

ABSTRACT
Mononuclear phagocytes and NK cells constitute the first line of innate immune defense. How these cells interact and join forces against cancer is incompletely understood. Here, we observed an early accumulation of slan+ (6-sulfo LacNAc) non-classical monocytes (slanMo) in stage I melanoma, which was followed by an increase in NK cell numbers in stage III. Accordingly, culture supernatants of slanMo induced migration of primary human NK cells in vitro via the chemotactic cytokine IL-8 (CXCL8), suggesting a role for slanMo in NK cell recruitment into cancer tissues. High levels of TNF-α and IFN-γ were produced in co-cultures of TLR-ligand stimulated slanMo and NK cells, whereas much lower levels were contained in cultures of slanMo and NK cells alone. Moreover, TNF-α and IFN-γ concentrations in slanMo/NK cell co-cultures exceeded those in CD14+ monocyte/NK cell and slanMo/T cell co-cultures. Importantly, TNF-α and IFN-γ that was produced in TLR-ligand stimulated slanMo/NK cell co-cultures induced senescence in different melanoma cell lines, as indicated by reduced melanoma cell proliferation, increased senescence-associated β-galactosidase expression, p21 upregulation, and induction of a senescence-associated secretory phenotype (SASP). Taken together, we identified a role for slanMo and NK cells in a collaborative innate immune defense against melanoma by generating a tumor senescence-inducing microenvironment. We conclude that enhancing the synergistic innate immune crosstalk of slanMo and NK cells could improve current immunotherapeutic approaches in melanoma.
KEYWORDS: slanMo, NK cell, melanoma, senescence, cytokines
Introduction
Despite recent success with T cell-based checkpoint inhibitor immunotherapies, a high number of melanoma patients still do not respond to current treatments. For these patients, an optimal stimulation of innate immune cell populations, representing the first line of immune defense against pathogens and tumors, might reestablish functional immune responses and reduce tumor growth.
Monocytes are an abundant blood leukocyte population that develop into macrophages in tissues and exert both homeostatic and inflammatory functions. slan+ monocytes (slanMo) are part of the non-classical monocyte (CD14dim/CD16+) subset in humans. They are defined by a carbohydrate modification on P-selectin glycoprotein-ligand 1 (PSGL-1), which can be specifically recognized by the monoclonal antibody M-DC8.1 slanMo share several characteristics with dendritic cells, which distinguish them from conventional CD14+ monocytes, including high cytokine production and efficient T cell priming.2,3 It was reported that tumor cells can activate slanMo and stimulate TNF-α production.4 slanMo can be detected at tumor-draining lymph nodes of patients with different solid tumors as well as non-Hodgkin lymphoma.5,6 The precise role and function of slanMo in cancer tissues is still under investigation, but recent reports have suggested the presence of a macrophage-like and a DC-like phenotype in tissues.6,7 slanMo can increase proliferation, expression of activation markers such as CD69, NKG2D, Nkp46, and CD95, IFN-γ cytokine production and cytotoxicity of NK cells.4,8-10 In turn, NK cells can promote slanMo maturation and IL-12 production, thereby facilitating adaptive immune responses.8 So far, it is unclear whether the crosstalk between slanMo and NK cells can result in an improved anti-tumor response.
The composition of the tumor microenvironment (TME) is dictated by several factors, including the need for appropriate chemotactic stimuli targeting the different immune subsets. In the murine system, patrolling monocytes were recently suggested to guide NK cell recruitment into tumors. In a mouse model of melanoma lung metastasis mice lacking patrolling monocytes (Nr4a1 super enhancer −/-) showed impaired NK cell recruitment and a reduced anti-metastatic activity.11,12 Human blood NK cells are attracted by various chemokines, including CX3CL1 (fractalkine), CCL2, CCL3, CCL4, CCL5, CXCL8 (IL-8), CXCL10.13 For instance, the IL-8 chemokine receptors CXCR1 and CXCR2 are expressed on CD56dimCD16+ NK cells and mediate recruitment into tissues.14,15 NK cells are known for exerting direct perforin/granzyme B-mediated cytotoxicity against tumor cells in the tissue. Alternatively, it was recently reported that NK cell cytokine secretion can arrest tumor cell growth through sensing of PDGF-DD via Nkp44 on NK cells.16 However, it is largely unknown which cell types are capable of recruiting human NK cells to tumor sites.
Cellular senescence in tumors is a common response to oncogene-mediated transformation, termed oncogene-induced senescence.17 However, other forms of senescence induction are possible, including homeostatic triggers such as telomere-mediated replicative senescence or the DNA-damage response. Those triggers can lead to activation of the p53/p21 (CDKN1A) or the Retinoblastoma (Rb)/p16ink4a pathways, resulting in a persistent growth arrest.18,19 Recent reports suggest that the proinflammatory cytokines TNF-α and IFN-γ can induce senescence, termed cytokine-induced senescence.20–23 In a β-cancer mouse model, it was demonstrated that antigen-specific T cells were able to limit tumor progression via TNF-α/IFN-γ-dependent senescence induction.24 This mechanism was reliant on both TNF-α and IFN-γ mediated signaling, as the individual cytokines alone were not sufficient to induce senescence. The contribution of innate immunity to tumor senescence is still incompletely understood.
In this study, we investigated how slanMo and NK cells collaboratively affect melanoma growth. Since the slanMo/NK cell crosstalk relies on those cells being present in the TME, we initially analyzed patient samples for the presence of slanMo and NK cells. Additionally, we performed migration studies in order to elucidate a potential chemotactic mechanism for R848-activated slanMo recruiting NK cells to the TME. Finally, we determined the mechanism of melanoma growth arrest by inflammatory cytokines produced in slanMo/NK co-cultures. Taken together, the results gained here are of important relevance for therapies targeting innate anti-tumor immunity.
Results
slanMo and NK cells co-localize in the melanoma immune microenvironment
To investigate a contribution of a potential slanMo/NK cell crosstalk in anti-melanoma immune responses, we determined the presence and distribution of slanMo and NK cells in formalin-fixed paraffin embedded (FFPE) samples from all four stages of melanoma and in healthy skin samples (Figure 1(a)). Using anti-slan staining with the monoclonal antibody DD2, we frequently found slanMo in stage I melanoma lesions, whereas their numbers were reduced in stage II and remained decreased over the rest of disease progression, with the exception of stage III cutaneous melanoma (Figure 1(a,b)). NK cells, identified by CD56, were present in comparatively low numbers in stages I and II, but increased in frequency in stage III (Figure 1(a,c)). Frequencies of NKT cells that express CD56, but also CD3, were very low compared to NK cells in melanoma tissues (data not shown). Of note, CD56+ NK cells could be clearly distinguished by morphology from melanoma cells that occasionally expressed low levels of CD56 (Fig. S1). We could not detect NK cells and rarely slanMo in skin from healthy donors (Figure 1(a-c)). When focusing on stage III lymph node metastasis (III LN), we observed similar numbers of slanMo and NK cells with a significant positive correlation (Figure 1(a)).
Figure 1.
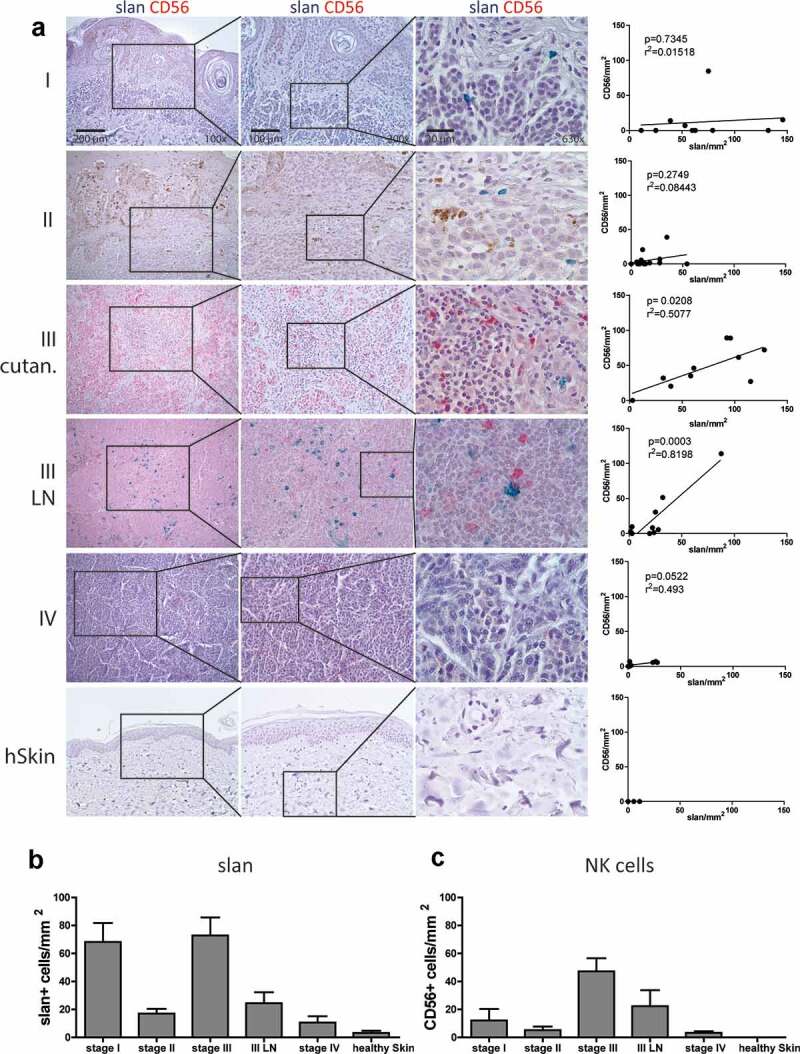
slanMo and NK cells infiltrate human melanoma. (A) slanMo (slan – green) and NK cells (CD56 – red) were analyzed in FFPE samples of melanoma patients from stage I, stage II, stage III cutaneous lesions (III cutan.), stage III Lymphnode (III LN) metastasis, stage IV distant metastasis, and healthy skin (hSkin) (n = 7). Based on 3 images (200x magnification) of double-stained (slan + CD56) samples, the density of cells/mm2 was analyzed and densities of slanMo were correlated with CD56+ NK cells. Density of (B) slanMo (slan) and (C) CD56+ NK cells displayed as cells/mm2 over all stages of melanoma development and in healthy skin. Number of patients for stage I (n = 10), stage II (n = 16), stage III (n = 10), stage III LN (n = 10), stage IV (n = 8), and healthy skin (n = 7). Original magnification x100 (left column), x200 (middle column), x630 (right column).
slanMo induce NK cell migration via IL-8 production
Since high numbers of slanMo in melanoma stage I preceded the accumulation of NK cells in melanoma stage III, we asked whether slanMo are capable of recruiting NK cells. We therefore analyzed the expression and secretion of chemokines in supernatants from slanMo, non-stimulated or stimulated with the TLR7/8 agonist R848. Our results show that several chemokines were expressed by slanMo and were upregulated after R848 stimulation, most notably CCL3, CCL4, and IL-8 (Figure 2(a,b)). In order to quantify specific migration of NK cells in response to slanMo-derived chemokines, we established an in vitro migration assay, in which R848-activated slanMo-conditioned medium (slanMo CM) was provided in the bottom chamber. NK cells showed significant-specific migration (9% of total input cells normalized to control) toward R848-stimulated slanMo CM (Figure 2(c)), which was strongly reduced using supernatant from slanMo left unactivated (Fig. S2A). R848 alone was not sufficient to induce NK cell migration (Fig. S2A). Next, we tested the ability of NK cells to migrate in response to slanMo-associated CCL3, CCL4, and IL-8 (Fig. S2A). Only very few NK cells responded to CCL3 and CCL4, whereas IL-8 triggered a dose-dependent specific NK cell migration comparable to SDF-1 (CXCL12), which is known to be a potent inducer of NK cell migration (Fig. S2B). Since IL-8 was highly present in the chemotactic slanMo CM, we neutralized IL-8 during NK cell migration. This resulted in a significant reduction in NK cell migration in response to slanMo CM (Figure 2(c)). IL-8 is known to bind to the chemokine receptors CXCR1 and CXCR2, mediating internalization and directed migration.25,26 Consistent with previous findings, we observed that NK cells expressed CXCR1 and CXCR2 (Fig. S2C). Migration toward slanMo CM significantly reduced expression of both CXCR1 and CXCR2, which is indicative of receptor engagement (Figure 2(d)). In contrast, NK cells migrating under medium control conditions expressed high levels of both CXCR1 and CXCR2 (Figure 2(d)). Moreover, IL-8 neutralization in the slanMo CM significantly abolished receptor downregulation (Figure 2(d,e)). These data demonstrate that slanMo are capable of recruiting NK cells via IL-8.
Figure 2.
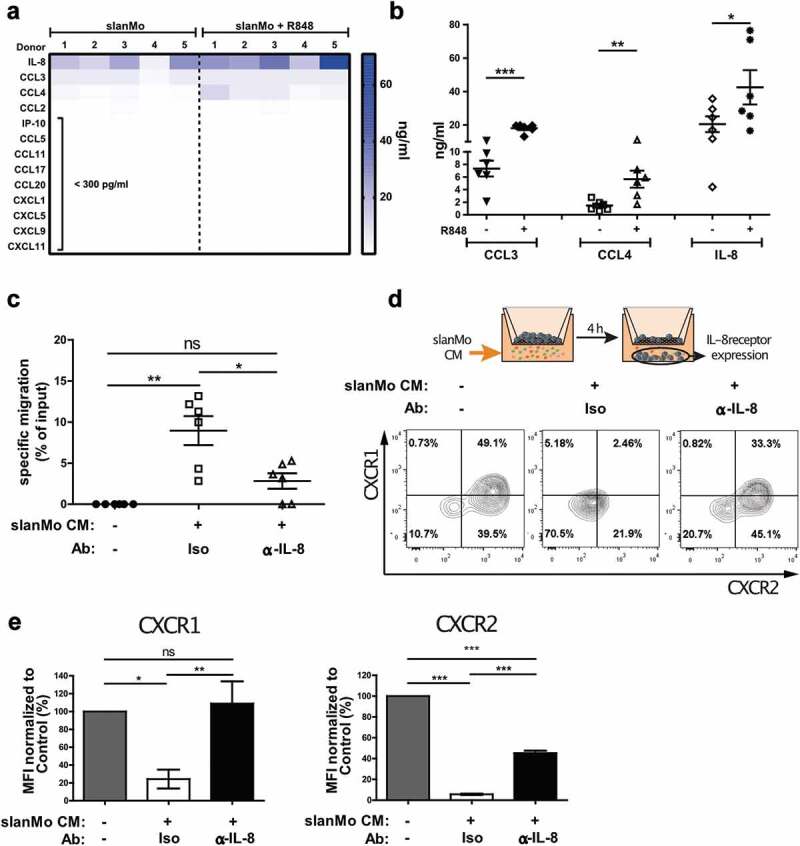
slanMo induce specific NK cell migration via IL-8 production. (A) slanMo were cultured for 24 h and cell-free supernatants were analyzed regarding chemokine production. Cells were either treated with 1 µg/ml R848 6 h after seeding or left untreated. For each condition, five different healthy donors are displayed. (B) Concentrations of CCL3, CCL4, and IL-8 as determined in the chemokine screen. (C) NK cells were used in a migration assay with 5 µm pore size with R848-stimulated slanMo supernatant (slan conditioned medium (slanMo CM)) in the presence of an anti-IL-8 neutralizing antibody or respective isotype control. Migrated NK cells were quantified by measuring ATP levels in the bottom chamber after 2 h. Cumulative data from 6 donors. (D) Experimental set-up for transwell experiment analyzing receptor expression on migrated NK cells. Migrated NK cells were analyzed for CXCR1 and CXCR2 expression by flow cytometry. Representative data out of five donors. (E) Mean fluorescence intensity values for receptor expression of CXCR1 and CXCR2 as shown in (D).
slanMo and NK cells synergistically elicit a cytokine-induced growth arrest in melanoma cells
We next investigated whether the cytokine milieu generated by slanMo and NK cells affects melanoma cells, as it could occur in the TME. We incubated the melanoma cell line SK-Mel-28 for a period of 3–4 d with conditioned medium (CM) harvested from co-cultures of R848-stimulated slanMo and NK cells. After culture in CM, the melanoma cells were replated and equivalent cell numbers were cultured in the absence of CM. We observed greatly impaired growth of melanoma cells initially exposed to CM, whereas cells cultured in normal medium or with R848 (corresponding to concentrations used for slanMo/NK cell co-culture assays) grew exponentially (Figure 3(a) and Fig. S3A). In contrast, incubation with medium harvested from individual slanMo or NK cell cultures had minor or no influence on cell growth (Figure 3(a)). Thus, the observed growth arrest required the combination of slanMo and NK cells for the production of soluble factors mediating the melanoma growth-limiting effect.
Figure 3.
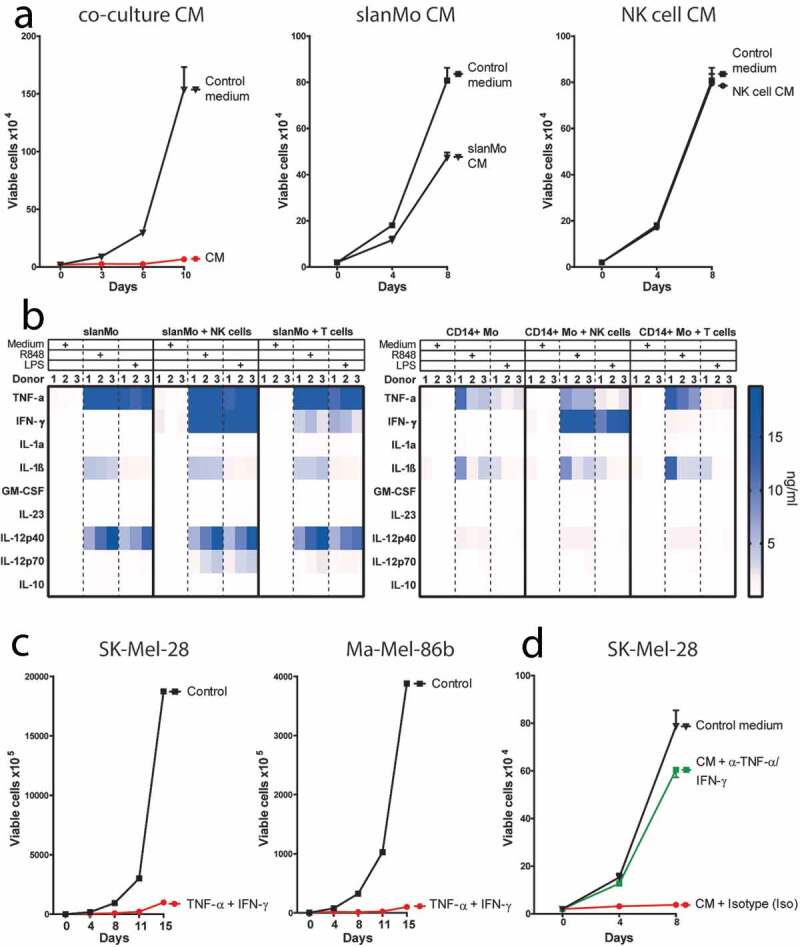
The conditioned medium (CM) from slanMo and NK cell co-cultures leads to a growth arrest in melanoma cells. (A) Growth curve of SK-Mel-28 untreated (Control medium), or treated with R848-activated slanMo/NK cell co-culture conditioned medium (CM), R848-activated slanMo-conditioned medium (slanMo CM), or R848-activated NK cell-conditioned medium (NK cell CM). The melanoma cells were incubated for 3–4 d with the respective CM. After CM treatment, the cells were washed and cultured in normal control medium. Cumulative data from 2 (CM) or 3 donors (slanMo CM and NK cell CM). (B) Cytokine screen of conditioned medium from different cultures either unstimulated or stimulated for 18 hours with 1 µg/ml R848 or 100 ng/ml LPS. (C) Growth curve of SK-Mel-28 and Ma-Mel-86b after treatment with recombinant TNF-α and IFN-γ (10 ng/ml and 100 ng/ml, respectively). (D) Growth curve of SK-Mel-28 after incubation with co-culture CM with neutralizing antibodies against TNF-α/IFN-γ (α-TNF-α/IFN-γ) or respective isotype controls (Iso) (each 10 µg/ml). Cumulative data from 12 donors.
To investigate potential factors mediating the growth arrest, we analyzed the cytokine secretion profile in the CM of slanMo/NK cell co-cultures relative to mono- and co-cultures of other mononuclear cells that can be present in the TME. We observed that R848-stimulation and LPS-stimulation strongly upregulated TNF-α, IL-1β, and IL-12 secretion in slanMo mono-cultures (Figure 3(b)), exceeding the cytokine concentrations reached in corresponding CD14+ monocyte (CD14+ Mo) mono-cultures. In contrast, NK cells, as well as T cells, alone produced only negligible cytokine levels <200 pg/ml (data not shown). slanMo/NK cell co-cultures contained higher TNF-α, IFN-γ, and IL-12 levels compared to CD14+ monocyte (CD14+ Mo)/NK cell co-cultures (Figure 3(b)). Moreover, the slanMo/NK cell co-culture yielded higher levels of IFN-γ, TNF-α, and IL-12 compared to slanMo/T cell as well as CD14+ Mo/T cell co-cultures, highlighting the pro-inflammatory cytokine production by the slanMo/NK cell crosstalk. Despite the fact that both slanMo and NK cells are capable of producing TNF-α, we observed that the TNF-α produced in our co-cultures predominantly derived from slanMo, as evidenced by intracellular staining (Fig. S3B). Since TNF-α and IFN-γ were expressed at particularly high levels in TLR-activated slanMo/NK cell co-cultures, we incubated melanoma cells with recombinant TNF-α and IFN-γ. Indeed, exposure of melanoma cells to TNF-α and IFN-γ significantly inhibited proliferation in several melanoma cell lines (Figure 3(c) and S3C). Neutralization of TNF-α and IFN-γ in slanMo/NK cell CM completely abolished the growth arrest and restored exponential cell growth (Figure 3(d)).
Growth inhibition induced by CM was quantified by analyzing the proliferative capacity of melanoma cells with Ki67 Immunofluorescence staining. The percentage of Ki67-positive cells was reduced by ~35% when the cells were cultured in CM (Figure 4(a)). Similar results were obtained when using recombinant TNF-α and IFN-γ (Figure 4(b)). This was supported by a 3H-Thymidine incorporation assay showing that proliferation was reduced to 66% of the untreated control (Figure 4(c)). The results were dependent on CM from co-cultures and consistent for multiple melanoma cell lines (Fig. S3D and S3E). As expected, the growth reduction was confirmed by using recombinant TNF-α and IFN-γ (Figure 4(d)). In both proliferation methods, a reversal of the anti-proliferative effect upon TNF-α and IFN-γ neutralization was observed (Figure 4(a,c)). Together, our data demonstrate that TNF-α and IFN-γ produced in slanMo/NK cell co-cultures affect melanoma cell growth by significantly reducing cell proliferation.
Figure 4.
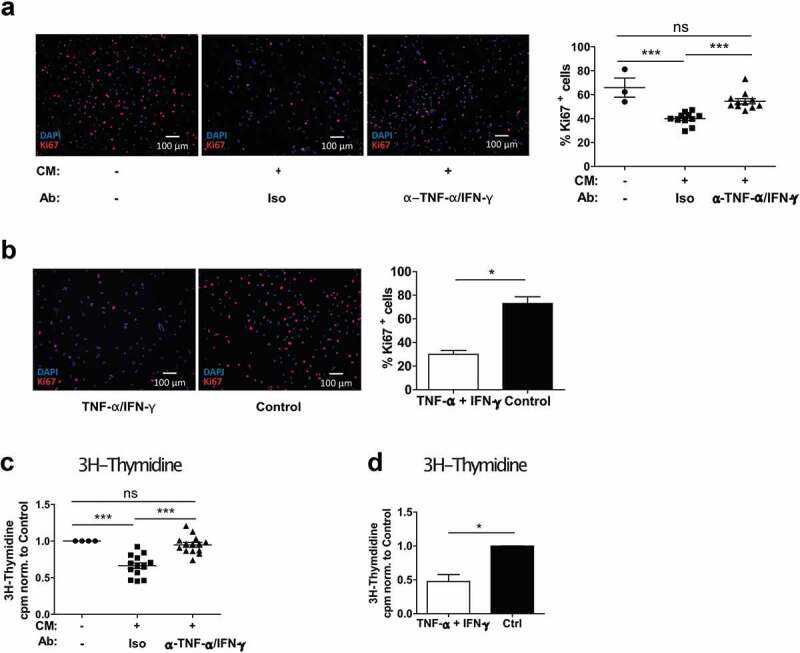
The co-culture conditioned medium (CM) from slanMo and NK cells reduces the proliferation of melanoma cells. (A) Ki67 staining of SK-Mel-28 cultured with CM as described before. Quantification of Ki67+ cells. Representative image and cumulative data out of 11 donors. (B) Ki67 staining of SK-Mel-28 treated with recombinant TNF-α and IFN-γ and quantification of positive cells. Representative image out of 3 experiments. (C) 3H-Thymidine incorporation assay with SK-Mel-28 cells treated with CM as described before. 1 d after reseeding, the cells were pulsed with 3H-Thymidine for 12 h. Cumulative data out of 14 donors. (D) 3H-Thymidine incorporation assay with SK-Mel-28 cells treated with recombinant TNF-α and IFN-γ. Cumulative data from 4 experiments.
slanMo and NK cells induce senescence in melanoma cells
To investigate the cellular characteristics of the growth arrest, we determined tumor cell growth by microscopy after culture in CM harvested from slanMo/NK cell co-cultures. We observed the accumulation of morphologically distinct melanoma cells, displaying enlarged, flattened, and often multinucleated adherent cell features (Fig. S4A inlet). Since these characteristics are reminiscent of a senescence phenotype, we analyzed senescence-associated β-galactosidase (SA β-Gal), which is a common marker for senescent cells.27 After culture of melanoma cells with co-culture CM, the fraction of SA β-Gal+ cells was increased ~6 fold compared to untreated cells, whereas no increase was observed upon culture with CM from slanMo or NK cell monocultures (Figure 5(a), S4B). TNF-α and IFN-γ neutralization significantly reduced SA β-Gal activity, whereas addition of recombinant TNF-α/IFN-γ resulted in higher cell numbers positive for SA β-Gal (Figure 5(a), b).
Figure 5.
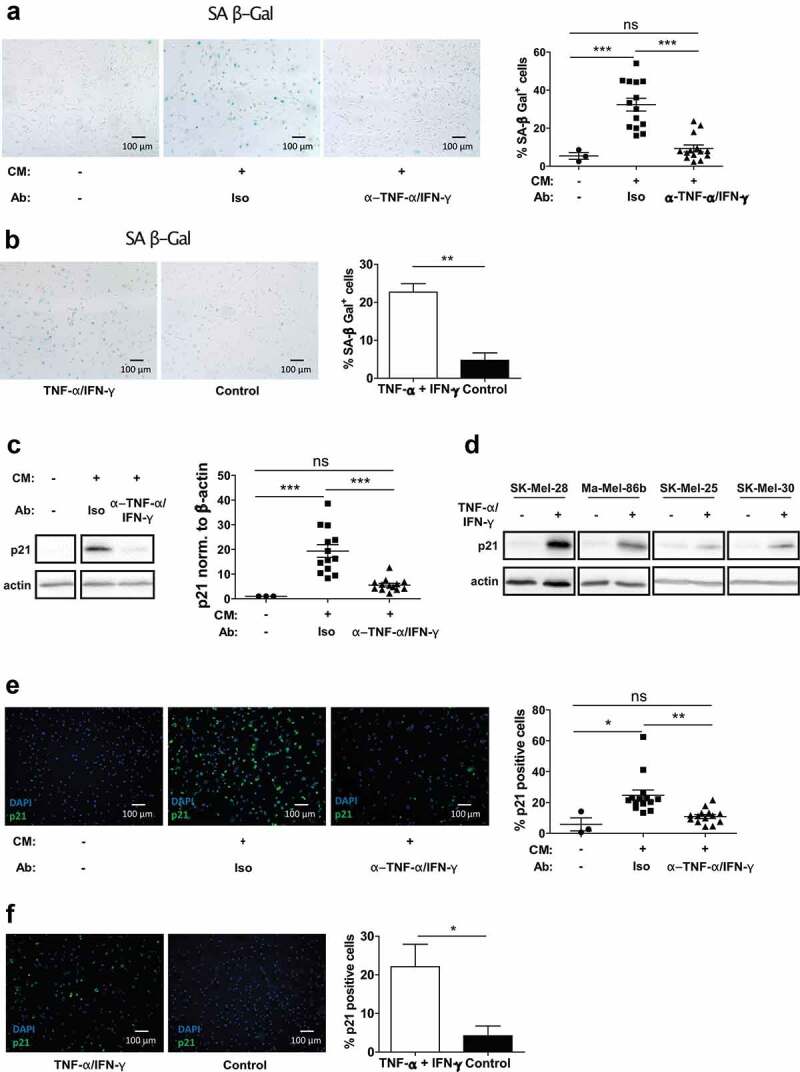
The slanMo/NK cell cross-talk induces melanoma cell senescence characterized by p21 upregulation. (A) SK-Mel-28 were treated with co-culture CM, as described before. After 4 d incubation, the cells were reseeded and stained for senescence-associated β-Galactosidase (SA β-Gal) activity. Depicted is a representative image and quantification of positive cells includes cumulative data from 14 donors. (B) SK-Mel-28 were treated with TNF-α and IFN-γ and stained for SA β-Gal and quantified. Cumulative data from 3 experiments. (C) Western Blot for p21 of SK-Mel-28 cells treated as described before with conditioned medium (CM) with TNF-α and IFN-γ neutralization (each 10 µg/ml) or respective isotype controls (Iso). Quantification of p21 signal intensity. Representative image and cumulative data from 13 donors. (D) Western Blot of p21 expression in SK-Mel-28, Ma-Mel-86b, SK-Mel-25, and SK-Mel-30 untreated or treated with recombinant TNF-α/IFN-γ (10 ng/ml/100 ng/ml) for 4 d. Representative images out of 2 experiments. (E) SK-Mel-28 were treated as described before, reseeded and analyzed for p21 in Immunofluorescence. Quantification of p21 positive cells. Cumulative data out of 14 donors. (F) SK-Mel-28 were treated with TNF-α and IFN-γ and stained for p21 by Immunofluorescence and quantified. Representative image out of 3 experiments.
To substantiate the senescent phenotype, we analyzed expression of p21 (CDKN1A) as a downstream target of p53 in the p53 senescence pathway. While treatment of melanoma cells with CM significantly upregulated p21 expression, TNF-α and IFN-γ neutralization normalized p21 levels (Figure 5(c)). Recombinant TNF-α and IFN-γ induced p21 in different melanoma cell lines such as SK-Mel-28, Ma-Mel-86b, SK-Mel-25, and SK-Mel-30 (Figure 5(d)), indicating that p21 represents a common regulator of non-proliferative, senescence states in melanoma cells. Moreover, p21 was detected by immunofluorescence after fixation of CM-treated melanoma cells grown on microscopic slides. p21 increased after exposure to CM and recombinant TNF-α and IFN-γ and could be reverted by neutralization of TNF-α and IFN-γ in the CM (Figure 5(e, f)). When staining p21 in stage II and stage III cutaneous melanoma samples by immunohistochemistry, we observed increasing p21 positive cells with incremental tumor progression (Fig. S4C).
In summary, these data demonstrate that the growth arrest observed in response to slanMo/NK cell co-cultures is linked to a TNF-α/IFN-γ dependent senescence state in melanoma cells characterized by high p21 levels.
Melanoma cells display a senescence-associated secretory phenotype upon exposure to slanMo/NK cell co-cultures
Senescence is known to induce changes in the cells’ secretome, termed senescence-associated secretory phenotype (SASP).28 After treatment with slanMo/NK cell co-culture CM, upregulation of SASP factors such as IL-6, IL-8, IL-1α, IL-1β was detected by qPCR analysis (Figure 6(a)). Other significantly upregulated markers included the matrix metalloproteinases (MMPs) 1, 2, 3, 9, the serpins PAI-1 and PAI-2, the chemokines CXCL1, CXCL2, CCL2, and CCL7, the growth factor VEGF-A, TGF-β, and ICAM-1. In all cases except for CXCL1 and TGF-β, the mRNA levels were significantly reduced upon TNF-α and IFN-γ neutralization.
Figure 6.
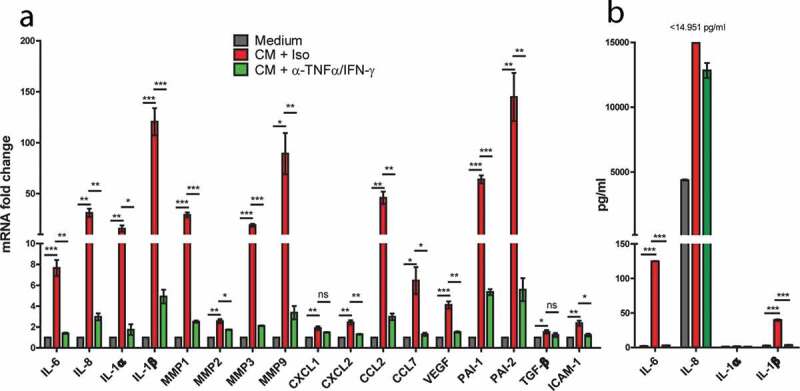
Senescence-associated secretory phenotype (SASP) induction in response to slanMo/NK cell co-culture CM. (A) SK-Mel-28 cells were treated as described before with co-culture conditioned medium (CM) and TNF-α and IFN-γ neutralization (α-TNF-α/IFN-γ). A panel of SASP-associated markers was analyzed by qPCR. Cumulative data from 3 biological replicates. (B) The cell-free-conditioned medium from SK-Mel-28 cells reseeded after treatment with slanMo/NK cell CM was analyzed for several soluble factors by Legendplex analysis. Cumulative data from three biological replicates. Data are presented as mean ±SEM and p values were assessed using unpaired t-test with *p < .05, **p < .01, ***p < .001.
Furthermore, we analyzed the expression of IL-6, IL-8, IL-1α, IL-1β at protein level. In line with the mRNA results, IL-6, IL-8, and IL-1β were significantly upregulated after exposure to co-culture CM, for IL-8 even above the maximal detection limit of the assay (Figure 6(b)). This effect was abolished or reduced after neutralization of TNF-α and IFN-γ. Together, our data provide evidence that slanMo and NK cells cooperate in a synergistic feedback loop that generates high levels of TNF-α and IFN-γ that drive melanoma cells into senescence and trigger the secretion of senescence-associated factors with the potential to further shape the tumor microenvironment.
Discussion
Here, we discovered that TLR-ligand stimulated slanMo and NK cells interact in a synergistic crosstalk generating high levels of pro-inflammatory TNF-α and IFN-γ that are able to induce melanoma cell senescence. Thereby, we uncover a novel senescence eliciting role for innate immunity extending the previous concept of T helper 1 (Th1) cell-mediated cytokine-induced senescence.24 The slanMo/NK cell-induced melanoma senescence phenotype manifested in severely reduced proliferation, increased SA β-Gal expression, and upregulation of common SASP factors. Additionally, our results reveal induction of p21 in senescent melanoma cells. We further demonstrate the relevance of the melanoma senescence phenotype by detecting slanMo and NK cells in the TME in all stages of melanoma development and by suggesting an IL-8-mediated recruitment mechanism for migration of NK cells toward TLR-activated slanMo.
Our histological analysis revealed that slanMo and NK cells can infiltrate primary and metastatic melanoma. We observed a high density of slanMo, particularly in stage I melanoma, which cannot be considered a homeostatic cutaneous condition since healthy skin was virtually devoid of slanMo. These results extend previous reports that described high numbers of slanMo in metastatic tumor-draining lymph nodes of carcinoma patients but absence of slanMo in the primary carcinoma.5 Increased slanMo numbers in early melanoma development suggest immediate recruitment to the tumor site. This would appear consistent with the reported patrolling function of human slanMo as a subpopulation of non-classical monocytes.29,30 In this model, the local chemokine production by slanMo could trigger the recruitment of other immune cells, such as NK cells. NK cells can be detected in various human tissues, such as LN, lung, liver, or kidney, and are known to traffic to sites of inflammation.31,32 In our histological analysis, NK cells were sparse in stages I and II, but increased in numbers in stage III for both satellite tumors and LN metastasis. This strong NK cell influx in stage III was accompanied by a significant correlation of slanMo and NK cell numbers, suggesting a connected migration mechanism. This is intriguing when considering the hierarchical migration routes described by Hanna et al., where Ly6Clow monocytes recruited NK cells after tumor encounter.11 We observed substantial migration of human NK cells toward conditioned medium from R848-activated slanMo, which we found to be mostly dependent on IL-8 being produced in particularly high levels by slanMo. This mechanism is consistent with the literature and our results, where expression of the IL-8 receptors CXCR1 and CXCR2 is reported for CD56dim NK cells coupled to the ability to migrate toward IL-8 derived from different sources such as dendritic cells.14,15 In contrast, there is evidence that CD56bright NK cells are the major NK cell population in tumor tissue.33 However, with limited knowledge about how CD56 expression changes after extravasation from the blood into tissue, it is difficult to compare migration of blood NK cells with cells detected in the tissue. On the other hand, IL-8 has been shown to have pro-tumorigenic effects through recruitment of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) or neutrophils.34–36 The slanMo-mediated recruitment of other immune population could be the subject of future studies. The discovery of a novel potential innate migration axis is highly relevant for the understanding and initiation of immune responses in tissues. In this context, Böttcher et al.37 demonstrated that melanoma infiltrating NK cells recruit cDC1 via XCL1 and CCL5, which facilitated the initiation of the adaptive immune responses through recruitment and priming of T cells. In this line of thought, our results showing early slanMo infiltration followed by subsequent increase in NK cell numbers could suggest a sequence, in which slanMo extravasate at the site of newly formed tumors or metastasis and then recruit NK cells to initially limit tumor progression and foster adaptive immune responses.
The TME generally plays an important role in tumor growth, progression, and metastasis. We observed that the CM generated from TLR-activated slanMo and NK cell co-cultures reduced the proliferation of melanoma cell lines. Together with TNF-α and IFN-γ neutralization experiments, this demonstrated that the slanMo/NK cell co-culture is required to facilitate IFN-γ and TNF-α production-reaching levels capable of limiting melanoma growth. This is consistent with the established concept of the synergistic slanMo/NK cell crosstalk, in which slanMo-derived IL-12 stimulates IFN-γ production by NK cells and vice versa, which generates a positive feedback loop reaching very high levels of IL-12, TNF-α, and IFN-γ.8 When investigating the cellular basis for the growth arrest, we discovered that slanMo/NK cell co-culture CM upregulated the senescence marker p21 in melanoma cells, suggesting activation of the p53/p21 pathway. Similarly, p21 was also increased in stage III cutaneous melanoma samples compared to stage II, which mirrored the augmented influx of slanMo and NK cells in stage III. Although p21 staining of melanoma samples in situ by itself offers only limited information with respect to senescence in the tumor tissue, our finding suggests that a slanMo/NK cell crosstalk cytokine milieu might drive p21 upregulation and possibly senescence in melanoma in situ. Of note, in Th1-mediated cytokine-induced senescence, senescence was triggered by the p16Ink4a/Rb pathway.24 However, melanoma cells regularly carry mutations in the CDKN2A locus, which is encoding for p16Ink4a, arguing for the possibility of a p21-mediated alternative mechanism.38 p21 acts downstream of p53; however, it can also function in a p53-independent manner.19,39,40 Since also p53 is commonly mutated in melanoma cell lines and in particular in the cell lines SK-Mel-28 and SK-Mel-30 used in this study (Broad Institute Cancer Cell Line Encyclopedia), it appears that p21 is upregulated independent of p53 in our TNF-α + IFN-γ triggered melanoma senescence model.
Oncogene-induced senescence is established as an important counter-mechanism against initial mutagenic events by limiting proliferation of tumorigenic clones.17 This cell-intrinsic mechanism is propagated by SASP-mediated release of factors triggering senescence in neighboring cells.41 Moreover, chemotactic SASP factors recruit innate immune cells supporting the clearance of senescent tumor cells42 and increased secretion of inflammatory mediators such as IL-1β potentially stimulates anti-tumor immune responses.43 However, senescence in tumor cells can also be associated with tumor-promoting functions mediated by pro-tumorigenic SASP factors.28 We discovered that the TNF-α/IFN-γ induced senescence increased the secretion of common SASP factors, most prominently IL-8, IL-6, IL-1β, in addition to different MMPs and chemokines (Figure 6(a,b)). Importantly, tumors can utilize SASP-associated chemokines to recruit immune regulatory myeloid cells to the TME, which dampen local tumor-specific immune responses.44 Additionally, the increased mRNA levels for MMPs might skew the melanoma cells toward higher metastatic potential.45–47 Besides SASP-mediated non-cell-autonomous functions, senescence in tumors can act autonomously through affecting the tumorigenic and invasive potential of cancer cells.48,49 This suggests that the innate immune cell-induced senescence phenotype observed in our study might promote tumor progression and invasiveness through cell-autonomous and non-cell-autonomous mechanisms.28 Of note, NK cells were reported to recognize and eliminate senescent cells.50–52 This is enabled through expression of stress-induced ligands such as DNAX accessory molecule-1 (DNAM-1) ligands or UL16 binding proteins (ULBP) on the surface of senescent tumor cells. In line with this, the slanMo/NK cell crosstalk may synergize with controlling melanoma cells in two ways: By inducing senescence-mediated growth arrest of melanoma cells and by augmentation of the melanoma-directed cellular cytotoxicity.8
Despite the likely presence of immunogenic cell death products like nuclear acid fragments or nuclear proteins like HMGB1 that can trigger TLR ligands and activate monocytes, it can be assumed that slanMo are not always optimally stimulated in the TME. This is intriguing in the light of TLR 7/8 ligand therapeutics such as imiquimod, which is therapeutically applied for cutaneous melanoma lesions and metastasis.53 The use of topically applied imiquimod has been successfully studied for the treatment of primary superficial melanoma and for cutaneous melanoma metastasis.54–56 Since slanMo express TLR 7 and 8, imiquimod therapy can potentially activate those cells optimally in the TME and thereby initiate a senescence-inducing slanMo/NK cell crosstalk. This is relevant for current clinical trials examining whether checkpoint inhibitor therapy resistance can be overcome by combination treatment with TLR agonists, e.g., for treatment of tilsotolimod with ipilimumab in PD-1 refractory melanoma patients (NCT03445533).
Taken together, our results uncover an innate immune crosstalk as inducer of cytokine-induced senescence of melanoma cells. Our data support a concept that TLR 7/8 ligand therapies (e.g., imiquimod) in melanoma patients might strongly activate slanMo, fuel the recruitment of NK cells and lead to the formation of a slanMo/NK cell synergistic crosstalk that effectively hampers tumor progression through TNF-α/IFN-γ-mediated senescence induction.
Materials and methods
Cell lines
The human melanoma cell lines SK-Mel-28 (ATCC), Ma-Mel-86b (from Dr. Annette Paschen, University Clinics Essen), SK-Mel-25 (ATCC), SK-Mel-30 (ATCC), were cultured in RPMI 1640 (Biochrom) supplemented with 10% FCS (Sigma-Aldrich), 2 mM L-glutamine, and 1% penicillin/streptomycin. Cultured cells were tested negative for mycoplasma contamination (Multiplexion GmbH) and cell line identity was confirmed by MCA assay (Multiplexion GmbH) in 2017 for SK-Mel-28, Ma-Mel-86b, and in 2019 again for SK-Mel-30, SK-Mel-25. All cell lines were cultured for 1 week prior to experiments.
Tissue specimen
Formalin-fixed paraffin-embedded tissue sections of melanoma patients from all stages (I–IV) were retrieved from the National Center for Tumor Diseases. Characteristics of patients are presented in table S1. Skin samples from healthy donors (n = 7) were included as control tissue. Patient’s written informed consent was obtained prior to sample acquisition and use of samples was reviewed by the Ethic Board of the University of Heidelberg (S-091/2011; S-306/2010).
Immunohistochemistry (IHC)
Paraffin-embedded samples were cut into sections of 2–5 microns. After rehydration, antigen retrieval was performed by boiling the samples for 30 minutes in citrate buffer pH 6 (Dako), followed by blocking with 5% goat serum and incubation with the primary anti-human antibodies: slan (Miltenyi, clone DD2, individual hybridoma supernatant, 50 µg/ml), CD56 (Thermo Scientific, clone 123c3, 1:75), CD3 (Abcam, clone SP7, 1:250), CDKN1A/p21 (abcam, clone EPR362, 1:200). Single color stainings were developed using the Dako Real Detection System (Dako) according to the manufacturer’s instructions. For double color immunohistochemistry, after staining the first antigen as described for single staining, the slides were blocked for endogenous peroxidase activity for 5 min with 3% H2O2; then, the biotin-Streptavidin sites from the first reaction were blocked with the Avidin/Biotin Blocking Kit (Vector Laboratories), followed by overnight incubation with purified anti-slan or anti-CD3. The mouse IgM antibody clone DD2 was then detected with a goat anti-mouse IgM-specific detection system and rabbit anti-human CD3 with a goat anti-rabbit specific system (Vector Laboratories). This was followed by chromogen development with Histogreen (Linaris), hematoxylin counterstain, and embedding in xylene-based solution.
Based on three representative 100× magnification images of areas with immune infiltrate and tumor cell interactions of double-stained (slan + CD56) FFPE samples, the density of cells/mm2 was analyzed blindly by two independent scientists and correlated for slanMo and CD56+ NK cells. p and r2 values were calculated using linear regression in Graph Pad Prism. The density of p21 positive cells was quantified analogously with p21 single-stained sections.
Isolation of slanMo, NK cells, CD14 monocytes, and T cells
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy adult donors by density gradient purification with Ficoll (Biochrom). slanMo were isolated as described previously.3 Briefly, PBMCs were incubated in diluted M-DC8 hybridoma supernatant for 15 minutes at 4°C. After washing, the cells were incubated with rat anti-mouse IgM microbeads (Miltenyi) for 15 minutes at 4°C, washed again and then slanMo were purified by positive selection with an autoMACS (Miltenyi). Untouched NK cells from the remaining PBMCs were further purified using the human NK cell Isolation Kit (Miltenyi Biotech) according to the manufacturer’s instructions via autoMACS negative selection. The purity of slan+CD3− slanMo and of CD56+CD3− NK cells was ≥95%. Freshly isolated slanMo and NK cells were cultured in complete RPMI 1640 containing 2 mM L-glutamine, 1% penicillin/streptomycin, 1% non-essential amino acids (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich), and 10% heat-inactivated pooled human AB serum (Sigma-Aldrich).
CD14 monocytes were isolated with CD14 microbeads (Miltenyi) by positive selection according to the protocol. T cell isolation was performed with the CD4 + T cell isolation kit (Miltenyi) by following the instructions and a negative selection autoMACS protocol.
Immune cell co-cultures for supernatant generation and cytokine screening
Freshly purified slanMo were seeded in 6-well or 12-well plates at a concentration of 5 × 105 cells/ml and a density of 3 × 105 cells/cm2. After 6 hours of incubation at 37°C and 5% CO2, purified NK cells were added to the well at 1 × 106 cells/ml and 6 × 105 cells/cm2, resulting in a final ratio of 1:2 slanMo:NK cells. At the time of NK cells seeding, the co-culture was stimulated with 1 µg/ml R848 (Invitrogen). Twenty-four hours after slanMo seeding, the supernatant was removed, cleared of cellular parts by centrifugation and then either used directly or stored at −80°C. Alternatively, slanMo were cultured alone and stimulated with 1 µg/ml R848 after 6 h for harvesting of supernatants after 24 hours complete incubation.
For analysis of cytokine production, slanMo or CD14 monocytes were seeded in the same densities as described before for slanMo, and then co-cultured with NK cells or T cells, seeded as described for NK cells. Co-cultures were stimulated 6 hours after slanMo/CD14 monocyte seeding with 1 µg/ml R848 or 100 ng/ml LPS. Supernatants were removed after 24 hours total culture and stored at −20°C.
Cytokine assays
The supernatants of different co-cultures were screened for cytokine levels using the Legendplex pro-inflammatory Chemokine panel, human Cytokine panel 2, and anti-virus response panel (Biolegend).
NK cell migration assay
Purified NK cells were incubated overnight and then used in a 96 well migration system with a pore size of 5 µm (Neuroprobe). First, conditioned medium from slanMo R848-stimulated or unstimulated mono-cultures (slanMo CM), or recombinant R848 (1 µg/ml), was added to the bottom well. Anti-IL-8 neutralizing antibody (R&D, clone 6217, 20 µg/ml) or control mouse IgG1 isotype control (20 µg/ml, BioXcell) was added directly to the slanMo CM. After fixation of the membrane, 5 × 104 NK cells were added on the membrane and incubated for 2,5 hours. The cells that migrated to the bottom chamber were transferred and measured via total ATP content with the Cell Titer Glo Luminescent cell viability assay (Promega). To calculate cell numbers, a standard curve was generated by measuring defined numbers of NK cells. Specific migration was defined as:
((Number of cells [sample] – number of cells [control])/number of cells [input])*100
Alternatively, 2 × 105 NK cells were seeded on a 24 well-permeable support and the slanMo CM with anti-IL-8 antibody or isotype control (each 10 µg/ml) was added as chemoattractant. After 4 hours of incubation at 37°C, the migrated cells from the bottom well were resuspended, stained for CXCR1 (clone 8F1, Biolegend) and CXCR2 (clone 5E8, Biolegend), and analyzed by flow cytometry.
Melanoma cell growth inhibition with conditioned-medium (CM)
SK-Mel-28, Ma-Mel-86b, SK-Mel-25, and SK-Mel-30 were seeded at a density of 1 × 104 cells/cm2 and 2.5 × 104 cells/ml in diluted CM (1:5), to reduce potential growth-inhibiting effects originating from already exhausted co-culture medium. In order to prevent premature confluency of the control cultures during the passages, the overall growth was limited by using RPMI containing 5% instead of 10% FCS. Neutralizing antibodies against human TNF-α (AbbVie, Adalimumab, 10 µg/ml) and human IFN-γ (BioXcell, clone B133.5, 10 µg/ml) or the respective isotype controls hIgG1 (BioXcell, 10 µg/ml) and mIgG1 (BioXcell, MOPC-21, 10 µg) were added to the CM. As control, the cells were also incubated without stimulus, with recombinant TNF-α (PromoKine, 10 ng/ml) and IFN-γ (ImmunoTools, 100 ng/ml), and with recombinant R848 (1 µg/ml). After 4 d of culture, the melanoma cells were harvested and either seeded as before for prolonged culture or seeded at 2 × 104 cells/cm2 and 4 × 104 cells/ml in appropriate plates for downstream analysis.
Alternatively, SK-Mel-28 cells were seeded in 96 well flat bottom plates and treated with CM + TNF-α/IFN-γ neutralization as described before. The plate was imaged every 2 h over a period of 4 d with 100x magnification.
3H-thymidine assay, senescence-associated β-galactosidase (SA β-Gal) assay
For 3H-Thymidine proliferation assay, the cells were treated as described before and 1 d after reseeding in 96 well plates, the adherent cells were pulsed for 12 hours with 3H Thymidine solution (Hartmann Analytic). After incubation, the cells were transferred to a membrane Filtermat (Perkin Elmer). The membrane was washed, developed with Betaplate Scint solution (Perkin Elmer) and measured in a Luminescence Counter (MicroBeta TriLux, Perkin Elmer).
SA β-Gal staining was performed on melanoma cells reseeded for 1 d after CM exposure with the SA β-Gal staining kit (United States Biological) according to the manufacturer’s instructions.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific), following manufacturers’ instructions. RNA was treated with RNase-free DNase I, followed by EDTA deactivation for 10 min at 65°C (both from Thermo Fisher Scientific). First-strand cDNA was synthesized from 2000 ng of total RNA with random hexamer primers, using dNTP mix and RevertAid H Minus Reverse transcriptase (all from Thermo Fisher Scientific). qRT-PCR was performed in a Stratagene Mx3500P qPCR machine (Agilent) using SYBR Select Master Mix containing SYBR GreenE dye (Applied Biosystems). The relative quantity of cDNA was estimated using the ΔΔCT method and data were normalized to GAPDH as a housekeeping gene. The following human forward and reverse primers, purchased from Metabion, were used for qRT-PCR (Table S2)
Immunofluorescence (IF) staining of CM-treated melanoma cells
CM-treated melanoma cells were seeded on glass diagnostic slides (Menzel) at 2 × 104 cells/cm2. After 1 d of culture, the cells were fixed with 4% paraformaldehyde (PFA) for 20 minutes at room temperature and then permeabilized with 0.1% (w/v) Triton-X 100 for 10 minutes at room temperature. The primary antibodies CDKN1A/p21 (abcam, clone EPR362, 1:500) and Ki67 (Dako, clone MIB-1, 1:75) were incubated overnight at 4°C, followed by the secondary goat anti-mouse A647 and goat anti-rabbit A488 (both Dianova). Ki67 and p21 positive cells were identified and counted based on fluorescence intensity with the software ImageJ.
Western blot
After 4 d of culture in CM, melanoma cells were lysed by heating in hot lysis buffer (10 mM Tris-HCl pH 8.0, 1% SDS, 1 mM Na-Orthovanadate) for 15 minutes in a heat block at 100°C with regular mixing. The solution was centrifuged at 14.000 rpm at 4°C and the supernatant was analyzed by BCA assay (Thermo Scientific) for protein concentration.
The protein samples were diluted in SDS buffer and run on a 15% SDS gel. After blocking in 5% BSA, the membrane was incubated overnight with anti-p21 (abcam, EPR362, 1:2000) primary antibody, followed by incubation with secondary goat anti-rabbit HRP antibody. Protein levels were normalized to β-actin.
Statistical analysis
Data were analyzed in GraphPad Prism and are presented as mean ±SEM. p Values were assessed using one-way ANOVA with Bonferroni’s multiple comparison post hoc test for groups of 3 and paired t-test for groups of 2, unless otherwise indicated. *p < .05, **p < .01, ***p < .001.
Supplementary Material
Acknowledgments
We thank Stefan Meisel, Jasmin Roth, and Annette Arnold for technical assistance, and Fareed Ahmad for valuable discussions.
Funding Statement
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 259332240 / RTG2099 (to F. Funck, K. Schäkel and A. Cerwenka), GRK 2099/2_AOBJ 654560 (to A. Cerwenka), SFB-TRR 156 Project B10 (to A. Cerwenka) and Project C02 (to K. Schäkel), the state of Baden-Württemberg foundation special program “Angioformatics single cell platform” (to A. Cerwenka), and SCHA 1693/1-1 (to K. Schäkel).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Schakel K, Mayer E, Federle C, Schmitz M, Riethmuller G, Rieber EP.. A novel dendritic cell population in human blood: one-step immunomagnetic isolation by a specific mAb (M-DC8) and in vitro priming of cytotoxic T lymphocytes. Eur J Immunol. 1998;28:4084–12. doi:. [DOI] [PubMed] [Google Scholar]
- 2.Schakel K, Kannagi R, Kniep B, Goto Y, Mitsuoka C, Zwirner J, Soruri A, von Kietzell M, Rieber EP. 6-Sulfo LacNAc, a novel carbohydrate modification of PSGL-1, defines an inflammatory type of human dendritic cells. Immunity. 2002;17(3):289–301. doi: 10.1016/S1074-7613(02)00393-X. [DOI] [PubMed] [Google Scholar]
- 3.Schakel K, von Kietzell M, Hansel A, Ebling A, Schulze L, Haase M, Semmler C, Sarfati M, Barclay AN, Randolph GJ. Human 6-sulfo LacNAc-expressing dendritic cells are principal producers of early interleukin-12 and are controlled by erythrocytes. Immunity. 2006;24:767–777. doi: 10.1016/j.immuni.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 4.Schmitz M, Zhao S, Deuse Y, Schakel K, Wehner R, Wohner H, Hölig K, Wienforth F, Kiessling A, Bornhäuser M, et al. Tumoricidal potential of native blood dendritic cells: direct tumor cell killing and activation of NK cell-mediated cytotoxicity. J Immunol. 2005;174(7):4127–4134. doi: 10.4049/jimmunol.174.7.4127. [DOI] [PubMed] [Google Scholar]
- 5.Vermi W, Micheletti A, Lonardi S, Costantini C, Calzetti F, Nascimbeni R, Bugatti M, Codazzi M, Pinter PC, Schäkel K, et al. slanDCs selectively accumulate in carcinoma-draining lymph nodes and marginate metastatic cells. Nat Commun. 2014;5(1):3029. doi: 10.1038/ncomms4029. [DOI] [PubMed] [Google Scholar]
- 6.Vermi W, Micheletti A, Finotti G, Tecchio C, Calzetti F, Costa S, Bugatti M, Calza S, Agostinelli C, Pileri S. slan(+) Monocytes and macrophages mediate CD20-dependent B-cell lymphoma elimination via ADCC and ADCP. Cancer Res. 2018;78:3544–3559. doi: 10.1158/0008-5472.CAN-17-2344. [DOI] [PubMed] [Google Scholar]
- 7.Micheletti A, Finotti G, Calzetti F, Lonardi S, Zoratti E, Bugatti M, Stefini S, Vermi W, Cassatella MA. slanDCs/M-DC8+ cells constitute a distinct subset of dendritic cells in human tonsils [corrected]. Oncotarget. 2016;7:161–175. doi: 10.18632/oncotarget.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wehner R, Lobel B, Bornhauser M, Schakel K, Cartellieri M, Bachmann M, Rieber EP, Schmitz M. Reciprocal activating interaction between 6-sulfo LacNAc + dendritic cells and NK cells. Int J Cancer. 2009;124(2):358–366. doi: 10.1002/ijc.23962. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz M, Zhao S, Schakel K, Bornhauser M, Ockert D, Rieber EP. Native human blood dendritic cells as potent effectors in antibody-dependent cellular cytotoxicity. Blood. 2002;100:1502–1504. doi: 10.1182/blood.V100.4.1502.h81602001502_1502_1504. [DOI] [PubMed] [Google Scholar]
- 10.Tufa DM, Ahmad F, Chatterjee D, Ahrenstorf G, Schmidt RE, Jacobs R. IL-1beta limits the extent of human 6-sulfo LacNAc dendritic cell (slanDC)-mediated NK cell activation and regulates CD95-induced apoptosis. Cell Mol Immunol. 2017;14:976–985. doi: 10.1038/cmi.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, Herrley E, Rasquinha N, McArdle S, Wu R. Patrolling monocytes control tumor metastasis to the lung. Science. 2015;350(6263):985–990. doi: 10.1126/science.aac9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narasimhan PB, Eggert T, Zhu YP, Marcovecchio P, Meyer MA, Wu R, Hedrick CC. Patrolling monocytes control NK cell expression of activating and stimulatory receptors to curtail lung metastases. J Immunol. 2020;204(1):192–198. doi: 10.4049/jimmunol.1900998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castriconi R, Carrega P, Dondero A, Bellora F, Casu B, Regis S, Ferlazzo G, Bottino C. Molecular mechanisms directing migration and retention of natural killer cells in human tissues. Front Immunol. 2018;9:2324. doi: 10.3389/fimmu.2018.02324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lima M, Leander M, Santos M, Santos AH, Lau C, Queiros ML, Gonçalves M, Fonseca S, Moura J, Teixeira MDA. Chemokine receptor expression on normal blood CD56 + NK-cells elucidates cell partners that comigrate during the innate and adaptive immune responses and identifies a transitional NK-cell population. J Immunol Res. 2015;2015:839684. doi: 10.1155/2015/839684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vujanovic L, Ballard W, Thorne SH, Vujanovic NL, Butterfield LH. Adenovirus-engineered human dendritic cells induce natural killer cell chemotaxis via CXCL8/IL-8 and CXCL10/IP-10. Oncoimmunology. 2012;1(4):448–457. doi: 10.4161/onci.19788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, Bohl B, Bando JK, Kim AH, Walker J, Andahazy M. Natural killer cells control tumor growth by sensing a growth factor. Cell. 2018;172(3):e19. doi: 10.1016/j.cell.2017.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–346. doi: 10.1016/S0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 19.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Zhou M, Lin F, Liu D, Hong W, Lu L, Zhu Y, Xu A. Interferon-gamma induces senescence in normal human melanocytes. PLoS One. 2014;9:e93232. doi: 10.1371/journal.pone.0093232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar PS, Shiras A, Das G, Jagtap JC, Prasad V, Shastry P. Differential expression and role of p21cip/waf1 and p27kip1 in TNF-alpha-induced inhibition of proliferation in human glioma cells. Mol Cancer. 2007;6:42. doi: 10.1186/1476-4598-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kandhaya-Pillai R, Miro-Mur F, Alijotas-Reig J, Tchkonia T, Kirkland JL, Schwartz S. TNFalpha-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging (Albany NY). 2017;9:2411–2435. doi: 10.18632/aging.101328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wieder T, Brenner E, Braumuller H, Bischof O, Rocken M. Cytokine-induced senescence for cancer surveillance. Cancer Metastasis Rev. 2017;36:357–365. doi: 10.1007/s10555-017-9667-z. [DOI] [PubMed] [Google Scholar]
- 24.Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494(7437):361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 25.Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2. Evidence for time-dependent signal generation. J Biol Chem. 1998;273(37):23830–23836. doi: 10.1074/jbc.273.37.23830. [DOI] [PubMed] [Google Scholar]
- 26.Nasser MW, Raghuwanshi SK, Grant DJ, Jala VR, Rajarathnam K, Richardson RM. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J Immunol. 2009;183:3425–3432. doi: 10.4049/jimmunol.0900305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collison JL, Carlin LM, Eichmann M, Geissmann F, Peakman M. Heterogeneity in the locomotory behavior of human monocyte subsets over human vascular endothelium in vitro. J Immunol. 2015;195:1162–1170. doi: 10.4049/jimmunol.1401806. [DOI] [PubMed] [Google Scholar]
- 30.Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, Puel A, Biswas SK, Moshous D, Picard C. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellora F, Castriconi R, Dondero A, Carrega P, Mantovani A, Ferlazzo G, Moretta A, Bottino C. Human NK cells and NK receptors. Immunol Lett. 2014;161(2):168–173. doi: 10.1016/j.imlet.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 32.Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V, Cipollone G, Navarra G, Mingari MC, Moretta L. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J Immunol. 2014;192:3805–3815. doi: 10.4049/jimmunol.1301889. [DOI] [PubMed] [Google Scholar]
- 33.Levi I, Amsalem H, Nissan A, Darash-Yahana M, Peretz T, Mandelboim O, Rachmilewitz J. Characterization of tumor infiltrating natural killer cell subset. Oncotarget. 2015;6:13835–13843. doi: 10.18632/oncotarget.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, Alignani D, Labiano S, Azpilikueta A, Rodriguez-Paulete A. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. 2016;22(15):3924–3936. doi: 10.1158/1078-0432.CCR-15-2463. [DOI] [PubMed] [Google Scholar]
- 35.Ogawa R, Yamamoto T, Hirai H, Hanada K, Kiyasu Y, Nishikawa G, Mizuno R, Inamoto S, Itatani Y, Sakai Y, et al. Loss of SMAD4 promotes colorectal cancer progression by recruiting tumor-associated neutrophils via the cxcl1/8-cxcr2 axis. Clin Cancer Res. 2019;25:2887–2899. doi: 10.1158/1078-0432.CCR-18-3684. [DOI] [PubMed] [Google Scholar]
- 36.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6(237):237ra67. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis E Sousa C. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castellano M, Pollock PM, Walters MK, Sparrow LE, Down LM, Gabrielli BG, Parsons PG, Hayward NK. CDKN2A/p16 is inactivated in most melanoma cell lines. Cancer Res. 1997;57:4868–4875. [PubMed] [Google Scholar]
- 39.Cayrol C, Knibiehler M, Ducommun B. p21 binding to PCNA causes G1 and G2 cell cycle arrest in p53-deficient cells. Oncogene. 1998;16:311–320. doi: 10.1038/sj.onc.1201543. [DOI] [PubMed] [Google Scholar]
- 40.Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novák B, Bakal C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat Commun. 2017;8(1):14728. doi: 10.1038/ncomms14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang T-W, Lasitschka F, Andrulis M. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15(8):978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce J, Krizhanovsky V. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153(2):449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30(4):533–547. doi: 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim YH, Choi YW, Lee J, Soh EY, Kim JH, Park TJ. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun. 2017;8:15208. doi: 10.1038/ncomms15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu D, Hornsby PJ. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007;67:3117–3126. doi: 10.1158/0008-5472.CAN-06-3452. [DOI] [PubMed] [Google Scholar]
- 47.Angelini PD, Zacarias FMF, Pedersen K, Parra-Palau JL, Guiu M, Bernado Morales C, et al. Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res. 2013;73:450–458. doi: 10.1158/0008-5472.CAN-12-2301. [DOI] [PubMed] [Google Scholar]
- 48.Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553(7686):96–100. doi: 10.1038/nature25167. [DOI] [PubMed] [Google Scholar]
- 49.Yang L, Fang J, Chen J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017;3(1):17049. doi: 10.1038/cddiscovery.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013;210:2057–2069. doi: 10.1084/jem.20130783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011;71:5998–6009. doi: 10.1158/0008-5472.CAN-10-3211. [DOI] [PubMed] [Google Scholar]
- 52.Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, Golani O, Polic B, Krizhanovsky V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany NY). 2016;8(2):328–344. doi: 10.18632/aging.100897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellis LZ, Cohen JL, High W, Stewart L. Melanoma in situ treated successfully using imiquimod after nonclearance with surgery: review of the literature. Dermatol Surg. 2012;38:937–946. doi: 10.1111/j.1524-4725.2012.02362.x. [DOI] [PubMed] [Google Scholar]
- 54.Steinmann A, Funk JO, Schuler G, von den Driesch P. Topical imiquimod treatment of a cutaneous melanoma metastasis. J Am Acad Dermatol. 2000;43:555–556. [PubMed] [Google Scholar]
- 55.Fan Q, Cohen S, John B, Riker AI. Melanoma in situ treated with topical imiquimod for management of persistently positive margins: a review of treatment methods. Ochsner J. 2015;15:443–447. [PMC free article] [PubMed] [Google Scholar]
- 56.Tio D, van Montfrans C, Ruijter CGH, Hoekzema R, Bekkenk MW. Effectiveness of 5% Topical Imiquimod for Lentigo Maligna Treatment. Acta Derm Venereol. 2019;99:884–888. doi: 10.2340/00015555-3241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.