

Abstract
The vascular endothelium provides the crucial interface between the blood compartment and tissues, and displays a series of remarkable properties that normally maintain homeostasis. This tightly regulated palette of functions includes control of haemostasis, fibrinolysis, vasomotion, inflammation, oxidative stress, vascular permeability, and structure. While these functions participate in the moment-to-moment regulation of the circulation and coordinate many host defence mechanisms, they can also contribute to disease when their usually homeostatic and defensive functions over-reach and turn against the host. SARS-CoV-2, the aetiological agent of COVID-19, causes the current pandemic. It produces protean manifestations ranging from head to toe, wreaking seemingly indiscriminate havoc on multiple organ systems including the lungs, heart, brain, kidney, and vasculature. This essay explores the hypothesis that COVID-19, particularly in the later complicated stages, represents an endothelial disease. Cytokines, protein pro-inflammatory mediators, serve as key danger signals that shift endothelial functions from the homeostatic into the defensive mode. The endgame of COVID-19 usually involves a cytokine storm, a phlogistic phenomenon fed by well-understood positive feedback loops that govern cytokine production and overwhelm counter-regulatory mechanisms. The concept of COVID-19 as an endothelial disease provides a unifying pathophysiological picture of this raging infection, and also provides a framework for a rational treatment strategy at a time when we possess an indeed modest evidence base to guide our therapeutic attempts to confront this novel pandemic.
Keywords: Endothelium, Thrombosis, Cytokine, Inflammation, Microvasculature
Introduction
The vascular endothelium provides the crucial interface between the blood compartment and tissues. The endothelial monolayer that lines the intima of arteries, veins, and microvessels measures up to 7000 m2 in surface area.1 The endothelium possesses a series of remarkable properties that contribute capitally to homeostasis (Figure 1, left). The endothelium furnishes one of the only surfaces, either natural or synthetic, that under physiological conditions maintains blood in a liquid state during prolonged contact. The endothelium displays a tightly regulated palette of functions that control vasomotion, inflammation, oxidative stress, vascular permeability, and structure.2 The endothelial cells also provide a crucial interface in host defences, forming the front line of encounter with bloodborne pathogens, thus sensing danger threatening the organism in a concerted fashion, sending early warning signals of infection, invasion, or injury.3 While these functions participate in the moment-to-moment regulation of the circulation and coordinate many host defence mechanisms, they can also contribute to disease when their usually homeostatic and defensive functions over-reach and turn against the host (Figure 1, middle and right). 4 , 5
Figure 1.

The left side of the diagram depicts a resting endothelial monolayer with the endothelial cells of squamous morphology resting on an intact basement membrane. The homeostatic mechanisms displayed by the resting endothelium include the listed properties as detailed in the text. When the endothelial cells undergo the cytopathic effect of a viral infection such as SARS-CoV-2, or encounter pathogen-associated molecular patterns (PAMPs) derived from viruses or bacteria such as lipopolysaccharide, proinflammatory cytokines such as IL-1 or TNF, or damage-associated molecular patterns (DAMPs) derived from dead or dying cells, the endothelial cells become activated. The endothelial cells display more columnar morphology. They can express adhesion molecules that attract leucocytes and chemokines that direct their migration into the subendothelial space. Sloughing of endothelial cells uncovers the thrombogenic basement membrane. Adherent neutrophils can undergo formation of neutrophil extracellular traps that provide an amplifier for endothelial damage mediated in part by IL-1α. Inflammatory activation of endothelial cells can disrupt VE-cadherin largely responsible for the integrity of the endothelial barrier function.62 Activated endothelial cells can also express matrix metalloproteinases that can degrade the basement membrane and further interrupt endothelial barrier function. In small vessels, such as those that embrace alveoli in the lung, this impaired barrier function can lead to capillary leak. These various disturbances in endothelial function, depicted in the middle part of the diagram, lead to end organ damage including adult respiratory distress syndrome and thrombosis in the lungs, predispose to plaque rupture and thrombosis in coronary arteries, and affect the microvasculature leading to myocardial ischaemia and damage. The thrombotic diathesis provoked by endothelial dysfunction can also predispose towards strokes. Microvascular as well macrovascular injury can potentiate acute renal failure. Hepatic dysfunction can also result from microvascular thrombosis among other mechanisms. Deep venous thrombosis can occur as endothelial disfunction represents an important part of Virchow’s triad, and sets the stage for pulmonary embolism. Thus, loss of the endothelial protective and unleashing of the mechanisms depicted can lead to multiorgan system failure that characterizes the advanced stages of COVID-19.
SARS-CoV-2, the aetiological agent of COVID-19, causes the current pandemic. It produces protean manifestations ranging from head to toe, wreaking seemingly indiscriminate havoc on multiple organ systems, in particular the lungs, heart, brain, kidney, and vasculature. This essay will explore the hypothesis that COVID-19, particularly in the later complicated stages, represents an endothelial disease. This concept not only provides a unifying pathophysiological picture of this raging infection but also furnishes a framework for a rational treatment strategy at a time when we possess an indeed modest evidence base to guide our therapeutic attempts to confront this novel pandemic.
The endothelium participates pivotally in thrombosis and fibrinolysis
The normal endothelial surface owes its remarkable haemocompatibility to a tightly orchestrated set of functions.6 Heparan sulfate proteoglycans decorate the surface of the endothelium. These molecules bind antithrombin III, as do heparinoids that we use daily in practice as an anticoagulant. The endothelial surface bears thrombomodulin, which binds thrombin and stimulates the protein C–protein S anticoagulant axis.1 , 3 The endothelial cell can also express a tissue factor pathway inhibitor that can antagonize triggering of thrombosis by the potent procoagulant protein tissue factor.7
Endothelial cells possess an endogenous mechanism for combatting platelet activation. This function depends on endothelial surface expression of an ecto-ADPase, CD39, as well as the release of nitric oxide and prostacyclin.8 , 9 Together, this array of anticoagulant and antithrombotic properties accounts for much of the ability of the endothelial cell to combat intravascular blood clot formation under normal circumstances. Should a stray thrombus form on the intimal lining of a blood vessel, the endothelial cells can express plasminogen activators that can boost endogenous fibrinolysis.10 Endothelial cells can produce both tissue-type plasminogen activator (tPA) and urokinase plasminogen activator (uPA),11 and, through the release of nitric oxide by platelet-derived substances, inhibit platelet function and increase local blood flow to flush away an evolving clot.
Although the normal endothelium possesses this palette of anticoagulant, antithrombotic, and profibrinolytic attributes, the balance between these salutary functions and an opposite panel of properties that promote thrombus accumulation can change on a dynamically regulated basis. The endothelial cell usually possesses little procoagulant potential. However, when stimulated by proinflammatory cytokines, pathogen-associated molecular patterns such as bacterial endotoxins, or neutrophil extracellular traps (NETs; see below), the endothelial cell can express and in turn exert tissue factor activity.12 , 13 Tissue factor activates the coagulation system by amplifying many-fold the enzymatic capacity of factors VII and X, triggering thrombin generation and clot formation.14 The endothelial cell also stores pre-formed von Willebrand factor (vWf) in intracellular granules called Weibel–Palade bodies. Upon activation, the endothelial cells can release this large protein that in higher molecular weight multimers provides a potent bridge for platelet aggregates and thrombus assembly, favouring formation of an organized clot.15
While under usual circumstances the antiaggregatory arachidonate product prostacyclin (PGI2) dominates endothelial vasoactive prostanoid production, the endothelial cell can also produce thromboxane, a pro-platelet aggregatory and vasoconstrictor prostaglandin.16 The activated endothelial cell can also manufacture plasminogen activator inhibitor-1 (PAI-1), which can antagonize the endogenous fibrinolytic properties conferred upon the endothelial surface by the expression of uPA and tPA, as noted above. Thus, while ordinarily programmed to combat blood clotting and thrombus accumulation, the endothelium—when activated by inflammatory or infectious signals—can exert an opposite battery of functions. While critically important in staunching haemorrhage or other injury, during disease the endothelial surface can promote clotting of arteries, microvessels, and veins, contributing critically to thrombo-embolism.
The endothelial vasodilator/vasoconstrictor balance
Under normal conditions, the endothelial cells promote tonic vasodilatation through the well-known mechanism of production of the vasodilatory gas nitric oxide from l-arginine via the activity of endothelial nitric oxide synthase.17 The endothelial cell can also elaborate diverse hyperpolarizing factors that promote relaxation of smooth muscle and hence dilatation of muscular arteries. As noted above, the normal endothelial cells also secrete PGI2 that, in addition to its antiaggregatory effects on platelets, potently vasodilates.18 This array of vasodilatory actions can also modulate moment-to-moment local blood flow in a paracrine fashion. Numerous mechanisms can interfere with endothelial-dependent vasodilatation. These mechanisms range from impaired nitric oxide synthase expression19 and/or activity to inactivation of nitric oxide or its conversion to highly pro-oxidant compounds by encountering pro-oxidant species such as superoxide anion, yielding the potent pro-oxidant peroxynitrate.20 , 21 Moreover, the endothelial cell can produce one of the most potent vasoconstrictors known, endothelin-1, in response to angiotensin II, thrombin, or oxidized LDL.22 , 23 While key in maintaining normal vascular homeostasis, during disease the salutary endothelium’s functions can give way to inappropriate vasoconstriction contributing to tissue ischaemia.
The endothelial inflammatory balance
Positioned at the key interface between the blood and tissues, the endothelium normally resists prolonged contact with the leucocytes that abound in blood that bathes the intimal surface.3 , 24 Stationed as the sentinel, the endothelium serves as the portal governing the entry of leucocytes into tissues to combat invaders, microbial or viral, and to help repair injury and heal wounds. The interplay of the endothelium with leucocyte mediators of innate and adaptive immunity depends on a series of leucocyte adhesion molecules expressed at negligible levels under physiological circumstances. Members of the selectin class of leucocyte adhesion molecules slow the transit of blood leucocytes past the endothelial surface by mediating rolling of these cells. E-selectin (CD62E) causes polymorphonuclear leucocytes to tarry on the endothelial surface. P-selectin (CD62P) and L-selectin (CD62L) also mediate interaction of the endothelial surface with various classes of blood leucocytes. The elevated expression of these endothelial–leucocyte adhesion molecules depends upon irritative stimuli, principally proinflammatory cytokines such as interleukin-1α (IL-1α) and IL-1β or tumour necrosis factor-α (TNF-α).
The firm binding of leucocytes to the activated endothelial surface depends upon adhesion molecules of the IgG superfamily. These molecules include intercellular adhesion molecule-1 (ICAM-1, CD54) and vascular cell adhesion molecule-1 (VCAM-1, CD106). Integrins associated with the endothelial surface also participate in these adhesive interactions and furnish cognate ligands for the adhesion molecules.25 Once tightly bound to the endothelial surface, chemoattractant cytokines of various classes can beckon the bound cells to traverse the endothelial monolayer and enter tissues where they can combat invaders or contribute to tissue repair.26
Antioxidant/pro-oxidant balance in the endothelium
The endothelial cell possesses a number of defence mechanisms that lower local oxidative stress. When subjected to normal laminar shear stress, the endothelium produces superoxide dismutase that scavenges the important reactive oxygen species .24 The endothelial cell can also express glutathione peroxidases that can mitigate oxidative stress.27 Likewise, haem oxygenase provides another mechanism by which the endothelial cell can resist local oxidative stress.28 , 29 In contrast, when stimulated by proinflammatory cytokines and other agonists, the endothelial cell can mobilize NADPH oxidases that generate superoxide anions, contributing to local oxidative stress.30 As with other beneficial properties, the endothelium can also contribute to disease through impaired antioxidant defences or actual generation of reactive oxygen species, as is the case in hypertension,31 hyperlipidaemia, and diabetes,32 among other cardiovascular conditions.
Endothelial barrier function
Under physiological circumstances, the endothelial gateway selectively regulates endothelial permeability and fosters vascular integrity. An intact endothelial barrier depends on myriad mechanisms including vascular endothelial-cadherin (VE-cadherin, CD144).33 A number of derangements can threaten the integrity of this single-cell layer that stands between the blood compartment and tissues. Impaired endothelial viability can promote sloughing of endothelial cells and their death by various mechanisms including pyroptosis and apoptosis.34 , 35 Among the stimuli for these pathways of programmed cell death are proinflammatory cytokines and reactive oxygen species. Endothelial cells can also perish due to accidental cell death or oncosis. Regardless of the mechanism of endothelial injury, breaches in the physical integrity of the monolayer can lead to capillary leak in the microvasculature, overturning the usually semi-permeable properties of the endothelium and contributing to inappropriate leakage of vascular contents into the tissue compartment and extracellular space.36 , 37
Cytokine storm: a perfect storm in COVID-19
As noted in each of the foregoing sections, proinflammatory cytokines conspire to elicit from endothelial cells a change from their homeostatic functions to those that can contribute to thrombosis and local tissue injury. Cytokines such as IL-1α and IL-1β, IL-6, and TNF-α, among others, contribute critically to normal host defences, but when produced inappropriately or in excess they can perturb all of the carefully orchestrated protective functions of the normal endothelium and potentiate pathological processes. The untrammelled production of proinflammatory cytokines contributes to a condition termed a cytokine storm (Figure 2). The pathophysiological mechanisms of a cytokine storm depend on phenomena described in the 1980s that centre on autoinduction of the primordial proinflammatory cytokine IL-1. IL-1 can induce its own gene expression, providing an amplification loop that can instigate a cytokine storm.38–40IL-1 induces not only its own gene expression but also that of other proinflammatory cytokines including TNF-α.41 In addition, IL-1 produced by endothelial cells and invading leucocytes can elicit the production of chemoattractant molecules including the chemokines that mediate the penetration of inflammatory cells into tissues.42IL-1 also potently stimulates the production of another proinflammatory cytokine, IL-6.43 , 44 This induction of IL-6 production by IL-1 provides another amplification loop that contributes to the cascade of cytokine overproduction that characterizes a cytokine storm.
Figure 2.
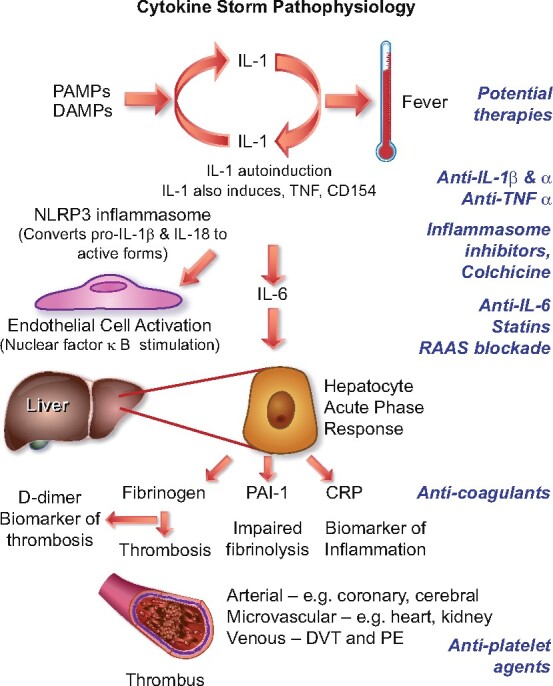
Cytokine storm. Proinflammatory cytokines such as IL-1 and TNF-α induce each other’s gene expression, unleashing an amplification loop that sustains the cytokine storm. The endothelial cell is a key target of cytokines, as they induce action of a central proinflammatory transcriptional hub, nuclear factor-κB. IL-1 also causes substantial increases in production by endothelial and other cells of IL-6, the instigator of the hepatocyte acute phase response. The acute phase reactants include fibrinogen, the precursor of clot, and PAI-1, the major inhibitor of our endogenous fibrinolytic system. C-reactive protein, commonly elevated in COVID-19, provides a readily measured biomarker of inflammatory status. The alterations in the thrombotic/fibrinolytic balance due to the acute phase response promotes thrombosis in arteries, in the microvasculature including that of organs such as the myocardium and kidney, and in veins, causing deep vein thrombosis and predisposing towards pulmonary embolism. Thus, the very same cytokines that elicit abnormal endothelial functions can unleash the acute phase response which together with local endothelial disfunction can conspire to cause the clinical complications of COVID-19. The right side of this diagram aligns therapeutic agents that attack these mechanisms of the cytokine storm and may thus limit its devastating consequences.
In addition to local effects, IL-6 provides a proximal stimulus to the acute phase response.45 This programne of protein synthesis elicited in the hepatocyte by IL-6 boosts the synthesis of fibrinogen, the precursor of clots, of PAI-1, a major inhibitor of our endogenous fibrinolytic mediators, and of C-reactive protein, a biomarker of inflammation that rises consistently in COVID-19.46 The havoc wreaked by the cytokine storm thus not only affects local endothelial function but can also provoke a prothrombotic and antifibrinolytic imbalance in blood that favours thrombus accumulation. The complications of COVID-19 follow very closely the consequences of excessive cytokine actions on endothelial cells outlined above and depicted in Figure 1.
The initial characterization of COVID-19 as a pneumonitis incorporates the notion of disordered endothelial function. While initial infection of type I and II pneumocytes and alveolar macrophages no doubt participates in the initiation of infection, disordered endothelial function certainly contributes to the ongoing ravages of SARS-CoV-2 in the lung as elsewhere. Impaired endothelial barrier function can contribute to protein accumulation in the alveolar space and fluid accumulation and impaired oxygenation of the blood. IL-1 stimulation reduces VE-cadherin, dubbed the guardian of integrity of the endothelium. This finding links a cytokine storm directly to capillary leak, and aggravation of the adult respiratory syndrome (ARDS) picture that advanced COVID-19 presents.33 , 47 The deranged balance in the prothrombotic/antithrombotic properties of the endothelium can certainly contribute to thrombosis in situ in the pulmonary vasculature, as occurs in COVID-19.48 Impaired gateway function of the endothelium for traversal of leucocytes into tissues clearly participates in pneumonitis.
However, we now recognize that SARS-CoV-2’s destructive actions range far and wide beyond the pulmonary parenchyma. Alterations in endothelial thrombotic/fibrinolytic balance can predispose to thrombosis not only in the pulmonary circulation but also in peripheral veins and arteries of the cerebral circulation, causing unheralded strokes in apparently healthy young people and doubtless contributing to the local and patchy embarrassment of blood flow in ‘COVID toes’ that probably represent microvascular dysfunction with tissue ischaemia. In between the brain and distal lower extremities, thromboses can occur in all arterial beds within the microvasculature, including that of the coronary circulation, and that of the kidneys. Venous thrombosis and pulmonary embolism also commonly complicate COVID-19, pathological processes that clearly depend on deranged endothelial functions.49 NETs induced by inflammatory cytokines activate procoagulant functions of endothelial cells, and contribute to coagulation and the formation of the typically tightly organized thrombi in COVID-19. Thus, disordered endothelial homeostasis provoked by cytokines provides a common thread in numerous complications of COVID-19.13 , 50–52
Endothelial functions as a therapeutic target
To combat the adverse balance between thrombotic and fibrinolytic properties of the endothelium, numerous anticoagulant and antiplatelet therapies are under evaluation in ongoing and planned clinical trials in COVID-19. Key questions that require an answer in this domain are which agents to give to whom and in what doses, given the narrow therapeutic window of such agents, and the common concomitant conditions that elevate bleeding risk in many COVID-19 patients.
In view of the central role of inflammatory mediators in the complications of COVID-19, anti-inflammatory therapies merit careful clinical evaluation. Glucocorticoids and colchicine exert generalized anti-inflammatory actions and show promise in the treatment of patients with advanced COVID-19.53 , 54 Statins have direct anti-inflammatory effects beyond their lipid-lowering actions, mediated by inhibition of prenylation of small G proteins or induction of transcription factors such as KLF-2 that promote homeostatic endothelial functions.55Non-randomized treatment with statins yielded preliminary retrospective evidence of improved outcomes in COVID-19, as well as reductions in biomarkers of inflammation.56
Targeted inhibition of cytokines, major effectors of endothelial activation, represents a more focused approach than generalized anti-inflammatory agents. IL-1 not only induces leucocyte adhesion molecules but, by reducing VE-cadherin production, can contribute to impaired endothelial barrier function and thus capillary leak, a major issue that complicates COVID-19 pneumonitis.47 Agents that inhibit the inflammasome–IL-1β–IL-6 pathway may thus comprise a more endothelial-directed approach to treatment of COVID-19. Some clinical trials that use such strategies have already yielded preliminary results; some, but not all, indicate signals of efficacy. Colchicine may act in part as an inhibitor of the assembly of the inflammasome. Small, non-randomized studies of a recombinant form of the endogenous IL-1 receptor antagonist, anakinra, have furnished sufficient encouragement to merit further definitive investigation.57 , 58 Anakinra blocks both IL-1α and IL-1β, and requires daily dosing. Canakinumab, a selective IL-1β antibody, has a much longer biological half-life than anakinra, rendering it less readily reversible. Several studies investigating canakinumab in COVID-19 are underway (NCT04362813 and NCT04365153.)
Downstream of IL-1, antibodies that interfere with IL-6 signalling have also shown signs of benefit in some but not all preliminary studies, although this as well as other anticytokine therapies may entail an increased risk of superinfection.59 , 60 Other anti-IL-6 strategies also warrant consideration.61 Upon inflammatory stimulation, vascular endothelial and smooth muscle cells produce large amounts of IL-6; thus, blocking signalling of this distal mediator can limit local vascular amplification of inflammatory responses, including in the lung. IL-6 also triggers the acute phase response, boosting fibrinogen and PAI-1 production, and thus favours clot formation and persistence (Figure 2).
The pivotal roles of these proinflammatory mediators in host defences render these initial results plausible and promising. Yet, rigorous, controlled, and prospective clinical trials must evaluate the balance between the potential benefits by forestalling the consequences of cytokine storm versus the potential of lowering defences against bacterial superinfections that commonly complicate individuals with impaired pulmonary protective functions and remain subject to the rigours of mechanical ventilation and endotracheal intubation.
In sum, we can envisage COVID-19 as a disease of the endothelium, certainly with respect to its complications. This unifying hypothesis can help to understand the complex pathophysiology of this current plague and may also help to inform our therapeutic approaches to combatting the consequences of SARS-CoV-2 infection.
Funding
P.L. receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), and the RRM Charitable Fund.
Conflict of interest: P.L. is an unpaid consultant to, or involved in clinical trials for, Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion, Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Merck, Novartis, Pfizer, and Sanofi-Regeneron. He is a member of the scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech, Inc. His laboratory has received research funding in the last 2 years from Novartis. He is on the Board of Directors of XBiotech, Inc. and has a financial interest in Xbiotech, a company developing therapeutic human antibodies. His interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. T.L. has no conflicts to declare.
Contributor Information
Peter Libby, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Thomas Lüscher, Heart Division, Royal Brompton & Harefield Hospital and National Heart and Lung Institute, Imperial College, London, UK.
References
- 1. Aird WC. Endothelium. In: Kitchens CS, Kessler CM, Konkle BA, eds. Consultative Hemostasis and Thrombosis, 3rd ed. Philadelphia. PA: W.B. Saunders; 2013. p33–41. [Google Scholar]
- 2. Libby P. The vascular biology of atherosclerosis. In: Zipes DP, Libby P, Bonow RO, Mann DL, Tomaselli GF, eds. Braunwald’s Heart Disease, 11th ed. Philadelphia, PA: Elsevier; 2018. p859–875. [Google Scholar]
- 3. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007;7:803–815. [DOI] [PubMed] [Google Scholar]
- 4. Libby P. The active roles of cells of the blood vessel wall in health and disease. Mol Aspects Med 1987;9:499–567. [DOI] [PubMed] [Google Scholar]
- 5. Libby P, d Birinyi LK. The dynamic nature of vascular endothelial functions. In: Zilla P, Fasol R, Deutsch M, eds. Endothelialization of Vascular Grafts. Basel: Karger; 1987. p80–99. [Google Scholar]
- 6. d’Alessandro E, Becker C, Bergmeier W, Bode C, Bourne JH, Brown H, Buller HR, ten Cate-Hoek AJ, ten Cate V, van Cauteren YJM, Cheung YFH, Cleuren A, Coenen D, Crijns HJGM, de Simone I, Dolleman SC, Klein CE, Fernandez DI, Granneman L, van t Hof A, Henke P, Henskens YMC, Huang J, Jennings LK, Jooss N, Karel M, van den Kerkhof D, Klok FA, Kremers B, Lämmle B, Leader A, Lundstrom A, Mackman N, Mannucci PM, Maqsood Z, van der Meijden PEJ, van Moorsel M, Moran LA, Morser J, van Mourik M, Navarro S, Neagoe RAI, Olie RH, van Paridon P, Posma J, Provenzale I, Reitsma PH, Scaf B, Schurgers L, Seelig J, Siegbahn A, Siegerink B, Soehnlein O, Soriano EM, Sowa MA, Spronk HMH, Storey RF, Tantiwong C, Veninga A, Wang X, Watson SP, Weitz J, Zeerleder SS, ten Cate H, Scientific Reviewer C, Badimon L, Binder C, Hoylaerts M, Karlheinz P, Koenen R, Lip GYH, Massberg S, von Hundelshausen P, Weber C, Wojta J. Thrombo-inflammation in cardiovascular disease: an expert consensus document from the Third Maastricht Consensus Conference on Thrombosis. Thromb Haemost 2020;120:538–564. [DOI] [PubMed] [Google Scholar]
- 7. Steffel J, Lüscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113:722–731. [DOI] [PubMed] [Google Scholar]
- 8. Marcus A, Broekman M, Drosopoulos JHF, Islam N, Alyonycheva T, Safier L, Hajjar K, Posnett D, Schoenborn M, Schooley K, Gayle R, Maliszewski C. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J. Clin Invest 1997;99:1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang ZH, Stulz P, von Segesser L, Bauer E, Turina M, Lüscher TF. Different interactions of platelets with arterial and venous coronary bypass vessels. Lancet 1991;337:939–943. [DOI] [PubMed] [Google Scholar]
- 10. Sawdey MS, Loskutoff DJ. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J Clin Invest 1991;88:1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levin EG, Loskutoff DJ. Cultured bovine endothelial cells produce both urokinase and tissue-type plasminogen activators. J Cell Biol 1982;94:631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol 2007;14:55–61. [DOI] [PubMed] [Google Scholar]
- 13. Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, Nakamura M, Newton G, Luscinskas FW, Libby P. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1α and cathepsin G. Arterioscler Thromb Vasc Biol 2018;38:1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Polgar J, Matuskova J, Wagner DD. The P-selectin, tissue factor, coagulation triad. J Thromb Haemost 2005;3:1590–1596. [DOI] [PubMed] [Google Scholar]
- 15. Wagner DD. The Weibel–Palade body: the storage granule for von Willebrand factor and P-selectin. Thromb Haemost 1993;70:105–110. [PubMed] [Google Scholar]
- 16. Lüscher TF, Vanhoutte PM. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension 1986;8:344–348. [DOI] [PubMed] [Google Scholar]
- 17. Furchgott RF. Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide (Nobel Lecture). Angew Chem Int Edn 1999;38:1870–1880. [DOI] [PubMed] [Google Scholar]
- 18. Vane JR. Nobel lecture. Adventures and excursions in bioassay—the stepping stones to prostacyclin. Postgrad Med J 1983;59:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oemar BS, Tschudi MR, Godoy N, Brovkovich V, Malinski T, Lüscher TF. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation 1998;97:2494–2498. [DOI] [PubMed] [Google Scholar]
- 20. Pennathur S, Heinecke JW. Mechanisms for oxidative stress in diabetic cardiovascular disease. Antiox Redox Signal 2007;9:955–969. [DOI] [PubMed] [Google Scholar]
- 21. van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med 2000;192:1731–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gupta RM, Libby P, Barton M. Linking regulation of nitric oxide to endothelin-1: the Yin and Yang of vascular tone in the atherosclerotic plaque. Atherosclerosis 2020;292:201–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boulanger CM, Tanner FC, Béa ML, Hahn AW, Werner A, Lüscher TF. Oxidized low density lipoproteins induce mRNA expression and release of endothelin from human and porcine endothelium. Circ Res 1992;70:1191–1197. [DOI] [PubMed] [Google Scholar]
- 24. Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mestas J, Ley K. Monocyte–endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med 2008;18:228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Noels H, Weber C, Koenen RR. Chemokines as therapeutic targets in cardiovascular disease. Arterioscler Thromb 2019;39:583–592. [DOI] [PubMed] [Google Scholar]
- 27. Lubos E, Kelly NJ, Oldebeken SR, Leopold JA, Zhang Y-Y, Loscalzo J, Handy DE. Glutathione peroxidase-1 deficiency augments proinflammatory cytokine-induced redox signaling and human endothelial cell activation. J Biol Chem 2011;286:35407–35417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nagy E, Eaton JW, Jeney V, Soares MP, Varga Z, Galajda Z, Szentmiklosi J, Mehes G, Csonka T, Smith A, Vercellotti GM, Balla G, Balla J. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler Thromb 2010;30:1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quan S, Yang L, Abraham NG, Kappas A. Regulation of human heme oxygenase in endothelial cells by using sense and antisense retroviral constructs. Proc Natl Acad Sci USA 2001;98:12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pennathur S, Heinecke JW. Oxidative stress and endothelial dysfunction in vascular disease. Curr Diab Rep 2007;7:257–264. [DOI] [PubMed] [Google Scholar]
- 31. Camici GG, Sudano I, Noll G, Tanner FC, Lüscher TF. Molecular pathways of aging and hypertension. Curr Opin Nephrol Hypertens 2009;18:134–137. [DOI] [PubMed] [Google Scholar]
- 32. Paneni F, Costantino S, Battista R, Castello L, Capretti G, Chiandotto S, Scavone G, Villano A, Pitocco D, Lanza G, Volpe M, Lüscher TF, Cosentino F. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet 2015;8:150–158. [DOI] [PubMed] [Google Scholar]
- 33. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 2013;26:441–454. [DOI] [PubMed] [Google Scholar]
- 34. Hansson GK, Chao S, Schwartz SM, Reidy MA. Aortic endothelial cell death and replication in normal and lipopolysaccharide-treated rats. Am J Pathol 1985;121:123–127. [PMC free article] [PubMed] [Google Scholar]
- 35. Kavurma MM, Tan NY, Bennett MR. Death receptors and their ligands in atherosclerosis. Arterioscler Thromb 2008;28:1694–1702. [DOI] [PubMed] [Google Scholar]
- 36. Franck G, Even G, Gautier A, Salinas M, Loste A, Procopio E, Gaston AT, Morvan M, Dupont S, Deschildre C, Berissi S, Laschet J, Nataf P, Nicoletti A, Michel JB, Caligiuri G. Haemodynamic stress-induced breaches of the arterial intima trigger inflammation and drive atherogenesis. Eur Heart J 2019;40:928–937. [DOI] [PubMed] [Google Scholar]
- 37. Libby P. Once more unto the breach: endothelial permeability and atherogenesis. Eur Heart J 2019;40:938–940. [DOI] [PubMed] [Google Scholar]
- 38. Dinarello CA, Ikejima T, Warner SJC, Orencole SF, Lonnemann G, Cannon JG, Libby P. Interleukin-1 induces interleukin-1. I. Induction of circulating interleukin-1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol 1987;139:1902–1910. [PubMed] [Google Scholar]
- 39. Warner SJC, Auger KR, Libby P. Interleukin-1 induces interleukin-1. II. Recombinant human interleukin-1 induces interleukin-1 production by adult human vascular endothelial cells. J Immunol 1987;139:1911–1917. [PubMed] [Google Scholar]
- 40. Warner SJC, Auger KR, Libby P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J Exp Med 1987;165:1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warner SJ, Libby P. Human vascular smooth muscle cells. Target for and source of tumor necrosis factor. J Immunol 1989;142:100–109. [PubMed] [Google Scholar]
- 42. Wang J, Sica A, Peri G, Walter S, Martin-Padura I, Libby P, Ceska M, Lindley I, Colotta F, Mantovani A. Expression of monocyte chemotactic protein and interleukin-8 by cytokine-activated human vascular smooth muscle cells. Arterioscler Thromb 1991;11:1166–1174. [DOI] [PubMed] [Google Scholar]
- 43. Loppnow H, Libby P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell Immunol 1989;122:493–503. [DOI] [PubMed] [Google Scholar]
- 44. Loppnow H, Libby P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin-6. J Clin Invest 1990;85:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kang S, Narazaki M, Metwally H, Kishimoto T. Historical overview of the interleukin-6 family cytokine. J Exp Med 2020;217:e20190347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wright FL, Vogler TO, Moore EE, Moore HB, Wohlauer MV, Urban S, Nydam TL, Moore PK, McIntyre RC Jr. Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 infection. J Am Coll Surg 2020;doi: 10.1016/j.jamcollsurg.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, Zhong M, Wu W, Ye Z, Gao X, Rao G, Mehta D, Rehman J, Malik AB. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest 2020;130:3684–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 2020;383:120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wichmann D, Sperhake J-P, Lütgehetmann M, Steurer S, Edler C, Heinemann A, Heinrich F, Mushumba H, Kniep I, Schröder AS, Burdelski C, de Heer G, Nierhaus A, Frings D, Pfefferle S, Becker H, Bredereke-Wiedling H, de Weerth A, Paschen H-R, Sheikhzadeh-Eggers S, Stang A,, Schmiedel S, Bokemeyer C, Addo MM, Aepfelbacher M, Püschel K, Kluge S. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med 2020;doi: 10.7326/M20-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood 2014;123:2768–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, Daßler-Plenker J, Guerci P, Huynh C, Knight JS, Loda M, Looney MR, McAllister F, Rayes R, Renaud S, Rousseau S, Salvatore S, Schwartz RE, Spicer JD, Yost CC, Weber A, Zuo Y, Egeblad M. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med 2020;217:e20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair CN, Weber A, Barnes BJ, Egeblad M, Woods RJ, Kanthi Y, Knight JS. Neutrophil extracellular traps in COVID-19. JCI Insight 2020;5:e138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Horby P, Lim WS, Emberson J, Mafham M, Bell J, Linsell L, Staplin N, Brightling C, Ustianowski A, Elmahi E, Prudon B, Green C, Felton T, Chadwick D, Rege K, Fegan C, Chappell LC, Faust SN, Jaki T, Jeffery K, Montgomery A, Rowan K, Juszczak E, Baillie JK, Haynes R, Landray MJ. Effect of dexamethasone in hospitalized patients with COVID-19: preliminary report. medRxiv 2020:2020.06.22.20137273. [Google Scholar]
- 54. Deftereos SG, Giannopoulos G, Vrachatis DA,, Siasos GD, Giotaki SG, Gargalianos P, Metallidis S, Sianos G, Baltagiannis S, Panagopoulos P, Dolianitis K, Randou E, Syrigos K,, Kotanidou A, Koulouris NG, Milionis H, Sipsas N,, Gogos C, Tsoukalas G, Olympios CD, Tsagalou E, Migdalis I, Gerakari S, Angelidis C, Alexopoulos D, Davlouros P, Hahalis G, Kanonidis I, Katritsis D, Kolettis T, Manolis AS, Michalis L, Naka KK,, Pyrgakis VN, Toutouzas KP, Triposkiadis F, Tsioufis K, Vavouranakis E, Martinèz-Dolz L, Reimers B, Stefanini GG, Cleman M, Goudevenos J, Tsiodras S, Tousoulis D, Iliodromitis E, Mehran R, Dangas G, Stefanadis C, GRECCO-19 Investigators. Effect of colchicine vs standard care on cardiac and inflammatory biomarkers and clinical outcomes in patients hospitalized with coronavirus disease 2019: the GRECCO-19 randomized clinical trial. JAMA Netw Open 2020;3:e2013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Libby P, Aikawa M, Jain MK. Vascular endothelium and atherosclerosis. Handb Exp Pharmacol 2006;176:285–306. [DOI] [PubMed] [Google Scholar]
- 56. Zhang X-J, Qin J-J, Cheng X, Shen L, Zhao Y-C, Yuan Y, Lei F, Chen M-M, Yang H, Bai L, Song X, Lin L, Xia M, Zhou F, Zhou J, She Z-G, Zhu L, Ma X, Xu Q, Ye P, Chen G, Liu L, Mao W, Yan Y, Xiao B, Lu Z, Peng G, Liu M, Yang J, Yang L, Zhang C, Lu H, Xia X, Wang D, Liao X, Wei X, Zhang B-H, Zhang X, Yang J, Zhao G-N, Zhang P, Liu PP, Loomba R, Ji Y-X, Xia J, Wang Y, Cai J, Guo J, Li H. In-hospital use of statins is associated with a reduced risk of mortality among individuals with COVID-19. Cell Metab 2020;doi: 10.1016/j.cmet.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, Oltolini C, Castiglioni B, Tassan Din C, Boffini N, Tomelleri A, Farina N, Ruggeri A, Rovere-Querini P, Di Lucca G, Martinenghi S, Scotti R, Tresoldi M, Ciceri F, Landoni G, Zangrillo A, Scarpellini P, Dagna L. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol 2020;2:e325–e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, Sacco E, Naccache J-M, Bézie Y, Laplanche S, Le Berre A, Le Pavec J, Salmeron S, Emmerich J, Mourad J-J, Chatellier G, Hayem G. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol 2020;2:e393–e400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu X, Han M, Li T, Sun W, Wang D, Fu B, Zhou Y, Zheng X, Yang Y, Li X, Zhang X, Pan A, Wei H. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci USA 2020;117:10970–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Somers EC, Eschenauer GA, Troost JP, Golob JL, Gandhi TN, Wang L, Zhou N, Petty LA, Baang JH, Dillman NO, Frame D, Gregg KS, Kaul DR, Nagel J, Patel TS, Zhou S, Lauring AS, Hanauer DA, Martin ET, Sharma P, Fung CM, Pogue JM. Tocilizumab for treatment of mechanically ventilated patients with COVID-19. medRxiv 2020:2020.05.29.20117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Magro G. SARS-CoV-2 and COVID-19: is interleukin-6 (IL-6) the ‘culprit lesion’ of ARDS onset? What is there besides Tocilizumab? SGP130Fc. Cytokine X 2020;2:100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G, Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J 2015;36:1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]