT26 and MC38 cells, the Wnt responsive element and mutant control sequences were cloned in front of the ZsGreen gene in a lentiviral vector from Super 8X TOPflash or FOPflash plasmid, respectively. Wnt/β-catenin activity was calculated as mean fluorescence intensity (MFI) of ZsGreen normalized to that of SV40 promoter-driven mCherry in the same lentivector.
Syngeneic tumor models and treatments
Seven- to eight-week-old female BABL/c mice were injected with a single cell suspension of 0.5 × 106 CT26 or CT26-OVA per mouse subcutaneously on the right flank. MC38-S33Y.β-cat cells (1 × 106/mouse) were subcutaneously injected to DEREG (Foxp3-DTR/EGFP) mice (MMRRC Stock No: 32050-JAX). When the average tumor size reached around 100 mm3, mice received intraperitoneal (i.p.) injections of iCRT14 at a dose of 80 mg/kg every other day. iCRT14 was freshly dissolved in 20% DMSO, 40% PEG, 10% Tween 80 and 30% saline before i.p. injection. For tumor vaccine treatment, mice with CT26-OVA tumors were given i.p. injections of 100 µg OVA protein and 100 µg poly(I:C) (Sigma-Aldrich). To deplete Treg cells, diphtheria toxin (DT) (Merck) was administered i.p. at a dose of 15 µg/kg of body weight every 4 d to DEREG mice. Tumor size was measured using a caliper to determine the diameter and tumor volume was calculated as the formula: 0.5×length×width2 (length was the longer dimension). All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) and the Bioethics Committee of School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, China.
Flow cytometry
Subcutaneous tumors were dissected and dissociated into single-cell suspension using mouse tumor dissociation kit (Miltenyi Biotec). The cell suspension was incubated with Zombie Dyes (BioLegend) to exclude dead cells and then stained with antibodies for cell surface markers. For staining of cytoplasmic proteins, the mice were i.p. injected with 250 µg brefeldin A (BFA) (Selleck) 4 h before euthanasia. Cytoplasmic staining was performed using Cyto-Fast™ Fix/Perm Buffer Set (BioLegend). Nuclear proteins were stained using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience). Fluorescence data were collected on a BD LSRFortessa cell analyzer (BD Biosciences) and analyzed using FlowJo software.
Immunofluorescence
Subcutaneous tumor samples were cut into 5-µm sections. Immunofluorescent staining of tumor sections was performed using anti-CD4 [clone RM4-5], anti-CD8a [clone 53–6.7] and anti-NKp46 [clone 29A1.4] antibodies (BioLegend). Slides were mounted in anti-fade mounting medium with DAPI (ThermoFisher Scientific) and visualized under a Zeiss fluorescence microscope.
Quantitative PCR (qPCR)
RNA was isolated using the RNeasy Mini kit (Qiagen). The reverse transcription was conducted using the PrimeScript RT Reagent Kit (Takara). qPCR reaction was carried out using SYBR Green PCR Master Mix Kit (Takara) and ABI 7900HT Fast Real-Time PCR System (Applied Biosystems). Relative mRNA expression level was calculated by the 2−ΔΔ CT method and GAPDH was used as internal control.
ELISA
CT26 cell line was cultured treated with 25 μM iCRT14 for 24 h before cell culture supernatant was collected. CXCL10 concentration was measured using mouse CXCL10 ELISA kit (R&D Systems) according to the manufacturer’s instruction.
Western blot
Cell lysates were collected using RIPA buffer (ThermoFisher Scientific) supplemented with protease inhibitor cocktail (Roche).Protein concentration was determined using Pierce BCA Protein Assay Kit (ThermoFisher Scientific). Proteins (15 to 25 μg) were applied to 10% SDS-PAGE and transferred to PVDF membranes (Millipore). Images were developed using a chemiluminescent HRP substrate (Millipore).
Statistics
All data were represented as mean ± SEM. Statistical analysis was performed using GraphPad Prism. Two-way ANOVA was used to analyze tumor volumes, and two-tailed Student’s t-test was used to determine the significance of all the other assays. P < .05 was considered statistically significant.
Supplementary Material
Funding Statement
This work was supported by Ministry of Science and Technology of the People’s Republic of China [2017YFA0102900 to W.-Q.G.], National Natural Science Foundation of China [81602484 to B.M., 81872406 and 81630073 to W.-Q.G.], Science and Technology Commission of Shanghai Municipality [16JC1405700 to W.-Q.G.], Shanghai Jiao Tong University Scientific and Technological Innovation Funds [2019TPB07 to W.-Q.G.], and the Shanghai Young Eastern Scholar Funds [QD2016005 to B.M.].
Author contribution
B.M. and W.-Q.G. designed the research project; C.W., J.Y., P.Y., L.G., L.J., and B.M. performed the experiments; C.W., J.Y., B.M., and W.-Q.G. analyzed the data and wrote the manuscript.
Disclosure of potential conflicts of interest
The authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A.. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–7. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 4.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 5.Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44(3):698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 6.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–150. doi: 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, Luster AD. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. 2019;50(6):1498–512e5. doi: 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao Q, Wang S, Chen X, Cheng S, Zhang Z, Li F, Huang L, Yang Y, Zhou B, Yue D, et al. Cancer-cell-secreted CXCL11 promoted CD8+ T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J Immunother Cancer. 2019;7(1):42. doi: 10.1186/s40425-019-0511-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018;18(3):139–147. doi: 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 12.Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, Nowak JA, Nishihara R, Qian ZR, Inamura K, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8(6):730–749. doi: 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/beta-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25(10):3074–3083. doi: 10.1158/1078-0432.CCR-18-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas N . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morin PJ, Kinzler KW, Sparks AB. Beta-catenin mutations: insights into the APC pathway and the power of genetics. Cancer Res. 2016;76(19):5587–5589. doi: 10.1158/0008-5472.CAN-16-2387. [DOI] [PubMed] [Google Scholar]
- 16.Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J, Jeyaraj DA, Pendharkar V, Ghosh K, Virshup IH, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene. 2016;35(17):2197–2207. doi: 10.1038/onc.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying S. Soy isoflavone genistein upregulates epithelial adhesion molecule E-cadherin expression and attenuates beta-catenin signaling in mammary epithelial cells. Carcinogenesis. 2009;30(2):331–339. doi: 10.1093/carcin/bgn279. [DOI] [PubMed] [Google Scholar]
- 18.Huang S-MA, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 19.Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, Nagourney R, Cardozo T, Brown AMC, DasGupta R, et al. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A. 2011;108(15):5954–5963. doi: 10.1073/pnas.1017496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci U S A. 1997;94(19):10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji L, Qian W, Gui L, Ji Z, Yin P, Lin GN, Wang Y, Ma B, Gao W-Q. Blockade of beta-catenin-induced CCL28 suppresses gastric cancer progression via inhibition of treg cell infiltration. Cancer Res. 2020;80(10):2004–2016. doi: 10.1158/0008-5472.CAN-19-3074. [DOI] [PubMed] [Google Scholar]
- 22.Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massagué J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165(1):45–60. doi: 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pacella I, Cammarata I, Focaccetti C, Miacci S, Gulino A, Tripodo C, Ravà M, Barnaba V, Piconese S. Wnt3a neutralization enhances T-cell responses through indirect mechanisms and restrains tumor growth. Cancer Immunol Res. 2018;6(8):953–964. doi: 10.1158/2326-6066.CIR-17-0713. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Tian T, Kalland KH, Ke X, Qu Y. Targeting Wnt/beta-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. 2018;39(7):648–658. doi: 10.1016/j.tips.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Duchartre Y, Kim YM, Kahn M. The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol. 2016;99:141–149. doi: 10.1016/j.critrevonc.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Current Biol: CB. 2003;13(8):680–685. doi: 10.1016/S0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.