

Abstract
Evidence to date suggests that β-arrestins act beyond their role as adapter proteins. Arginine vasopressin (AVP) may be a factor in inflammation and fibrosis in the pathogenesis of heart failure. In the present study we investigated the effect of AVP on inflammatory cytokine IL-6 production in murine hearts and the impact of β-arrestin 2-dependent signaling on AVP-induced IL-6 production. We found that administration of AVP (0.5 U/kg, iv) markedly increased the levels of IL-6 mRNA in rat hearts with the maximum level occurred at 6 h. In β-arrestin 2 KO mouse hearts, deletion of β-arrestin 2 decreased AVP-induced IL-6 mRNA expression. We then performed in vitro experiments in adult rat cardiac fibroblasts (ARCFs). We found that AVP (10−9–10−6 M) dose-dependently increased the expression of IL-6 mRNA and protein, activation of NF-κB signaling and ERK1/2 phosphorylation, whereas knockdown of β-arrestin 2 blocked AVP-induced IL-6 increase, NF-κB activation and ERK1/2 phosphorylation. Pharmacological blockade of ERK1/2 using PD98059 diminished AVP-induced NF-κB activation and IL-6 production. The selective V1A receptor antagonist SR49059 effectively blocked AVP-induced NF-κB phosphorylation and activation as well as IL-6 expression in ARCFs. In AVP-treated mice, pre-injection of SR49059 (2 mg/kg, iv) abolished AVP-induced NF-κB activation and IL-6 production in hearts. The above results suggest that AVP induces IL-6 induction in murine hearts via the V1A receptor-mediated β-arrestin2/ERK1/2/NF-κB pathway, thus reveal a novel mechanism of myocardial inflammation in heart failure involving the V1A/β-arrestin 2/ERK1/2/NF-κB signaling pathway.
Keywords: myocardial inflammation, β-Arrestin 2, arginine vasopressin, IL-6, adult rat cardiac fibroblasts, ERK1/2
Introduction
GPCRs transduce various environmental signals and coordinate cellular responses to environmental stimuli. β-Arrestin 1 and 2 are adapter proteins that primarily terminate GPCR signaling via receptor desensitization and internalization [1, 2]. Recent evidence has demonstrated that these proteins also act as scaffold proteins for multiple signaling molecules. For example, these proteins link GPCR signaling to ERK1/2, Akt, and Src kinases [3] and interact with the transcriptional factor NF-κB [4, 5]. Therefore, β-arrestins have emerged as key regulators of cellular functions [6], including controlling GPCR-dependent chemotactic signals in immune cells during inflammatory reactions.
The IL-6 family of cytokines, including IL-6, has been implicated in cardiac pressure overload and postischemic remodeling [7–9]. Recent studies have demonstrated that IL-6 plasma levels are elevated during congestive heart failure [10], which suggests that IL-6 can be used as a marker of inflammation in acute and chronic myocardial injury [11]. The increased production of cytokines, particularly inflammatory cytokines, such as tumor necrosis factor alpha (TNFα), interleukin-1 (IL-1) and IL-6, is at least partially responsible for heart dysfunction in patients with heart failure and cardiac remodeling [12, 13].
Arginine vasopressin (AVP) is secreted in response to hypovolemic or cardiac stress. Accordingly, AVP receptors are also found in some immune cells, such as rat B lymphocytes and thymic epithelial cells [14]. Therefore, AVP may stimulate the production of cytokines and antibodies in an autocrine manner during inflammation. The physiological effects of AVP are mediated via binding to specific membrane receptors on target cells. Three vasopressin receptor subtypes (V1AR, V2R, and V1BR [also termed V3R]) have been identified in humans [15–17]. All of these receptors belong to the GPCR superfamily [18, 19]. However, only V1AR is found in cardiac myocytes [20] and cardiac fibroblasts [21, 22]. Gαq-coupled V1AR typically induces protein kinase C, which is an activator of gene programming for cardiac hypertrophy [23], but it also interacts with GPCR kinase (GRK) isoforms [24], primarily GRK2 [21, 25]. GRKs activate G protein-independent signaling pathways in addition to having a clearly defined role in receptor desensitization [26, 27]. These pathways are related to the regulation of cardiac hypertrophy and apoptosis [28] and the promotion of cardioprotective extracellular signal-regulated kinase 1/2 (ERK1/2) signal transduction via β-arrestin [29]. However, little is known about the inflammatory regulation of β-arrestin 2 in the heart. The present study investigated the effect of AVP on IL-6 production in murine hearts and the impact of β-arrestin 2-dependent signaling on AVP-mediated IL-6 production.
Materials and methods
Drug and reagents
The Animal Care and Use Committee of Nantong University approved all of the procedures. Adult Sprague-Dawley rats were obtained from the Animal Center of Nantong University (Nantong, China). β-Arrestin 2 KO mice were gifted by the laboratory of Dr. Lefkowitz (Duke University, Durham, NC, USA). Dulbecco’s modified Eagle’s medium (DMEM), penicillin and streptomycin were purchased from Invitrogen (Gaithersburg, MD, USA). Arginine vasopressin (AVP, V9879) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The V1AR selective antagonist SR49059 was purchased from Tocris Bioscience (Minneapolis, MN, USA). A rat IL-6 ELISA kit (#BMS625) was purchased from ThermoFisher Scientific (Waltham, MA, USA). The adenovirus containing GFP was a gift from Dr. Yibin Wang (University of California, Los Angeles, CA, USA). The adenovirus containing human β-arrestin 2 was created by Fubio (Shanghai, China). The lentivirus containing shRNA targeting β-arrestin 2 was purchased from Genechem (Shanghai, China). NF-κB luciferase (E8491) and renilla luciferase (E2231) were purchased from Promega (Madison, MI, USA). Anti-P-NF-κB (#3033) and anti-β-arrestin 2 (4674S) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against NF-κB (Sc-109) and GAPDH (Sc-25778) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
Animals
Male rats and mice (8–12 weeks old) were administered 0.5 U/kg AVP via the tail vein. Murine heart tissues were harvested for RNA and protein isolation at designated timepoints after AVP administration. The Animal Care and Use Committee of Nantong University approved all of the procedures.
Cell culture and adenoviral or lentiviral infection
ARCFs were prepared from the hearts of adult (8- to 12-week-old) Sprague-Dawley rats as previously described [30]. The cells were cultured in DMEM containing 10% FBS and 100-U/mL penicillin-streptomycin for 4–5 days before being passaged. When the cells reached 90% confluence, they were passaged at a ratio of 1:3 with 0.25% trypsin. Experiments were consistently performed on cells from passages 3–5. The serum was replaced with serum-free medium, and the ARCFs were transfected with an adenovirus containing β-arrestin 2 or a lentivirus containing shRNA targeting β-arrestin 2 at an MOI of 100. Medium with or without treatment agent was added after 24 h according to the experimental design.
Transient transfection and luciferase gene reporter assay
ARCFs (250 × 103 cells/well) were plated in 12-well plates. The reporter plasmid was transfected when the confluency of the growing cells reached 80%–90% using Lipofectamine 2000. Cotransfection was performed with 0.5 μg of an NF-κB luciferase plasmid and 0.5 μg of a renilla luciferase plasmid. The cells transfected with the NF-κB-luciferase plasmid (0.5 μg) and the renilla control vector (0.5 μg) but not incubated with AVP were used to measure basal activity. The transfected cells were further cultured for 6 h, serum-starved overnight and cultured in the presence of AVP for 24 h. A luminometer (ThermoFisher Scientific, Waltham, MA, USA) was used to measure luciferase activation based on the manufacturer’s instructions.
Quantitative PCR (qPCR) for analyzing IL-6 mRNA levels and RT-PCR for analyzing the levels of vasopressin receptor subtypes
RT-PCR Fresh tissue samples were collected from 12-week-old male Sprague-Dawley rats. The messenger RNA (mRNA) levels of V1AR, V1BR, and V2R in ARCFs and the neonatal rat brain were assessed using RT-PCR, as described previously [22]. An RNA extraction kit was used to extract total RNA from fresh tissues and cultured ARCFs. A reverse transcription synthesis kit (TaKaRa, Dalian, China) was used to prepare complementary DNA (cDNA). All procedures were performed based on the manufacturer’s instructions. The reverse-transcribed cDNA was amplified using PCR under standard conditions with specific primers. cDNA amplification was performed with an initial step of 3 min at 95 °C followed by 35 cycles of 30 s at 94 °C, 30 s at 55–60 °C, 1 min at 72 °C, and a final extension of 7 min at 72 °C. The PCR products were analyzed using agarose gel electrophoresis and scanning densitometry. Primers for the receptor subtypes and β-actin are shown in Table 1.
Table 1.
The primers for murine V1AR, V2R, V1BR, IL-6, GAPDH, and β-actin
| Gene | Primers | Sizes (bp) |
|---|---|---|
| V1AR (NM_053019.2) | F:5′-GTTGCTGGCTTCCTTGAACA-3′ | 181 |
| R:5′-TGGGCTCCGGTTGTTAGAAT-3′ | ||
| V1BR (NM_001289800.1) | F:5′-CTGCAACCCCTGGATCTACA-3′ | 191 |
| R:5′-TGAGGTTAAGGCTGAGTCGG-3′ | ||
| V2R (NM_019136.1) | F:5′-GCAGATGGTGGGCATGTATG-3′ | 217 |
| R:5′- AAACACCCCACTGCCATTTC-3′ | ||
| β-actin (NM_031144.3) | F:5′-TTGCCCTAGACTTCGAGCAA-3′ | 446 |
| R:5′-GATGTGGATCAGCAAGCAGG-3′ | ||
| IL-6 (NM_012589) | F:5′-AGTTGCCTTCTTGGGACTGA-3′ | 222 |
| R:5′- AGCGATGATGCACTGTCAGA-3 | ||
| GAPDH (M17701) | F:5′-TTCAATGGCACAGTCAAGGC-3′ | 101 |
| R:5′-TCACCCCATTTGATGTTAGCG-3′ | ||
| IL-6 (NM_031168.2) | F:5′-AGATACAAAGAAATGATGGATG-3′ | 190 |
| R:5′-TGGTTGAAGATATGAATTAGAG-3′ | ||
| β-actin (NM_007393.5) | F:5′-GGCTGTATTCCCCTCCATCG-3′ | 154 |
| R:5′-CCAGTTGGTAACAATGCCATGT-3′ |
qPCR ARCFs (1 × 106 cells/well) were plated in 10-cm culture dishes. After the cells reached quiescence at 90% confluency, total RNA was extracted from the cultured NRCFs using Trizol reagent (Promega, Madison, WI, USA) after stimulation with AVP and PD98059, according to the experimental design. cDNA was amplified using a Step-one Real Time PCR System (Applied Biosystems, Foster City, CA, USA). The double-stranded DNA-specific dye SYBRGreen I was incorporated into the PCR buffer provided in the QuantiTech SYBR PCR kit (Qiagen, Valencia, CA, USA) to allow for the quantitative detection of the PCR product in a 25-μL reaction volume. The primer sequences used to detect murine IL-6, GAPDH, and β-actin are shown in Table 1. The following temperature profile was used for the reaction: 95 °C for 1 min, 40 cycles of denaturation at 95 °C for 5 s, annealing at 55 °C for 1 min, and extension at 72 °C for 30 s. GAPDH and β-actin were used as RNA loading controls for rat and mouse IL-6, respectively. The data were analyzed using the ΔCT method (2-ΔΔCT) [31].
Western blotting
Cells were treated with AVP for 0–120 min, washed twice with ice-cold PBS and lysed with 250 μL of ice-cold lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, 5 mg·mL−1 leupeptin, 1 mM phenylmethylsulfonyl fluoride, 20 mg/mL aprotinin, 1 mM NaF, and 1 mM Na3VO4). After centrifugation at 12 000 × g for 15 min at 4 °C, equal amounts of total cell lysates (20 μg of protein) were loaded onto 4%–12% SDS-PAGE gels, and immunoblotting for phosphorylated NF-κB, NF-κB, β-arrestin 2, phosphorylated ERK1/2, and GAPDH was performed.
ELISA for IL-6
Heart homogenates or cell culture supernatant samples (50 μL) were used to measure IL-6 using an ELISA kit, as described in the kit manual. The analytical sensitivity of the kit was 12.0 pg/mL and 31.3–2000 pg/mL, respectively.
Statistical analysis
The data and statistical analyses complied with the recommendations on experimental design and analysis in pharmacology [32]. An experimenter who was blinded to the experimental protocol randomized the animals into groups in the animal experiments on AVP-induced IL-6 production. The number of rats in each group in the in vivo study was less than needed to reduce the number of euthanized animals because the difference between the control and treatment groups was significant in a preliminary study. Randomization was not applicable in any of the other experiments. Comparisons were performed using one- or two-way ANOVA followed by Bonferroni’s test. All values are presented as the means ± SEM. P < 0.05 was considered statistically significant.
Results
β-Arrestin 2 mediates AVP-induced IL-6 expression in murine hearts
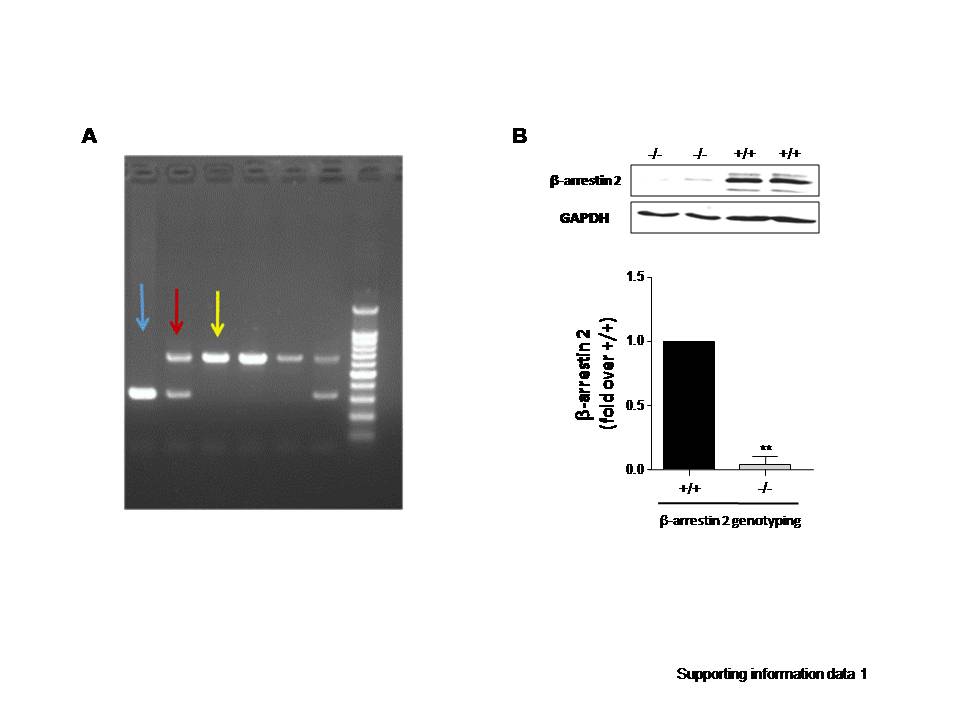
To determine whether AVP evokes IL-6 induction in the murine myocardium, AVP (0.5 U·kg−1) was administered via the tail veins of the rats. The level of troponin I in the serum was not increased 24 h after AVP administration (data not shown). AVP-evoked IL-6 expression in the heart, and the maximum expression of IL-6 mRNA and protein occurred 6 h (Fig. 1a, b) after AVP administration. Notably, the deletion of β-arrestin 2 reduced AVP-induced IL-6 mRNA expression (Fig. 1c) in β-arrestin 2 KO mouse hearts (Supporting Information Data S1).
Fig. 1.

Administration of AVP promotes the expression of IL-6 mRNA and protein in murine hearts. Animals were administered 0.5 U/kg AVP or 0.9% NaCl as vehicle via the tail vein. Total RNA and protein lysates were isolated from the myocardium of the left ventricle to measure IL-6 mRNA at 6 h after the administration of AVP. a, b AVP induced the elevation of IL-6 mRNA and protein levels in the rat myocardium. ELISA for IL-6 protein and qPCR for IL-6 mRNA were performed in triplicate to ensure the reliability of the individual values. c, Deletion of β-arrestin 2 reduced the AVP-induced IL-6 elevation in the mouse myocardium. The upper panels in (a, c) are representative images of the qPCR products, and the lower panels show the data expressed as the means ± SEM of four (rats) or five (mice) separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle, ##P < 0.01
To further investigate the mechanistic role of β-arrestin 2 in AVP-mediated IL-6 induction, we performed in vitro experiments using cultured adult rat cardiac fibroblasts (ARCFs). AVP-induced IL-6 expression in a dose- and time-dependent manner in cultured ARCFs (Fig. 2). The silencing of β-arrestin 2 with shRNA (Fig. 3a and Supporting Information Data 3) or the overexpression of β-arrestin 2 with a lentiviral vector (Fig. 3b and Supporting Information Data 2) were used to manipulate the levels of β-arrestin 2. The silencing of β-arrestin 2 suppressed the AVP-induced mRNA and protein levels of IL-6 in ARCFs (Fig. 3c, d). Notably, the overexpression of β-arrestin 2 did not alter the induction of IL-6 expression. These results suggest that β-arrestin 2 is necessary, but not sufficient, for the AVP-induced induction of IL-6 in murine hearts.
Fig. 2.

AVP induces IL-6 expression in cultured adult rat cardiac fibroblasts (ARCFs). a, b AVP induced the expression of IL-6 mRNA and protein in a dose-dependent manner in ARCFs. Starved cells in 6-well plates were stimulated with 10−10 M–10−6 M AVP for 12 h (a) or 24 h (b), and harvested for total RNA or cellular supernatant. c, d AVP induced IL-6 expression in a time-dependent manner in ARCFs. Starved cells in 6-well plates were incubated with 10−6 M AVP for 0–48 h and harvested for total RNA. IL-6 mRNA was measured by qPCR as described in the methods. The supernatant was collected to measure IL-6 using ELISA as described in the methods. The data are expressed as the means ± SEM of three separate experiments. $$P < 0.01 for time-course (repeated two-way ANOVA, n = 3). *P < 0.05, **P < 0.01 vs. control, n = 4
Fig. 3.

β-Arrestin 2 mediates AVP-induced IL-6 expression in ARCFs. a, b The silencing of β-arrestin 2 with shRNA (a) or the overexpression of β-arrestin 2 with a lentiviral vector (b) was used to manipulate the levels of β-arrestin 2. After the cells were transfected with lentiviral-shRNA for β-arrestin 2 or adenoviral containing β-arrestin 2 for 48 h, the cellular lysates were collected to determine the expression of β-arrestin 2 with western blot. c, d The silencing of β-arrestin 2 suppressed the AVP-induced mRNA and protein levels of IL-6 in ARCFs. After the cells were transfected with lentiviral or adenoviral for 48 h, the starved cells were stimulated with 10−6 M AVP for 12 h or 24 h, respectively and harvested for total RNA and supernatant. IL-6 mRNA was measured by qPCR as described in the Methods. The supernatant was collected to measure IL-6 using ELISA as described in the Methods. Notably, the overexpression of β-arrestin 2 did not alter the induction of IL-6 expression. *P < 0.05, **P < 0.01 vs. control, n = 4. ##P < 0.01. ns: no significance
β-Arrestin 2 is required for AVP-induced NF-κB activation
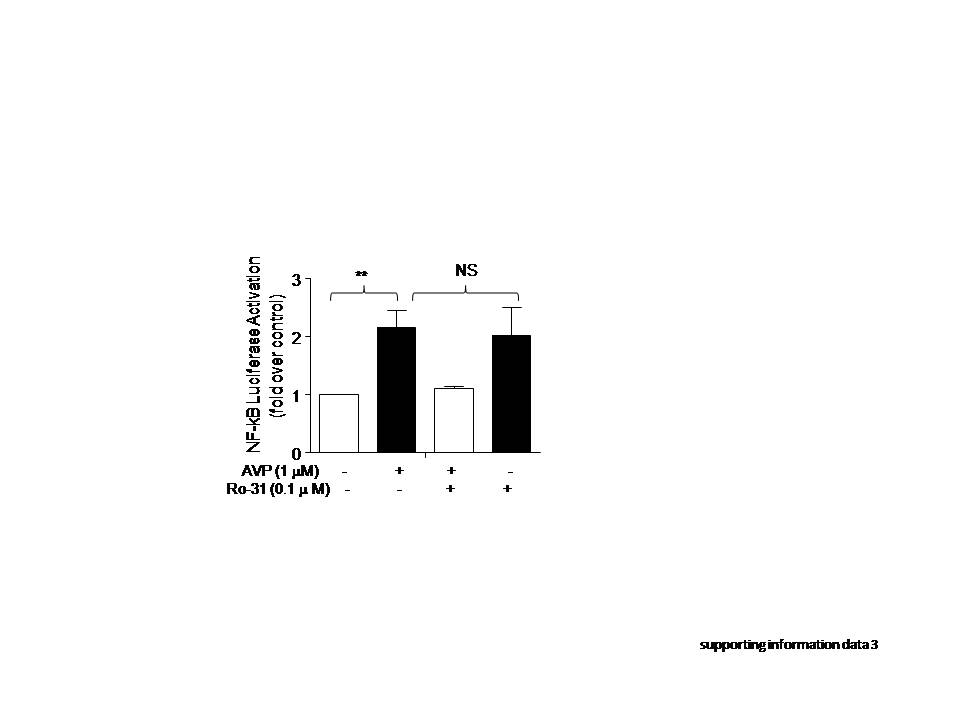
Our previous study demonstrated that AVP induces IL-6 production via NF-κB signaling in neonatal rat cardiac fibroblasts [33] and cultured ARCFs (Fig. 4). To determine whether β-arrestin 2 is involved in the AVP-mediated induction of NF-κB activation, we measured the levels of phosphorylated NF-κB and the luciferase activity of NF-κB under different conditions. We found that the silencing of β-arrestin 2 by shRNA efficiently suppressed AVP-induced NF-κB phosphorylation (Fig. 5a, b) and NF-κB luciferase activity (Fig. 5c). In contrast, the overexpression of β-arrestin 2 did not alter the activation or phosphorylation levels of NF-κB (Fig. 5b, c). Notably, AVP-evoked NF-κB activity in a PKC-independent manner, as a PKC inhibitor did not block AVP-induced NF-κB activity in ARCFs (Supporting information Data 3). These results suggest that β-arrestin 2 is necessary, but not sufficient, for AVP-evoked NF-κB signaling, which is consistent with its effect on AVP-induced IL-6 production.
Fig. 4.

AVP induced IL-6 production via NF-κB in ARCFs. a AVP evoked the activation of NF-κB in a dose- and time-dependent manner. Cells transfected with the NF-κB luciferase reporter plasmid were incubated with 10−6 M AVP for 0–36 h or with AVP for 24 h. Luciferase activation was measured as described in the kit manual. The data are the averages of three separate experiments. *P < 0.05, **P < 0.01 vs. control. b AVP induced the phosphorylation of NF-κB in a time-dependent manner. Starved cells were stimulated with 10−6 M AVP for 0–120 min. Cellular lysates were collected to measure the phosphorylation of NF-κB using an anti-phosphorylated NF-κB antibody, as described in the Methods. The upper panel is a representative blot, and the lower panel shows the average data of three separate experiments. #P < 0.05 for time-course (repeated two-way ANOVA, n = 3). *P < 0.05, **P < 0.01 vs. control (one-way ANOVA). c, d The inhibition of NF-κB by PDTC abolished the AVP-induced phosphorylation and activation of NF-κB-luciferase. Starved cells were pretreated with 50 µM PDTC for 1 h and further incubated with 1.0 µM AVP for 1 h. Cellular lysates were collected to measure NF-κB phosphorylation using an anti-phosphorylated NF-κB antibody, as described in the Methods. The activation of luciferase in cells treated with 1.0 µM of AVP for 24 h was assayed as described in the Methods. e The inhibition of NF-κB by PDTC suppressed the AVP-induced levels of IL-6 mRNA. Starved cells were pretreated with 50 µM PDTC for 1 h and further incubated with 1.0 µM AVP for 24 h. The supernatants were harvested to measure IL-6 mRNA. The average data are from four separate experiments. *P < 0.05, **P < 0.01 vs. control, ##P < 0.01 vs. AVP alone. Notably, PDTC alone had no effect on the AVP-induced phosphorylation and activation of NF-κB or on IL-6 mRNA levels
Fig. 5.

β-Arrestin 2 mediates AVP-induced NF-κB phosphorylation and activation in ARCFs. a, b The silencing of β-arrestin 2 using shRNA reduced the AVP-evoked phosphorylation of NF-κB, but overexpression did not further enhance the AVP-evoked phosphorylation of NF-κB. After lentiviral and adenoviral infection for 48 h, the starved cells were stimulated with 10−6 M AVP for 60 min. Cellular lysates were analyzed for NF-κB phosphorylation using an anti-phosphorylated NF-κB antibody. The upper images in a and b are representative images, and the lower panels in a and b show the average data from three separate experiments. *P < 0.05, **P < 0.01 vs. control. ns: no significance. c The effects of β-arrestin 2 on AVP-evoked changes in NF-κB luciferase activity. After lentiviral and adenoviral infection for 48 h, ARCFs were transfected with a plasmid containing NF-κB p65 luciferase, and the starved cells were stimulated with or without 1.0 µM AVP for 24 h. **P < 0.01 vs. control. ##P < 0.01 vs. lenti-scramble. ns: no significance
β-Arrestin 2-dependent ERK1/2 phosphorylation is essential for AVP-induced NF-κB and IL-6 production
β-Arrestin 2 is a critical component in the mediation of the β2-adrenergic stimulation of ERK1/2 signaling [34]. We next tested our hypothesis that β-arrestin 2-dependent ERK1/2 signaling is responsible for AVP activity in the heart. AVP induced the phosphorylation of ERK1/2 in a PD98059-sensitive manner in cultured ARCFs and intact rat hearts (Fig. 6). The deletion of β-arrestin 2 in β-arrestin 2 knockout mouse hearts or using β-arrestin 2 shRNA or β-arrestin 2 adenovirus reduced the AVP-evoked phosphorylation of ERK1/2, and the overexpression of β-arrestin 2 did not further enhance AVP-induced ERK1/2 activation. These results suggest that β-arrestin 2 is required for the AVP-induced phosphorylation of ERK1/2. Accordingly, β-arrestin 2 deficiency and ERK1/2 inhibition with PD98059 abolished the AVP-induced activation of NF-κB (Fig. 7a) and induction of IL-6 expression (Fig. 7b) in mouse hearts. A PD98059-sensitive pathway mediated AVP-induced IL-6 expression and NF-κB phosphorylation in mouse hearts, which was evidenced by the inhibition of NF-κB phosphorylation and the induction of IL-6 by PD98059 (Fig. 8). Taken together, these data support the hypothesis that β-arrestin 2-mediated ERK1/2 signaling contributes to the AVP-induced NF-κB/IL-6 inflammatory response in the heart.
Fig. 6.

AVP induces the phosphorylation of ERK1/2 via a PD98059-sensitive pathway in cultured adult cardiac fibroblasts (a, b) and rat heats (c). a, b Treatment with 0.1 µM AVP evoked the phosphorylation of ERK1/2 in a time-dependent manner (a). These effects were inhibited by 10 µM PD98059 (b). The upper panels in a and b are representative images and the lower panels show the data expressed as the means ± SEM of three separate experiments. P < 0.05 for time-course (repeated two-way ANOVA), *P < 0.05, **P < 0.01 vs. control. c AVP evoked phosphorylation of ERK1/2 in rat hearts. After AVP (0.5 U/kg) was injected into the tail vein for 0–5 h, the left ventricles were harvested to obtain ventricular lysates to detect phosphorylated and total ERK1/2. The upper panels are representative images, and the lower panels show the data expressed as the means ± SEM of three separate experiments. *P < 0.05, **P < 0.01 vs. control
Fig. 7.

β-Arrestin 2 is required for AVP phosphorylation in murine hearts and ARCFs. a β-Arrestin 2 deficiency reduced AVP-evoked ERK1/2 phosphorylation in murine hearts. After AVP (0.5 U/kg) was injected into the tail veins of the mice (wild-type (+/+), heterozygous (+/−) and homozygous (−/−) knockout mice) for 5 h, the left ventricles were harvested to obtain ventricular lysates to detect phosphorylated and total ERK1/2. The upper panels are representative blots, and the lower panels show the data expressed as the means ± SEM of five separate experiments. *P < 0.05 vs. control, ##P < 0.01. b, c The silencing of β-arrestin 2 diminished the AVP-evoked phosphorylation of ERK1/2 (b), and the overexpression of β-arrestin 2 did not enhance the phosphorylation of ERK1/2 (c). After the manipulation of β-arrestin 2 as described in Fig. 3, the cells were treated with 1.0 µM AVP for 15 min. The upper panels are representative blots, and the lower panels show the data expressed as the means ± SEM of three separate experiments. *P < 0.05, **P < 0.01 vs. control, ##P < 0.01. ns: no significance. Adeno-GFP vs. Adeno- β-arrestin 2 in the presence of AVP
Fig. 8.

PD98059-sensitive pathway mediates AVP-induced NF-κB phosphorylation and IL-6 expression in mouse hearts. Pretreatment with PD98059 or the deletion of β-arrestin 2 abolished AVP-induced NF-κB phosphorylation (a) and IL-6 expression (b). After pretreatment with 3 mg/kg PD98059 for 1 h, mice were administered 0.5 U/kg AVP via the tail vein. The hearts were harvested to measure mRNA and NF-κB phosphorylation at 6 h after the administration of AVP. The upper panels are representative blots, and the lower panels show the data expressed as the means ± SEM of five separate experiments. *P < 0.05 vs. vehicle, ##P < 0.01 vs. AVP alone
V1AR mediates AVP-induced NF-κB signaling and IL-6 production
We used RT-PCR and demonstrated that the V1A receptor is the only vasopressin receptor subtype in ARCFs (Fig. 9a). Notably, the V1A receptor-selective inhibitor SR49059 efficiently blocked AVP-induced NF-κB phosphorylation with an IC50 of 3.20 ± 0.13 nM (Fig. 9b), NF-κB activation with an IC50 of 1.25 ± 0.44 nM (Fig. 9c), and IL-6 expression with an IC50 of 0.55 ± 0.16 nM in ARCFs (Fig. 9d. Notably, 1 μM of SR49059 had no effect on basal IL-6 expression or NF-κB activation in ARCFs. Similarly, pretreating mice with 2 mg/kg SR49059 prior to AVP administration (0.5 U/kg) diminished NF-κB activation (Fig. 9f) and IL-6 production (Fig. 9e, g).
Fig. 9.

V1AR mediates AVP-induced NF-κB activation and IL-6 induction in ARCFs and rat hearts. a The V1A receptor subtype, but not the V1B or the V2 subtype, exists in ARCFs, as measured using RT-PCR. Total RNA was isolated from ARCFs and rat brains. RT-PCR was performed with primers designed to target each subtype of the AVP receptor. Similar results were reproduced at least three times. b–d The inhibition of the V1A subtype by SR49059 abolished AVP-evoked NF-κB phosphorylation (b) and activation (c) and IL-6 mRNA production (d) in ARCFs. Starved cells were pretreated with 0.1 nM-1.0 μM SR49059 (a V1A blocker) for 1 h and further stimulated with 1.0 μM AVP for 1 h (b), 24 h (c) and 12 h (d). The upper panel in b is a representative blot, and the lower panel shows the data expressed as the means ± SEM of three separate experiments. **P < 0.01 vs. control, ##P < 0.01 vs. AVP alone. e–g SR49059 blocked the AVP-induced expression of IL-6 and NF-κB phosphorylation in rat hearts. After being pretreated with SR49059 (2.0 mg·kg−1, ip) for 1 h, the animals were administered 0.5 U· kg−1 AVP via the tail vein for 6 h. The left ventricle was collected to measure NF-κB phosphorylation at 6 h after the administration of AVP. The data are expressed as the means ± SEM of four separate experiments. *P < 0.05 vs. vehicle, #P < 0.05, ##P < 0.01 vs. AVP alone. SR: SR49059
Discussion
The cytokine IL-6 is important for immune system regulation, inflammatory responses and cardiovascular remodeling. IL-6 has a low baseline mRNA expression level in cardiac fibroblasts and is absent in cardiomyocytes, but IL-6 levels are stimulated by β2AR [35], hypoxia [36] or coculture with macrophages [37]. The following results were found in the present study: (1) AVP increased the mRNA and protein levels of IL-6 in murine hearts; (2) the silencing or deletion of β-arrestin 2 reduced AVP-induced IL-6 production, NF-κB activation and ERK1/2 phosphorylation; (3) the pharmacological inhibition of ERK1/2 signaling diminished AVP-induced NF-κB activation and IL-6 production; and (4) the blockade of the V1A receptor by the selective antagonist SR49059 abolished AVP-evoked NF-κB phosphorylation and IL-6 induction in intact hearts and ARCFs.
AVP is an antidiuretic hormone that is secreted by the hypothalamus-pituitary-adrenal axis. AVP significantly affects vasoconstriction, immune regulation and body temperature regulation. Ventricular remodeling occurs when end-stage heart failure develops, and inflammation is an important mechanism for the development of ventricular remodeling. When heart failure occurs, the concentration of catecholamines increases, which induces cardiac fibroblasts to secrete the pro-inflammatory factor IL-6 via β2 adrenergic receptors [38]. IL-6 is involved in the remodeling of the extracellular matrix of cardiomyocytes, which induces or aggravates the development of ventricular remodeling, which promotes chronic heart failure [39]. The level of AVP may be used as an early warning sign of the disease, and it may provide new ideas for the treatment of cardiovascular diseases in the future.
GPCR- or stress-stimulated IL-6 secretion from cardiac fibroblasts may lead to cardiac inflammation, fibroblast proliferation and cardiac remodeling. Data from our and other studies have demonstrated that AVP promotes the proliferation of cardiac fibroblasts [21, 40–43]. We report for the first time that AVP induces IL-6 production in the murine myocardium. AVP-induced transforming growth factor-beta 1 (TGF-beta 1) secretion is responsible for cardiac fibroblast-myofibroblast transformation [44], but whether the induction of IL-6 plays a role in cardiac fibroblast proliferation requires further study.
A large number of studies have confirmed that β-arrestin is primarily involved in negative feedback regulation, such as the desensitization and internalization of GPCRs and seven transmembrane receptors (7TMRs), and recent studies have demonstrated that β-arrestin also participates in G protein-independent signaling pathways, which also mediate the physiological and pathological functions of cells. For example, β-arrestin 1 and β-arrestin 2 mediate protective β1-adrenergic signaling in cardiac myocytes [29]. Due to their various biochemical functions in the regulation of critical physiological and pathophysiological processes, including inflammation, β-arrestins have become a drug target in human diseases, such as heart failure. Heart failure is marked by increased AVP levels in the systemic circulation and local cardiac tissues [43]. Therefore, the present findings highlight IL-6 as the effector cytokine downstream of β-arrestin 2 that may link elevated AVP levels to inflammatory cardiac damage and, ultimately, cardiac remodeling.
β-Arrestin plays a regulatory role as a scaffold protein in a variety of cell models and pathological conditions. The cardioprotective effect of β-arrestin 2 may occur via GPCR-dependent [45] and GPCR-independent [46, 47] signaling pathways. It was recently reported that β-arrestin regultes the immune response in immune cells via the NF-κB signaling pathway. β-Arrestin 2 directly interacts with IκBα (an NF-κB inhibitor and the key molecule in innate and adaptive immunity) [5] and prevents the phosphorylation and degradation of IκBα [4, 5]. Consequently, β-arrestin 2 effectively modulates NF-κB activation and the expression of NF-κB target genes [4, 5]. Moreover, the stimulation of β2-adrenergic receptors significantly enhances the interaction between β-arrestin 2 and IκBα and greatly promotes the stabilization of IκBα by β-arrestin 2, which indicates that β-arrestin 2 mediates crosstalk between β2-adrenergic receptors and NF-κB signaling pathways [4]. This interaction suggests a novel mechanism for the regulation of the immune system by the sympathetic nervous system. In contrast, Witherow et al. found that the overexpression of β-arrestin 1 or β-arrestin 2 leads to a marked inhibition of NF-κB activity. Conversely, the suppression of the expression of β-arrestin 1, but not of β-arrestin 2, using RNA interference leads to a threefold increase in TNF-stimulated NF-κB activity [5]. The direct interaction between β-arrestin and NF-κB regulates NF-κB signaling, and the present study demonstrates that the phosphorylation of NF-κB is mediated by β-arrestin 2 signaling via ERK1/2 activation in cardiac cells.
It is well established that NF-κB regulates the expression of inflammatory factors (including IL-6) and the initiation and progression of myocardial fibrosis [48, 49]. The present study demonstrated that AVP, as an endogenous pro-inflammatory factor, activates NF-κB in fibroblasts, as evidenced by NF-κB phosphorylation, nuclear translocation and luciferase activity. PDTC is an inhibitor of NF-κB that inhibits the inflammatory effect of AVP to a certain extent via the inhibition of the activation of NF-κB p65 and alleviates AVP-induced inflammation and organ damage. Therefore, the proinflammatory effect of AVP depends on the regulation of NF-κB to some extent. By targeting the mechanisms of NF-κB regulation, we hope to develop new ideas for the treatment of inflammation and myocardial fibrosis in cardiovascular diseases.
There are three vasopressin receptor subtypes, namely, V1A, V1B, and V2. V1B is widely distributed in the central system, and V2 is concentrated in the renal collecting duct. Early studies by our laboratory revealed that only V1A receptors are present in neonatal rat cardiac fibroblasts. The distribution and function of the three receptors are different. The primary roles of the V1A receptor are vasoconstriction, platelet aggregation, blood pressure regulation, and hepatic glycogen metabolism [50]. The V1A receptor mediated the AVP-induced inflammatory response in this experiment, and its selective antagonist SR49059 significantly inhibited this response. Our data further demonstrate that a selective V1A receptor blocker efficiently abolishes AVP-induced NF-κB signaling and IL-6 production in intact hearts, which suggests that the V1A subtype mediates AVP-evoked inflammation in the murine myocardium.
β-Arrestin 2/ERK1/2/NF-κB signaling may not be specific to the V1A receptor because cardiomyocytes also contain other Gq-coupled receptors, which could induce the activation of such a pathway and participate in the inflammatory response in the heart. Notably, the Gq-PKC pathway was not involved in the V1A/AVP-mediated activation of NF-κB in our study (as shown in Supplementary Information Data 3) or in cellular protection via a PKC-independent pathway in our previous study [9]. However, the AVP-mediated activation of ERK1/2 has been previously demonstrated in H9c2 cells [9, 51] and cardiomyocytes [52]. Therefore, further study on whether different signaling mechanisms between Gq-PKC and β-arrestin mediates the diverse biological functions evoked by AVP is valuable because AVP is increased during the development of heart failure.
Conclusion
In summary, our data reveal an important role of β-arrestin 2 in the AVP-mediated inflammatory response, which highlights a novel inflammatory mechanism whereby AVP induces IL-6 production in intact murine hearts via a β-arrestin 2/ERK1/2/NF-κB pathway. These findings shed new light on our molecular understanding of inflammation in heart failure and possibly other cardiac stress conditions.
Supplementary Information
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank Dr. Yibin Wang (University of California, Los Angeles) for kindly providing the β-GFP adenovirus. This study was supported by the National Natural Science Foundation of China (No. 81370345, No. 81541008, No. 817790400) and the Nantong University Co-Innovation Program (NTU 2016-1).
Author contributions
SZS and WZZ contributed to the conception of the work, the interpretation of the data and the critical revision of the paper. SZS, NY, XFZ, QZ, LLZ, EAN and HC contributed to data acquisition. SZS and WZZ drafted and finalized the paper. All authors contributed to the final approval of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Shu-zhen Sun, Hong Cao
Supplementary information
The online version of this article (10.1038/s41401-019-0292-y) contains supplementary material, which is available to authorized users.
References
- 1.Zhang J, Ferguson SS, Barak LS, Aber MJ, Giros B, Lefkowitz RJ, et al. Molecular mechanisms of G protein-coupled receptor signaling: role of G protein-coupled receptor kinases and arrestins in receptor desensitization and resensitization. Recept Channels. 1997;5:193–9. [PubMed] [Google Scholar]
- 2.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–65. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 3.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 4.Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, et al. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell. 2004;14:303–17. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- 5.Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci USA. 2004;101:8603–7. doi: 10.1073/pnas.0402851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273:18677–80. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 7.Huang H, Deng X, He X, Yang W, Li G, Shi Y, et al. Identification of distinct c-terminal domains of the Bombyx adipokinetic hormone receptor that are essential for receptor export, phosphorylation and internalization. Cell Signal. 2011;23:1455–65. doi: 10.1016/j.cellsig.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Franklin JM, Carrasco GA. G-protein receptor kinase 5 regulates the cannabinoid receptor 2-induced up-regulation of serotonin 2A receptors. J Biol Chem. 2013;288:15712–24. doi: 10.1074/jbc.M113.454843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu W, Tilley DG, Myers VD, Coleman RC, Feldman AM. Arginine vasopressin enhances cell survival via a G protein-coupled receptor kinase 2/beta-arrestin1/extracellular-regulated kinase 1/2-dependent pathway in H9c2 cells. Mol Pharm. 2013;84:227–35. doi: 10.1124/mol.113.086322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasan RS, Sullivan LM, Roubenoff R, Dinarello CA, Harris T, Benjamin EJ, et al. Inflammatory markers and risk of heart failure in elderly subjects without prior myocardial infarction: the Framingham Heart Study. Circulation. 2003;107:1486–91. doi: 10.1161/01.cir.0000057810.48709.f6. [DOI] [PubMed] [Google Scholar]
- 11.Murray DR, Freeman GL. Proinflammatory cytokines: predictors of a failing heart? Circulation. 2003;107:1460–2. doi: 10.1161/01.cir.0000060808.79274.0c. [DOI] [PubMed] [Google Scholar]
- 12.Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–9. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- 13.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation ofgp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995;92:4862–6. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker C, Richards LJ, Dayan CM, Jessop DS. Corticotropin-releasing hormone immunoreactivity in human T and B cells and macrophages: colocalization with arginine vasopressin. J Neuroendocr. 2003;15:1070–4. doi: 10.1046/j.1365-2826.2003.01099.x. [DOI] [PubMed] [Google Scholar]
- 15.de Keyzer Y, Auzan C, Lenne F, Beldjord C, Thibonnier M, Bertagna X, et al. Cloning and characterization of the human V3 pituitary vasopressin receptor. FEBS Lett. 1994;356:215–20. doi: 10.1016/0014-5793(94)01268-7. [DOI] [PubMed] [Google Scholar]
- 16.Lolait SJ, O’Carroll AM, McBride OW, Konig M, Morel A, Brownstein MJ. Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature. 1992;357:336–9. doi: 10.1038/357336a0. [DOI] [PubMed] [Google Scholar]
- 17.Morel A, O’Carroll AM, Brownstein MJ, Lolait SJ. Molecular cloning and expression of a rat V1a arginine vasopressin receptor. Nature. 1992;356:523–6. doi: 10.1038/356523a0. [DOI] [PubMed] [Google Scholar]
- 18.Thibonnier M, Coles P, Thibonnier A, Shoham M. Molecular pharmacology and modeling of vasopressin receptors. Prog Brain Res. 2002;139:179–96. doi: 10.1016/s0079-6123(02)39016-2. [DOI] [PubMed] [Google Scholar]
- 19.Carmichael MC, Kumar R. Molecular biology of vasopressin receptors. Semin Nephrol. 1994;14:341–8. [PubMed] [Google Scholar]
- 20.Hiroyama M, Wang S, Aoyagi T, Oikawa R, Sanbe A, Takeo S, et al. Vasopressin promotes cardiomyocyte hypertrophy via the vasopressin V1A receptor in neonatal mice. Eur J Pharm. 2007;559:89–97. doi: 10.1016/j.ejphar.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Xu F, Zhang L, Wang X, Wang Y, Woo AH, et al. GRK2/beta-arrestin mediates arginine vasopressin-induced cardiac fibroblast proliferation. Clin Exp Pharm Physiol. 2016;44:285–93. doi: 10.1111/1440-1681.12696. [DOI] [PubMed] [Google Scholar]
- 22.Wasilewski MA, Myers VD, Recchia FA, Feldman AM, Tilley DG. Arginine vasopressin receptor signaling and functional outcomes in heart failure. Cell Signal. 2016;28:224–33. doi: 10.1016/j.cellsig.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Chan TO, Myers V, Chowdhury I, Zhang XQ, Song J, et al. Controlled and cardiac-restricted overexpression of the arginine vasopressin V1A receptor causes reversible left ventricular dysfunction through Galphaq-mediated cell signaling. Circulation. 2011;124:572–81. doi: 10.1161/CIRCULATIONAHA.111.021352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tilley DG, Zhu W, Myers VD, Barr LA, Gao E, Li X, et al. beta-adrenergic receptor-mediated cardiac contractility is inhibited via vasopressin type 1A-receptor-dependent signaling. Circulation. 2014;130:1800–11. doi: 10.1161/CIRCULATIONAHA.114.010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Wang X, Cao H, Chen Y, Chen X, Zhao X, et al. Vasopressin V1A receptor mediates cell proliferation through GRK2-EGFR-ERK1/2 pathway in A7r5 cells. Eur J Pharm. 2016;792:15–25. doi: 10.1016/j.ejphar.2016.10.023. [DOI] [PubMed] [Google Scholar]
- 26.Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–82. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 27.Huang ZM, Gold JI, Koch WJ. G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci. 2012;17:3047–60. doi: 10.2741/3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorn GW., 2nd GRK mythology: G-protein receptor kinases in cardiovascular disease. J Mol Med. 2009;87:455–63. doi: 10.1007/s00109-009-0450-7. [DOI] [PubMed] [Google Scholar]
- 29.Nijboer CH, Heijnen CJ, Willemen HL, Groenendaal F, Dorn GW, 2nd, van Bel F, et al. Cell-specific roles of GRK2 in onset and severity of hypoxic-ischemic brain damage in neonatal mice. Brain Behav Immun. 2010;24:420–6. doi: 10.1016/j.bbi.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Chen XF, Huang YJ, Chen QQ, Bao YJ, Zhu W. 2,3,4’,5-Tetrahydroxystilbene-2-O-beta-D-glucoside inhibits angiotensin II-induced cardiac fibroblast proliferation via suppression of the reactive oxygen species-extracellular signal-regulated kinase 1/2 pathway. Clin Exp Pharm Physiol. 2012;39:429–37. doi: 10.1111/j.1440-1681.2012.05692.x. [DOI] [PubMed] [Google Scholar]
- 31.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 32.Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al. Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol. 2015;172:3461–71. doi: 10.1111/bph.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu F, Sun S, Wang X, Ni E, Zhao L, Zhu W. GRK2 mediates arginine vasopressin-induced interleukin-6 production via nuclear factor-kappaB signaling neonatal rat cardiac fibroblast. Mol Pharm. 2017;92:278–84. doi: 10.1124/mol.116.107698. [DOI] [PubMed] [Google Scholar]
- 34.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–73. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 35.Thal DM, Homan KT, Chen J, Wu EK, Hinkle PM, Huang ZM, et al. Paroxetine is a direct inhibitor of g protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem Biol. 2012;7:1830–9. doi: 10.1021/cb3003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JH, Zhao L, Pan X, Chen NN, Chen J, Gong QL, et al. Hypoxia-stimulated cardiac fibroblast production of IL-6 promotes myocardial fibrosis via the TGF-beta1 signaling pathway. Lab Invest. 2016;96:839–52. doi: 10.1038/labinvest.2016.65. [DOI] [PubMed] [Google Scholar]
- 37.Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE. 2012;7:e35144. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White M, Ducharme A, Ibrahim R, Whittom L, Lavoie J, Guertin MC, et al. Increased systemic inflammation and oxidative stress in patients with worsening congestive heart failure: improvement after short-term inotropic support. Clin Sci. 2006;110:483–9. doi: 10.1042/CS20050317. [DOI] [PubMed] [Google Scholar]
- 39.Helmy A, Guilfoyle MR, Carpenter KLH, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J Cereb Blood Flow Metab. 2016;36:1434–48. doi: 10.1177/0271678X15620204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He YP, Zhao LY, Zheng QS, Liu SW, Zhao XY, Lu XL, et al. Arginine vasopressin stimulates proliferation of adult rat cardiac fibroblasts via protein kinase C-extracellular signal-regulated kinase 1/2 pathway. Acta Physiol Sin. 2008;60:333–40. [PubMed] [Google Scholar]
- 41.Yang XD, Zhao LY, Zheng QS, Li X. Effects of arginine vasopressin on growth of rat cardiac fibroblasts: role of V1 receptor. J Cardiovasc Pharmacol. 2003;42:132–5. doi: 10.1097/00005344-200307000-00020. [DOI] [PubMed] [Google Scholar]
- 42.Yan-ping H, Lian-you Z, Qiang-sun Z, Shao-wei L, Xiao-yan Z, Xiao-long L, et al. Mitogenic effect of arginine vasopressin on adult rat cardiac fibroblast: involvement of PKC-erk1/2 pathway. J Cardiovasc Pharmacol. 2008;52:72–81. doi: 10.1097/FJC.0b013e31817f36b8. [DOI] [PubMed] [Google Scholar]
- 43.Chen X, Lu G, Tang K, Li Q, Gao X. The secretion patterns and roles of cardiac and circulating arginine vasopressin during the development of heart failure. Neuropeptides. 2015;51:63–73. doi: 10.1016/j.npep.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Yan-Hong F, Hui D, Qing P, Lei S, Hai-Chang W, Wei Z, et al. Effects of arginine vasopressin on differentiation of cardiac fibroblasts into myofibroblasts. J Cardiovasc Pharmacol. 2010;55:489–95. doi: 10.1097/FJC.0b013e3181d706ae. [DOI] [PubMed] [Google Scholar]
- 45.Teixeira LB, ESLT Parreiras, Bruder-Nascimento T, Duarte DA, Simoes SC, Costa RM, et al. Ang-(1-7) is an endogenous beta-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci Rep. 2017;7:11903. doi: 10.1038/s41598-017-12074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCrink KA, Maning J, Vu A, Jafferjee M, Marrero C, Brill A, et al. beta-Arrestin2 improves post-myocardial infarction heart failure via sarco(endo)plasmic reticulum Ca2+-ATPase-dependent positive inotropy in cardiomyocytes. Hypertension. 2017;70:972–81. doi: 10.1161/HYPERTENSIONAHA.117.09817. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Hanada K, Staus DP, Makara MA, Dahal GR, Chen Q, et al. Galphai is required for carvedilol-induced beta1 adrenergic receptor beta-arrestin biased signaling. Nat Commun. 2017;8:1706. doi: 10.1038/s41467-017-01855-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neves MF, Amiri F, Virdis A, Diep QN, Schiffrin EL. Hypertension CMRGo. Role of aldosterone in angiotensin II-induced cardiac and aortic inflammation, fibrosis, and hypertrophy. Can J Physiol Pharmacol. 2005;83:999–1006. doi: 10.1139/y05-068. [DOI] [PubMed] [Google Scholar]
- 49.Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, et al. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H1337–44. doi: 10.1152/ajpheart.01175.2005. [DOI] [PubMed] [Google Scholar]
- 50.Kim PA, Voskresenskaya OG, Kamensky AA. Delayed nootropic effects of arginine vasopressin after early postnatal chronic administration to albino rat pups. Bull Exp Biol Med. 2009;147:687–90. doi: 10.1007/s10517-009-0604-1. [DOI] [PubMed] [Google Scholar]
- 51.Chen WC, Chen CC. Signal transduction of arginine vasopressin-induced arachidonic acid release in H9c2 cardiac myoblasts: role of Ca2+ and the protein kinase C-dependent activation of p42 mitogen-activated protein kinase. Endocrinology. 1999;140:1639–48. doi: 10.1210/endo.140.4.6654. [DOI] [PubMed] [Google Scholar]
- 52.Aharonovitz O, Aboulafia-Etzion S, Leor J, Battler A, Granot Y. Stimulation of 42/44 kDa mitogen-activated protein kinases by arginine vasopressin in rat cardiomyocytes. Biochim Biophys Acta. 1998;1401:105–11. doi: 10.1016/s0167-4889(97)00122-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.