

Abstract
Hyperlipidemia (HPL) characterized by metabolic disorder of lipids and cholesterol is one of the important risk factors for cardiovascular diseases. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a potent circulating regulator of LDL through its ability to induce degradation of the low-density lipoprotein cholesterol receptor (LDLR) in the lysosome of hepatocytes. Aloe-emodin (AE) is one of potentially bioactive components of Chinese traditional medicine Daming capsule. In this study we evaluated the HPL-lowering efficacy of AE in both in vivo and in vitro HPL models. High-fat diet-induced rats were treated with AE (100 mg/kg per day, ig) for 6 weeks. We found that AE administration significantly decreased the levels of total cholesterol (TC) and LDL in the serum and liver tissues. Moreover, AE administration ameliorated HPL-induced hepatic lipid aggregation. But AE administration did not significantly inhibit HMG-CoA reductase activity in the liver of HPL rats. A cellular model of HPL was established in human hepatoma (HepG2) cells treated with cholesterol (20 μg/mL) and 25-hydroxycholesterol (2 μg/mL), which exhibited markedly elevated cholesterol levels. The increased cholesterol levels could be reversed by subsequent treatment with AE (30 μM). In both the in vivo and in vitro HPL models, we revealed that AE selectively suppressed the sterol-regulatory element-binding protein-2 (SREBP-2) and hepatocyte nuclear factor (HNF)1α-mediated PCSK9 signaling, which in turn upregulated LDL receptor (LDLR) and promoted LDL uptake. This study demonstrates that AE reduces cholesterol content in HPL rats by inhibiting the hepatic PCSK9/LDLR pathway.
Keywords: aloe-emodin, hyperlipidemia, cholesterol, PCSK9, LDL receptor, HepG2 cells, atorvastatin
Introduction
Currently, the incidence of dyslipidemia has increased significantly in China and is closely related to the morbidity and mortality of cardiovascular diseases [1]. It is well recognized that multiple factors are involved in the increased risk of hyperlipidemia (HPL), including a high saturated or trans-fat diet, physical inactivity, and obesity [2]. Numerous clinical and experimental studies have revealed that HPL is associated with various cardiovascular diseases, including atherosclerosis, diabetes, and coronary artery diseases [3]. Recent studies have suggested that dyslipidemia, characterized by an increase in total cholesterol (TC) or low-density lipoprotein cholesterol (LDL), is an important risk factor for atherosclerotic cardiovascular diseases (ASCVDs) [4]. ASCVDs, including myocardial infarction, coronary heart disease, and ischemic stroke, contribute to a high mortality rate. It is thus imperative to take effective measures to control blood lipid abnormalities to prevent ASCVDs, stroke, diabetes, and fatty liver diseases. It has been widely recognized that optimal lipid-control drugs should not only control low-density lipoprotein cholesterol (LDL) and cholesterol levels but also control ASCVDs, reducing myocardial infarction, ischemic stroke, or sudden cardiac death [5, 6]. Statins, as the clinical preferred drugs in the primary and secondary prevention of ASCVDs, can significantly reduce cardiovascular events. However, statins are limited by intolerance and adverse effects. It has been reported that more than 30% of patients are statin intolerant [7, 8]. Several serious adverse effects, such as rhabdomyolysis, acute renal failure, abnormal liver enzymes, and pulmonary fibrosis, have been reported [8, 9]. Clearly, new therapeutic agents devoid of these side effects are needed for the treatment of HPL and ASCVDs.
Sterol-regulatory element-binding proteins (SREBPs) are master regulators of lipid homeostasis [10]. Mammals have two SREBP-encoding genes that express three SREBPs: SREBP-1a, SREBP-1c, and SREBP-2. SREBP-2 principally regulates genes involved in cholesterol metabolism, whereas genes involved in fatty acid metabolism are preferentially regulated by SREBP-1. They are synthesized as inactive precursors localized to the endoplasmic reticulum bound to SREBP cleavage-activating protein (SCAP). Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a potent circulating regulator of LDL through its ability to induce degradation of the LDLR in the lysosome of hepatocytes [11]. PCSK9 inhibitors and monoclonal antibodies have become increasingly popular in the therapy of HPL [12]. Growing evidence has demonstrated that PCSK9 inhibitors are effective at controlling LDL and reducing the rate of cardiovascular events [13–15]. Nevertheless, the safety and unexpected adverse effects of PCSK9 inhibitors need to be examined during their long-term clinical application.
Many studies have revealed that several traditional Chinese medicines have excellent hypolipidemic effects, among which the Daming capsule has been widely used in patients with HPL in China [16]. Our previous work successfully screened six anthraquinones with fast and high absorption as potentially bioactive components of Daming capsule, including emodin-O-glucoside, aurantio-obtusin, aloe-emodin (AE, 1,3,8-trihydroxyanthraquinone; Fig. 1a), rhein, emodin, and chrysophanol [17]. Previous studies have shown that AE and its derivatives have a variety of pharmacological effects, such as antitumor, antibacterial, and antioxidant actions [18–20]. However, the potential role of AE in HPL remains unclear. Based on both animal and cellular evidence, the present study reported that AE exerted cholesterol-lowering effects by inhibiting PCSK9 and eventually promoting LDL degradation.
Fig. 1.
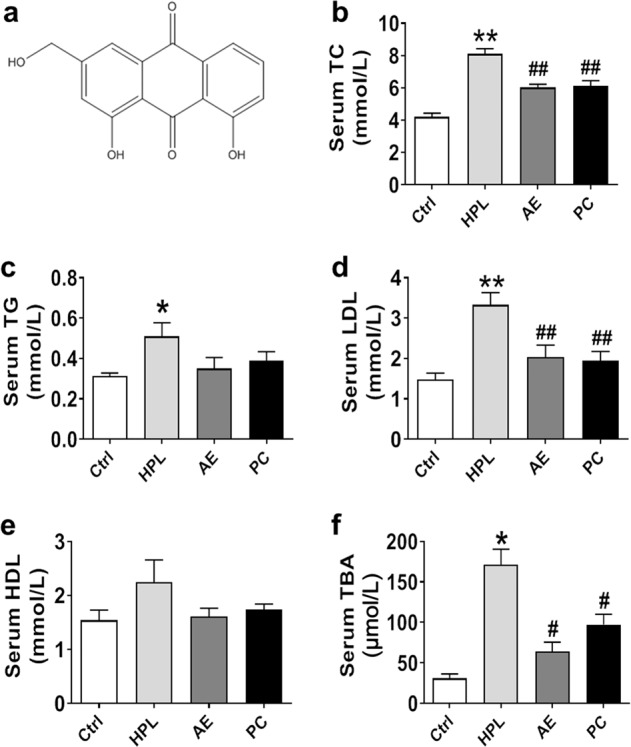
Effects of aloe-emodin (AE) on serum lipids in high-fat diet-induced hyperlipidemia (HPL) rats. a Chemical structure of AE. b Reduction in serum total cholesterol (TC) by AE at a dose of 100 mg/kg. c Serum triglyceride (TG). d Serum low-density lipoprotein (LDL). e Serum high-density lipoprotein (HDL). f Serum total bile acid (TBA). *P < 0.05, **P < 0.01 vs. Ctrl, #P < 0.05, ##P < 0.01 vs. HPL, n = 8. PC group: atorvastatin 7.2 mg/kg
Materials and methods
Animals and diet
All experiments were approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (SYDW2019-258) and were performed in accordance with the Guideline for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th edition, 2011). Healthy Wistar rats (male, ~7 weeks old, weight 200 ± 20 g) were provided by the Animal Center of the Second Affiliated Hospital of Harbin Medical University and were housed in a climate-controlled environment (22–24 °C). After 1 week of adaptation, the rats were randomly divided into four groups (n = 8 in each group): normal diet (Ctrl), HPL, AE treatment, and positive control treatment (PC, atorvastatin). Rats in the Ctrl group were fed a normal diet for 10 weeks. Rats in the HPL, AE, and PC groups were first fed a high-fat diet for 4 weeks to establish the HPL model. After validation of HPL, they were switched to a high-fat diet and treated with ddH2O, AE (100 mg/kg), or atorvastatin (7.2 mg/kg) for another 6 weeks. The composition of the high cholesterol diet was as follows: 77.6% carbohydrate, 10% fat, 10% protein, 2% cholesterol, 0.2% bile salt, and 0.2% methylthiouracil, which was purchased from Beijing Huafukang Biological Technology Co. Ltd (Beijing, China). The body weight was recorded every week. At the end of the experiments, animals were sacrificed, and blood samples and liver tissues were collected for subsequent examinations.
Measurement of lipid changes in rat serum, liver, and HepG2 cells
The collected blood samples were centrifuged at 620×g at 4 °C for 15 min. After centrifugation, the serum was transferred to a new centrifuge tube for measuring biochemical parameters. Serum levels of triglyceride (TG), TC, LDL, and high-density lipoprotein cholesterol (HDL) were measured by corresponding kits (Zhongsheng Beikong Biotechnology Co., Ltd, Beijing, China). Total bile acid (TBA) was analyzed according to the manufacturer’s protocol (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The TC content in HepG2 cells was analyzed by a kit according to the manufacturer’s instructions (Applygen Technology Inc., Beijing, China). TC and TG in rat livers were measured by kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Determination of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase
HMG-CoA reductase ELISA kit (Shanghai Enzyme-linked Biotechnology Co., Ltd, Shanghai, China) was used to determine the activity of HMG-CoA reductase according to the manufacturer’s instructions.
Hematoxylin and eosin staining
Liver tissues were fixed with 4% formaldehyde solution and cut into 4-μm-thick portions, which were stained with hematoxylin and eosin. Images were captured using a microscope (Olympus, Tokyo, Japan).
Cell culture and transfection
The HepG2 cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM containing penicillin (100 U/mL), streptomycin (100 mg/mL), and 10% fetal bovine serum (FBS) at 37 °C in a 5% humidified CO2 incubator. When cells reached approximately 80% confluence, the medium was switched to serum-free DMEM. HepG2 cells were incubated with AE at different concentrations (10, 20, 30, 50, 70, 90, and 110 μM) for 24 h. A concentration of AE of 30 μM was chosen for subsequent experiments. AE was dissolved in DMSO, and the DMSO concentration was kept below 0.05% in all cellular experiments to avoid possible toxic effects on cells. To investigate the role of AE in cholesterol metabolism, cells were randomly divided into the following groups: control group (Ctrl), TC group, and AE-treated group. In the TC group, cells were treated with cholesterol (20 μg/mL, dissolved in ethanol) and 25-hydroxycholesterol (2 μg/mL, dissolved in ethanol) for 6 h. In the AE-treated group, cells were first treated with cholesterol (20 μg/mL, dissolved in ethanol) and 25-hydroxycholesterol cholesterol (2 μg/mL, dissolved in ethanol) for 6 h and then treated with AE for another 24 h. Cells were harvested after all treatments for subsequent experiments. PCSK9-expressing plasmid (Changsha Yingrun Biotechnology Co., Ltd, Changsha, China) was transfected into HepG2 cells by X-tremeGENE siRNA transfection reagent (Roche, Pleasanton, CA, USA). Briefly, cells were trypsinized and seeded for 24 h before transfection. The transfection mixture was dissolved in Opti-MEM serum-free medium and added to the cells. After 24 h of transfection, the medium was replaced by fresh medium with or without AE. After AE treatment for 24 h, the cells were used for RNA extraction. To validate the inhibitory effect of AE on PCSK9, the PCSK9 inhibitor SBC-110736 (10 and 30 μM) was used.
LIVE/DEAD staining assay
The LIVE/DEAD® Viability/Cytotoxicity Assay Kit (Invitrogen, Carlsbad, CA, USA) was used to detect the amount of live and dead cells. The dye calcein acetoxymethyl ester (calcein-AM, 0.5 μL/mL) was mixed with ethidium homodimer-1 (EthD-1, 2 μL/mL) and added to cells followed by incubation for 15 min. The numbers of live and dead cells were detected by a laser scanning confocal microscope (FV1000, Olympus, Japan). The ratio of dead cells to total cells was calculated for quantitative comparisons.
Lactate dehydrogenase activity measurement
Lactate dehydrogenase (LDH) activity in the culture medium of HepG2 cells was tested according to the instructions (Beyotime Biotechnology, Haimen, China).
Western blot analysis
Total protein samples were isolated from rat liver and HepG2 cells, and the protein concentration was determined by a BCA kit (Beyotime Institute of Biotechnology, Haimen, China). Protein extracts were subjected to SDS-polyacrylamide gel electrophoresis for Western blotting analysis as previously described. Briefly, proteins were separated by 10% SDS-PAGE and then electrophoretically transferred onto nitrocellulose membranes (PALL Corporation, Ann Arbor, MI, USA). After blocking with 5% nonfat milk at room temperature for 2 h, the membranes were incubated at 4 °C overnight with anti-PCSK9 (1:500, Cat: bs-6060R, Biosynthesis Biotechnology Co. Ltd, Beijing, China), anti-SREBP-2 (1:500, Cat: 10007663, Cayman Chemical Company, Ann Arbor, MI, USA), anti-LDL receptor (1:200, Cat: ab30532, Abcam, Cambridge, UK), anti-SCAP (1:500, Cat: 13102, Cell Signaling, Boston, MA, USA), anti-peroxisome proliferator-activated receptor γ (PPARγ, 1:500, Cat: 16643-1-AP, Proteintech, Wuhan, China), anti-hepatocyte nuclear factor (HNF1α, 1:500, Cat: 22426-1-AP, Proteintech, Wuhan, China) or anti-GAPDH (1:1000, Cat: TA-08, ZSGB-Bio, Beijing, China) antibodies. Next, the membranes were incubated with a secondary antibody (1:5000, LI-COR Biosciences, Lincoln, NE, USA) for 1 h. Then, the membranes were captured by an Odyssey CLx (LI-COR Biosciences, Lincoln, NE, USA). Band densities were calculated by the Odyssey CLx software version 2.1 (LI-COR Biosciences, Lincoln, NE, USA).
Statistical analysis
Data are expressed as the mean ± SEM and analyzed by GraphPad Prism 5.0 software. Two-group comparisons were performed by Student’s t-test. Multiple-group comparisons were carried out using one-way ANOVA followed by Dunnett’s t-test. For the data with a control value of 1 and no SEM, randomized block ANOVA (repeated measures ANOVA) was used. P < 0.05 was considered statistically significant.
Reagents
AE (purity 98%) was provided by Tianfeng Biotechnology Co., Ltd (Xi’an, China). Lipitor® (atorvastatin calcium) was provided by Pfizer Pharmaceuticals Ltd (Suzhou, China). Cholesterol (purity 99%) and 25-hydroxycholesterol cholesterol (purity 98%) were obtained from Sigma (St. Louis, MO, USA). The PCSK9 inhibitor SBC-110736 (Cat: HY-101832, purity >99%) was purchased from MedChemExpress Company (Princeton, NJ, USA).
Results
Lipid-lowering effects of AE in both the serum and liver of high-fat rats
To verify the HPL model in rats induced by a high-fat diet for 4 weeks, we measured serum lipid concentrations, including TC, TG, LDL, and HDL. We found that TC, TG, and LDL were markedly increased in HPL rats compared to those in control rats, suggesting that the HPL model was successfully established (Supplementary Fig. S1). After that, rats continued on the high-fat diet and started to receive AE (100 mg/kg per day) for 6 more weeks. Serum lipid concentrations, including TG, TC, LDL, HDL, and TBA, were detected to evaluate the effects of AE. As shown in Fig. 1b–f, AE markedly reduced the levels of TC, LDL, and TBA but did not significantly alter TG and HDL levels. Atorvastatin was employed as a positive control, and our results showed that the impact of AE at 100 mg/kg was equivalent to that of atorvastatin at 7.2 mg/kg. Moreover, TC and TG in rat liver were examined. TC levels in rat livers were found to be significantly lower in AE-treated rats than in HPL rats, whereas no significant difference was observed in TG levels between the AE and HPL groups (Fig. 2a, b). Taken together, these findings supported that AE has promising cholesterol-lowering effects in both serum and liver.
Fig. 2.

Effects of AE on hepatic TC, TG, and the morphology of the rat liver. a TC in rat liver. b TG in rat liver. c Body weight after a 4-week high-fat diet. d Body weight during 6 weeks of treatment with AE or atorvastatin. e Liver weight. f H&E staining displayed the effect of AE on hepatic lipids, scale bar: 25 μm. *P < 0.05 vs. Ctrl, #P < 0.05 vs. HPL, n = 8
The body weight of rats fed a high-fat diet for 4 weeks was significantly higher than that of rats fed a normal diet (Fig. 2c). Moreover, body weight was dynamically changed during 6 weeks of treatment with AE or atorvastatin. After treatment for 6 weeks, no significant difference in body weight was found among these groups (Fig. 2d). In order to evaluate the effect of AE on the morphological changes in rat liver, rat livers were first weighed. Liver weight in HPL rats was higher than that in control rats; however, liver weight was reduced by AE treatment (Fig. 2e). Meanwhile, hematoxylin and eosin staining of liver tissues was conducted. As displayed in Fig. 2f, control rats had complete liver structure, clear hepatic lobule structure, orderly arrangement of hepatic cords, without fat vacuoles in the cytoplasm and fat degeneration or necrosis. However, in the HPL group, the structure of the hepatic lobule was destroyed, the hepatic cord was disordered, the liver cells were necrotic in spots and slices, and the size of liver cells became larger. Moreover, there was fatty degeneration to different degrees, and there were many fat vacuoles in the cytoplasm. After AE and atorvastatin treatment, the volume of hepatocytes and the number of fat vacuoles decreased significantly, and the hepatic cords were arranged neatly. Collectively, compared with the HPL group, the liver morphology of the AE and atorvastatin groups was significantly improved.
Next, we tested whether the alterations caused by AE in HPL rats could be reproduced in a cellular model. To this end, we treated HepG2 cells with cholesterol and 25-hydroxycholesterol to mimic HPL in vivo in rats. We then measured the cholesterol contents in HepG2 cells after AE treatment. Our results showed that cholesterol and 25-hydroxycholesterol treatment for 6 and 12 h significantly increased cholesterol levels (Fig. 3a), whereas 30 μM AE treatment abrogated such increases (Fig. 3b).
Fig. 3.
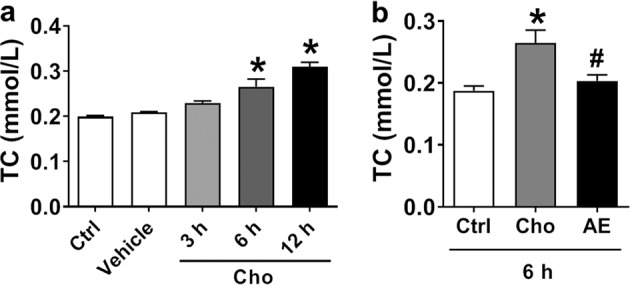
Effect of AE on TC in the HepG2 cell line as a cellular model of high cholesterol. a Cholesterol treatment for 3, 6, and 12 h. b Effect of AE (30 μM) on the cholesterol content after treatment for 6 h. *P < 0.05 vs. Ctrl, #P < 0.05 vs. Cho, n = 6
Lack of effect of AE on HMG-CoA reductase activity in HPL rats
It is known that statins reduce lipids and cholesterols primarily by inhibiting HMG-CoA reductase activity. Thus, there is a possibility that AE also acts through the same mechanism. To test this notion, we measured HMG-CoA reductase activity by enzyme-linked immunosorbent assay. As shown in Supplementary Fig. S2, the HPL-induced increase in HMG-CoA reductase activity was significantly inhibited by atorvastatin but not by AE. This result indicated that the mechanism underlying the cholesterol-lowering effects of AE may be different from atorvastatin.
Inhibition of the PCSK9 signaling pathway as a mechanism for the lipid-lowering property of AE
Recently, PCSK9 has been proposed to be another important enzyme for cholesterol metabolism [12]. Our data shown in Fig. 4a demonstrated that the level of PCSK9 in HPL rats was significantly increased compared to control rats and was restored by AE. By comparison, atorvastatin failed to inhibit PCSK9 protein expression. Consistently, HPL-induced upregulation of SREBP-2 and SCAP protein expression was decreased by AE treatment (Fig. 4b, c). Furthermore, the HPL-induced downregulation of LDLR expression was reversed by AE (Fig. 4d).
Fig. 4.

Effects of AE on the protein expression of PCSK9, SCAP, LDLR, and SREBP-2 in rat liver. a Western blot band of PCSK9 and statistical results. b Western blot band of SCAP and statistical results. c Western blot band of SREBP-2 and statistical results. d Western blot band of LDLR and statistical results. *P < 0.05 vs. Ctrl, #P < 0.05 vs. HPL, n = 7. PC group: atorvastatin 7.2 mg/kg
The same results were obtained from in vitro studies conducted in HepG2 cells. We found that 30 μM AE markedly inhibited the protein expression of SCAP and PCSK9 (Fig. 5a, b), leaving LDLR unaltered (Fig. 5c). To explore whether lower concentrations of AE still have the abovementioned effect, 10 and 20 μM AE were examined. As shown in Fig. 5a, b, 10 and 20 μM AE failed to inhibit the protein expression of SCAP and PCSK9. Based on these results, 30 μM was chosen as the appropriate concentration of AE. We further evaluated the role of AE in the PCSK9 pathway under high cholesterol conditions. As shown in Fig. 6a–c, SCAP and PCSK9 expression was significantly decreased by AE, while LDLR expression was increased after AE treatment. Moreover, AE inhibited SREBP-2 and HNF1α but not PPARγ (Fig. 6d–f). Additionally, no significant difference was found in the survival between control cells and cells treated with AE at concentrations of 30, 50, 70, 90, and 110 μM (Supplementary Fig. S3a, b). Moreover, similar results were obtained for LDH activity (Supplementary Fig. S3c). Finally, to validate the inhibitory role of AE in PCSK9 expression, the PCSK9 inhibitor SBC-110736 and the PCSK9 plasmid were employed. As shown in Fig. 7a, b, the protein level of PCSK9 was significantly decreased by AE and the PCSK9 inhibitor. Meanwhile, the effect of AE was similar to that of 10 μM PCSK9 inhibitor and weaker than that of 30 μM. Furthermore, the PCSK9 plasmid was transfected into HepG2 cells in the absence or presence of AE. We found that the mRNA level of PCSK9 increased dramatically compared with the control group, which was recovered by AE (Fig. 7c). Collectively, these findings supported that PCSK9 was a target of AE in our study.
Fig. 5.

Effects of AE at different concentrations ranging from 10 to 30 μM on the protein levels of SCAP, PCSK9, and LDLR in HepG2 cells. a Western blot band of SCAP and statistical results. b Western blot bands of PCSK9 and statistical results. c Western blot bands of LDLR and statistical results. *P < 0.05 vs. Ctrl, n = 6
Fig. 6.

Effects of AE on the protein levels of PCSK9, SCAP, LDLR, and SREBP-2 in cholesterol-treated HepG2 cells. a Western blot band of SCAP and statistical results. b Western blot band of PCSK9 and statistical results. c Western blot band of LDLR and statistical results. d Western blot band of SREBP-2 and statistical results. e Western blot band of hepatocyte nuclear factor (HNF)1α and statistical results. f Western blot band of peroxisome proliferator-activated receptor (PPAR)γ and statistical results. *P < 0.05, **P < 0.01 vs. Ctrl, #P < 0.05, ##P < 0.01 vs. Cho, n = 6
Fig. 7.

Validation of the effect of AE on PCSK9 signaling by a PCSK9 inhibitor or overexpression of PCSK9. a Western blot band of PCSK9. b Statistical results. *P < 0.05 vs. Ctrl, #P < 0.05 vs. Cho, n = 6. c Inhibition of PCSK9 mRNA levels by AE. *P < 0.05 vs. Ctrl, #P < 0.05 vs. PCSK9 group, n = 7
Discussion
Our previous study first screened AE as a potential lipid-lowering agent [17]. Our recent studies suggested that AE has a favorable therapeutic effect on high-fat diet-induced HPL by adjusting metabolic disorders by urinary metabolomics combined with pattern recognition analysis [21]. However, the exact molecular mechanisms of AE in regulating lipid metabolism remain unclear. Therefore, the present study mainly focused on this issue and demonstrated for the first time that AE significantly decreased TC and LDL in high-fat diet-induced HPL rats by inhibiting PCSK9 and thereby upregulating LDLR, eventually promoting LDL clearance. Both in vivo and in vitro studies indicated that AE successfully reduced cholesterol in HPL rats and high cholesterol in HepG2 cells. These findings provide new therapeutic candidates for HPL prevention and treatment.
Currently, statin treatment is one of the main drug therapies for patients with hypercholesterolemia. However, several questions remain unsolved. First, a certain number of patients are unable to tolerate statin therapy. Second, the residual cardiovascular risk is still high in patients with diabetes and atherosclerosis despite maximal statin therapy. Therefore, the LDL goal is unattainable with statin-alone therapy. Third, the adverse effects of statins should be seriously considered [22]. Under these circumstances, the novel approach of PCSK9 inhibitors contributes promising effects and relatively few side effects. Mounting evidence strongly supports that PCSK9 is an outstanding target for HPL treatment. Current available studies suggest that PCSK9 inhibitors have the following advantages: (1) they are an effective method for the management of hypercholesterolemia; (2) they provide dramatic lowering of LDL and related cardiovascular outcome benefits for high-risk patients; and (3) they provide an alternative therapeutic regimen for statin-intolerant patients [23]. However, several aspects of the current PCSK9 monoclonal antibody should be considered. For instance, subcutaneous injection of PCSK9 monoclonal (either alirocumab or evolocumab) may cause certain adverse effects. Moreover, its high cost may aggravate the patients’ economic burden [24]. Thus, new oral PCSK9 inhibitors with good bioavailability are urgently needed. Our study focused on this issue and uncovered AE as a potential lead compound with good PCSK9 inhibitory effects. Recent studies also reported that statins could increase circulating PCSK9 [25], which may provide indirect evidence for the unsatisfactory efficacy of statins. This finding indicated that it is necessary to use statins combined with other drugs, such as PCSK9 inhibitors, to better control LDL levels and cardiovascular events.
One interesting finding of the present study is that AE reduced both serum and hepatic cholesterol, whereas atorvastatin only decreased cholesterol in the serum. Moreover, AE affected both the formation and excretion of cholesterol. The serum HDL value was significantly increased in HPL rats and reduced after AE treatment, although no significance was found. Similarly, previous studies also reported higher serum HDL levels in high-fat diet-fed rats [26]. However, the underlying mechanisms remain unclear. Our data that AE failed to affect HMG-CoA reductase activity suggest that the mechanism by which AE lowered cholesterol is different from that of statins. It is well known that SCAP is a cholesterol-binding endoplasmic reticulum membrane protein. Many previous studies have revealed that SCAP is required to activate SREBP transcription factors, thereby regulating genes involved in lipid biosynthesis [27]. Moreover, SCAP was also found to regulate lipid clearance by modulating the expression of PCSK9 and LDLR [28]. In our study, AE was found to inhibit SREBP-2 and HNF1α, thereby repressing PCSK9 while upregulating LDLR expression in HPL rat livers. These results indicated that AE acted through regulating the SREBP-2- and HNF1α-dependent PCSK9 pathway. Similarly, previous studies showed that emodin ameliorates dyslipidemia and improves lipid and glucose metabolism in high-fat diet-induced obese mice by regulating the AMP-activated protein kinase and SREBP pathways [29]. However, it should be emphasized that the chemical structure of emodin is different from AE, although they belong to the same family. Similarly, berberine treatment inhibited HNF1α protein and PCSK9 mRNA and protein levels in HepG2 cells, which was blocked by ubiquitin-proteasome inhibitors [11]. Other studies also reported that berberine derivatives have similar effects regarding the treatment of HPL through the inhibition of PCSK9 and upregulation of LDLR expression [30]. These findings indicated that it is interesting to screen other PCSK9 inhibitors from pure chemicals of Traditional Chinese Medicine.
Previous studies reported that atorvastatin significantly reduced TG [31]. In contrast, as shown in Fig. 2b of our present study, AE did not significantly decrease hepatic TG levels, whereas atorvastatin had a tendency to decrease TG compared to the untreated HPL group. These results are consistent with the study by Micháliková et al. [32], which also did not detect a significant effect of atorvastatin on TG levels. Explanations for the controversial results may lie in the differences in diet and treatment duration and daily dose size. The relatively minor effects of atorvastatin may be due to the imbalance between the duration of high-fat diet administration and the duration of treatment in our study.
Generally, a high-fat diet usually increases body weight. Figure 2c shows that a high-fat diet for 4 weeks increased body weight compared to the control group in our present study. However, the body weight was dynamically changed during 6 weeks of treatment with AE or atorvastatin. As shown in Fig. 2d, we found that the body weight in HPL rats was higher than that in control rats in the first 3 weeks but decreased from the 4-week timepoint; thus, the body weight after 6 weeks of treatment was not significantly different among these groups. We have established a hyperlipidemic rat model using this high-fat diet for several batches of rats, which all successfully induced high serum triglyceride and cholesterol without increasing body weight at the endpoint [33]. We noticed that the food intake in high-fat diet rats, especially the last 2 weeks, was less than that in the control rats, which may be the reason for the reduction in body weight. This phenomenon was also reported in a previous study [34].
The present study has several limitations. First, several clinical studies have found that atorvastatin increased circulating PCSK9 levels dose-dependently in patients. However, our present study found that atorvastatin did not increase hepatic PCSK9 expression. SREBP-2 is an important factor in regulating both PCSK9 and LDLR, which contributes to a self-regulatory system preventing excessive cholesterol uptake to preserve cholesterol homeostasis. On the one hand, low intracellular cholesterol levels activate SREBP-2, leading to increased LDLR gene expression, which enhances LDL-C clearance from the circulation. On the other hand, SREBP-2 also induces the expression of PCSK9, leading to LDLR degradation, thus limiting hepatic cholesterol uptake and increasing circulating LDL-C. Therefore, the PCSK9 level depends on both LDLR and SREBP-2 expression [35]. We found that LDLR protein expression was slightly increased in the atorvastatin group compared with that in the HPL group, which may downregulate hepatic PCSK9 expression via feedback. Of course, concrete mechanisms should be explored in the future. Second, we found that both SREBP-2 and HNF1α were involved in the inhibition of PCSK9 by AE. However, the detailed regulation process between AE and SREBP-2 or HNF1α remains unclear. Nevertheless, these two signals both activated PCSK9, which is the key point of cholesterol degradation.
Our work represents the first to characterize the new pharmacological role of AE in regulating PCSK9 in HPL. More studies are needed to further evaluate the possibility of AE as a new lead compound in HPL drug therapy.
Conclusion
The present work demonstrated for the first time that AE significantly decreased TC and LDL in high-fat diet-induced HPL rats by downregulating PCSK9 and thereby upregulating LDLR, eventually leading to LDL uptake.
Supplementary information
Acknowledgements
This work was supported in part by the Major Program of the National Natural Science Foundation of China (81230081) and the Translational Fund of the Sino-Russian Medical Research Center, Heilongjiang Academy of Medical Sciences (CR201814).
Author contributions
ZMD and PZH designed the study and wrote the manuscript. ZLS, JH, YYZ, HQS, JG, and KYL performed experiments and analyzed the data.
Competing interests
The authors declare no competing interests.
Footnotes
These two authors contributed equally: Zhen-li Su, Peng-zhou Hang
Supplementary information
The online version of this article (10.1038/s41401-020-0392-8) contains supplementary material, which is available to authorized users.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karr S. Epidemiology and management of hyperlipidemia. Am J Manag Care. 2017;23:S139–48. [PubMed] [Google Scholar]
- 3.Nelson RH. Hyperlipidemia as a risk factor for cardiovascular disease. Prim Care. 2013;40:195–211. doi: 10.1016/j.pop.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin SS, Blaha MJ, Blankstein R, Agatston A, Rivera JJ, Virani SS, et al. Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: implications for statin therapy from the multi-ethnic study of atherosclerosis. Circulation. 2014;129:77–86. doi: 10.1161/CIRCULATIONAHA.113.003625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889–934. doi: 10.1016/j.jacc.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Vallejo-Vaz AJ, Robertson M, Catapano AL, Watts GF, Kastelein JJ, Packard CJ, et al. Low-density lipoprotein cholesterol lowering for the primary prevention of cardiovascular disease among men with primary elevations of low-density lipoprotein cholesterol levels of 190 mg/dl or above: analyses from the WOSCOPS (west of scotland coronary prevention study) 5-year randomized trial and 20-year observational follow-up. Circulation. 2017;136:1878–91. doi: 10.1161/CIRCULATIONAHA.117.027966. [DOI] [PubMed] [Google Scholar]
- 7.Banach M, Rizzo M, Toth PP, Farnier M, Davidson MH, Al-Rasadi K, et al. Statin intolerance—an attempt at a unified definition. Position paper from an international lipid expert panel. Arch Med Sci. 2015;11:1–23. doi: 10.5114/aoms.2015.49807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mancini GB, Baker S, Bergeron J, Fitchett D, Frohlich J, Genest J, et al. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian consensus working group update (2016) Can J Cardiol. 2016;32:S35–65. doi: 10.1016/j.cjca.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Bosch X, Poch E, Grau JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361:62–72. doi: 10.1056/NEJMra0801327. [DOI] [PubMed] [Google Scholar]
- 10.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology-divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710–30.. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 11.Dong B, Li H, Singh AB, Cao A, Liu J. Inhibition of PCSK9 transcription by berberine involves down-regulation of hepatic HNF1alpha protein expression through the ubiquitin-proteasome degradation pathway. J Biol Chem. 2015;290:4047–58. doi: 10.1074/jbc.M114.597229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Bartolo B, Scherer DJ, Brown A, Psaltis PJ, Nicholls SJ. PCSK9 inhibitors in hyperlipidemia: current status and clinical outlook. BioDrugs. 2017;31:167–74. doi: 10.1007/s40259-017-0220-y. [DOI] [PubMed] [Google Scholar]
- 13.Kazi DS, Moran AE, Coxson PG, Penko J, Ollendorf DA, Pearson SD, et al. Cost-effectiveness of PCSK9 inhibitor therapy in patients with heterozygous familial hypercholesterolemia or atherosclerotic cardiovascular disease. JAMA. 2016;316:743–53. doi: 10.1001/jama.2016.11004. [DOI] [PubMed] [Google Scholar]
- 14.Rosenson RS, Jacobson TA, Preiss D, Djedjos CS, Dent R, Bridges I, et al. Efficacy and safety of the PCSK9 inhibitor evolocumab in patients with mixed hyperlipidemia. Cardiovasc Drugs Ther. 2016;30:305–13. doi: 10.1007/s10557-016-6666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kastelein JJ, Hovingh GK, Langslet G, Baccara-Dinet MT, Gipe DA, Chaudhari U, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 monoclonal antibody alirocumab vs placebo in patients with heterozygous familial hypercholesterolemia. J Clin Lipidol. 2017;11:195–203 e4. doi: 10.1016/j.jacl.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 16.Jing A, Li-Mei Z, Yan-Jie L, Ben-Zhi C, Yong Z, Bao-Feng Y. A randomized, multicentre, open-label, parallel-group trial to compare the efficacy and safety profile of daming capsule in patients with hypercholesterolemia. Phytother Res. 2009;23:1039–42. doi: 10.1002/ptr.2654. [DOI] [PubMed] [Google Scholar]
- 17.An R, Li Y, Li M, Bai Y, Lu Y, Du Z. Plasma pharmacochemistry combined with pharmacokinetics and pattern recognition analysis to screen potentially bioactive components from Daming capsule using ultra high performance liquid chromatography with electrospray ionization quadrupole time-of-flight mass spectrometry. J Sep Sci. 2015;38:1507–14. doi: 10.1002/jssc.201401269. [DOI] [PubMed] [Google Scholar]
- 18.Lu GD, Shen HM, Chung MC, Ong CN. Critical role of oxidative stress and sustained JNK activation in aloe-emodin-mediated apoptotic cell death in human hepatoma cells. Carcinogenesis. 2007;28:1937–45. doi: 10.1093/carcin/bgm143. [DOI] [PubMed] [Google Scholar]
- 19.Hu B, Zhang H, Meng X, Wang F, Wang P. Aloe-emodin from rhubarb (Rheum rhabarbarum) inhibits lipopolysaccharide-induced inflammatory responses in RAW264.7 macrophages. J Ethnopharmacol. 2014;153:846–53. doi: 10.1016/j.jep.2014.03.059. [DOI] [PubMed] [Google Scholar]
- 20.Wu YY, Zhang JH, Gao JH, Li YS. Aloe-emodin (AE) nanoparticles suppresses proliferation and induces apoptosis in human lung squamous carcinoma via ROS generation in vitro and in vivo. Biochem Biophys Res Commun. 2017;490:601–7. doi: 10.1016/j.bbrc.2017.06.084. [DOI] [PubMed] [Google Scholar]
- 21.Ji H, Liu Y, He F, An R, Du Z. LC-MS based urinary metabolomics study of the intervention effect of aloe-emodin on hyperlipidemia rats. J Pharm Biomed Anal. 2018;156:104–15.. doi: 10.1016/j.jpba.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 22.Lin XL, Xiao LL, Tang ZH, Jiang ZS, Liu MH. Role of PCSK9 in lipid metabolism and atherosclerosis. Biomed Pharmacother. 2018;104:36–44. doi: 10.1016/j.biopha.2018.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Della Badia LA, Elshourbagy NA, Mousa SA. Targeting PCSK9 as a promising new mechanism for lowering low-density lipoprotein cholesterol. Pharmacol Ther. 2016;164:183–94. doi: 10.1016/j.pharmthera.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Reiss AB, Shah N, Muhieddine D, Zhen J, Yudkevich J, Kasselman LJ, et al. PCSK9 in cholesterol metabolism: from bench to bedside. Clin Sci (Lond) 2018;132:1135–53.. doi: 10.1042/CS20180190. [DOI] [PubMed] [Google Scholar]
- 25.Guo YL, Zhang W, Li JJ. PCSK9 and lipid lowering drugs. Clin Chim Acta. 2014;437:66–71. doi: 10.1016/j.cca.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 26.Tuzcu Z, Orhan C, Sahin N, Juturu V, Sahin K. Cinnamon polyphenol extract inhibits hyperlipidemia and inflammation by modulation of transcription factors in high-fat diet-fed rats. Oxid Med Cell Longev. 2017;2017:1583098. doi: 10.1155/2017/1583098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy BA, Tadin-Strapps M, Jensen K, Mogg R, Liaw A, Herath K, et al. siRNA-mediated inhibition of SREBP cleavage-activating protein reduces dyslipidemia in spontaneously dysmetabolic rhesus monkeys. Metabolism. 2017;71:202–12. doi: 10.1016/j.metabol.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Jensen KK, Tadin-Strapps M, Wang SP, Hubert J, Kan Y, Ma Y, et al. Dose-dependent effects of siRNA-mediated inhibition of SCAP on PCSK9, LDLR, and plasma lipids in mouse and rhesus monkey. J Lipid Res. 2016;57:2150–62.. doi: 10.1194/jlr.M071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Ding L, Song B, Xiao X, Qi M, Yang Q, et al. Emodin improves lipid and glucose metabolism in high fat diet-induced obese mice through regulating SREBP pathway. Eur J Pharmacol. 2016;770:99–109. doi: 10.1016/j.ejphar.2015.11.045. [DOI] [PubMed] [Google Scholar]
- 30.Wu C, Xi C, Tong J, Zhao J, Jiang H, Wang J, et al. Design, synthesis, and biological evaluation of novel tetrahydroprotoberberine derivatives (THPBs) as proprotein convertase subtilisin/kexin type 9 (PCSK9) modulators for the treatment of hyperlipidemia. Acta Pharm Sin B. 2019;9:1216–30.. doi: 10.1016/j.apsb.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao X, Nan Y, Zhao Y, Yuan Y, Ren B, Sun C, et al. Atorvastatin reduces lipid accumulation in the liver by activating protein kinase a-mediated phosphorylation of perilipin 5. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:1512–9. [DOI] [PubMed]
- 32.Micháliková D, Tyukos Kaprinay B, Lipták B, Švík K, Slovák L, Sotníková R, et al. Natural substance rutin versus standard drug atorvastatin in a treatment of metabolic syndrome-like condition. Saudi Pharm J. 2019;27:1196–202. doi: 10.1016/j.jsps.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai Y, Su Z, Sun H, Zhao W, Chen X, Hang P, et al. Aloe-emodin relieves high-fat diet induced QT prolongation via Mir-1 inhibition and Ik1 up-regulation in rats. Cell Physiol Biochem. 2017;43:1961–73.. doi: 10.1159/000484120. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Fan W, Zhang M, Zhang Q, Li L, Wang J, et al. Antiobesity, regulation of lipid metabolism, and attenuation of liver oxidative stress effects of hydroxy-alpha-sanshool isolated from zanthoxylum bungeanum on high-fat diet-induced hyperlipidemic rats. Oxid Med Cell Longev. 2019;2019:5852494. doi: 10.1155/2019/5852494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urban D, Poss J, Bohm M, Laufs U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013;62:1401–8. doi: 10.1016/j.jacc.2013.07.056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.