

Abstract
Pathologic features of Alzheimer's disease (AD) include accumulation of amyloid β (Aβ) and hyperphosphorylated tau protein. We have shown previously that the chemokine-like receptor 1 (CMKLR1) is a functional receptor for Aβ, and CMKLR1 contributes to the uptake of Aβ. However, it is unclear whether CMKLR1 ameliorates or aggravates the process of AD. Here, we show that deletion of the gene coding for CMKLR1 significantly increased Aβ deposits in brains of both male and female amyloid β precursor protein/presenilin-1 mice. However, it markedly decreased the mortality of these mice. Behavioral studies found that CMKLR1 deficiency improved cognitive impairment of male and female amyloid β precursor protein/presenilin-1 mice and intracerebroventricular-streptozotocin injection AD mice. We further explored the effect of CMKLR1 on tau pathology. We found that CMKLR1 deficiency or inhibition attenuated the hyperphosphorylation of tau in brains of AD mice in vivo and in the neuronal cells in vitro. The expression of CMKLR1 on the neurons affected tau phosphorylation by participating in tau seeding. Together, these results uncover a novel mechanism of CMKLR1 in the pathologic process of AD and suggest that inhibiting the promotion effect of CMKLR1 on tau seeding may provide a new strategy for the treatment of AD.
SIGNIFICANCE STATEMENT Evidence suggests that inflammation is involved in the pathologic progression of AD. The chemokine-like receptor 1 (CMKLR1), belonging to the family of GPCRs, is able to bind and uptake amyloid β. We show here, for the first time, that, although CMKLR1 deficiency increased amyloid β deposits in AD mice, it reduced the mortality and improved the cognitive deficits of AD mice. We furthermore show that CMKLR1 deficiency or inhibition attenuated tau hyperphosphorylation in brains of AD model mice in vivo and in neuronal cells in vitro. Finally, we first discovered that the expression of CMKLR1 on neurons affected tau phosphorylation by participating in tau seeding. These findings suggest that inhibition of CMKLR1 may provide a new strategy for the treatment of AD.
Keywords: amyloid β, Alzheimer's disease, chemokine-like receptor 1, tau phosphorylation, tau seeding
Introduction
Alzheimer's disease (AD), the most common form of dementia, is characterized by progressive memory loss and cognitive impairment (Querfurth and LaFerla, 2010). The neuropathology hallmarks of AD are the accumulation of extracellular senile plaques and intracellular neurofibrillary tangles (NFTs) composed of aggregated amyloid β (Aβ) and hyperphosphorylated tau, respectively (Iqbal et al., 2010; Querfurth and LaFerla, 2010). In addition to these hallmarks, neuroinflammation has also been found to contribute to AD (Wyss-Coray, 2006). The brain cells involved in AD-related neuroinflammation include microglia and astrocytes, which have been shown to be closely related to Aβ and phosphorylated tau. Both soluble and insoluble Aβ are neurotoxic and activate microglia and astrocytes to produce proinflammatory cytokines, which lead to progression of AD (Heneka et al., 2015). Compared with insoluble Aβ, soluble Aβ shows a stronger correlation with synaptic loss, neuronal death, and cognitive dysfunction (Yang et al., 2017). The activated microglia and astrocytes in turn clear soluble and insoluble Aβ through receptor-mediated phagocytosis and degradation (Ries and Sastre, 2016). Several receptors have been identified in glial cells as endogenous binding sites for Aβ, including the GPCRs, scavenger receptors, and Toll-like receptors (Le et al., 2001; Yu and Ye, 2015).
In addition, accumulating evidence suggests that cognitive dysfunction in AD is most closely related to the progression of tau pathology (Tracy et al., 2016; DeVos et al., 2017). Recent research has shown that tau pathology may be driven by a prion-like spread mechanism. The abnormally hyperphosphorylated or oligomeric tau can be secreted from intact neurons into the extracellular space. Then the extracellular tau, serving as the seed, can be taken up by normal neurons, which promotes the spread of pathologic tau (Takeda et al., 2015; DeVos et al., 2018). Further study has shown that treatment of tau antibodies block tau seeding in vitro, and inhibit the spread of tau pathology and improve cognition in vivo (Yanamandra et al., 2013). Recent research has also shown that immunization with tau antibody 43D to tau 6-18 can block Aβ pathologies promoted by tau seeding (Dai et al., 2018). These data suggest a potential therapeutic treatment for AD by preventing the seeding and spread of tau pathology.
The chemokine-like receptor 1 (CMKLR1, also termed ChemR23) is a chemoattractant receptor that belongs to the family of GPCRs. It is expressed on macrophages, immature dendritic cells, adipocytes, endothelial cells, and vascular smooth muscle cells (Kennedy and Davenport, 2018), indicating its potential roles in regulating immune cell function, obesity, and cancer. CMKLR1 is also expressed in the brain of humans, mice, and rats (Kennedy and Davenport, 2018). Our previous study found that Aβ binds specifically to CMKLR1 in stably transfected rat basophilic leukemia cells (CMKLR1-RBL) (Peng et al., 2015), indicating that CMKLR1 is a receptor for Aβ. Furthermore, Aβ induces CMKLR1-dependent cell migration and stimulates CMKLR1-RBL cells and primary glial cells to internalization of the Aβ-CMKLR1 complex (Peng et al., 2015). These data suggest a potential role for CMKLR1 in Aβ clearance. However, the role of CMKLR1 in tau pathology has not been investigated.
In this study, we first verified the scavenging capability of CMKLR1 on Aβ in vivo. We crossed amyloid β precursor protein/presenilin-1 (APP/PS1) transgenic mice and CMKLR1 KO (CMKLR1−/−) mice to generate APP/PS1-CMKLR1−/− mice. CMKLR1 deficiency induced a significant increase of Aβ deposits compared with APP/PS1 mice, as we expected. However, CMKLR1 deficiency markedly decreased the mortality of APP/PS1 mice, and improved the spatial reference learning and/or memory of APP/PS1 mice and intracerebroventricular (ICV)-streptozotocin (STZ) injection AD mice, which is one of the well-known experimental models for sporadic AD. Since the cognitive dysfunction of AD is closely related to tau pathology, we further explored the effect of CMKLR1 on tau pathology. CMKLR1 deficiency or inhibition attenuated the hyperphosphorylation of tau in the brain of AD mouse models in vivo and in the neuronal cells in vitro. CMKLR1 expressed on neurons affected tau phosphorylation by participating in tau seeding.
Material and Methods
Antibodies and reagents
Primary antibodies used in this study are listed in Table 1. DMEM, neurobasal-A, B-27, and trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA) were purchased from Invitrogen. DMEM with low glucose was purchased from Tianjin Haoyang Biological Manufacture. The BCA protein assay kit, NGS, and DAPI were from Beyotime Institute of Biotechnology. Chemerin15 (C15) peptide was custom synthesized at Anhui Guoping Pharmaceutical and purified to >95% homogeneity. Mouse and rabbit control IgGs were obtained from Santa Cruz Biotechnology. AlexaFluor-568-conjugated anti-rabbit IgG secondary antibody, AlexaFluor-488-conjugated anti-rabbit IgG secondary antibody, AlexaFluor-568-conjugated anti-mouse IgG secondary antibody, and AlexaFluor-594-conjugated anti-mouse IgG antibody were from Invitrogen. IRDye 800CW secondary antibodies and IMDye 800CW secondary antibodies were from LI-COR Biosciences. Other chemicals were purchased from Sigma Millipore.
Table 1.
Primary antibodies used in this study
| Antibody | Type | Specificity | Phosphorylation sites | Reference/source |
|---|---|---|---|---|
| Tau5 | Monoclonal | Tau (total) | Affinity Bioreagents | |
| pT199 | Polyclonal | P-tau | Ser199 | Invitrogen |
| pT205 | Polyclonal | P-tau | Thr205 | Invitrogen |
| pS396 | Polyclonal | P-tau | Ser396 | Invitrogen |
| pS404 | Polyclonal | P-tau | Ser404 | Invitrogen |
| Tau1 | Monoclonal | Tau (non-phosphorylated) | Merck | |
| Tau-13 | Monoclonal | Human tau | Tau15-25 | Biolegend |
| GFAP-Cy3TM | Monoclonal | GFAP | Sigma Millipore | |
| anti-β-actin | Polyclonal | β-actin | Sigma Millipore | |
| Iba1 | Polyclonal | Iba1 | FUJIFILM Wako Pure Chemical | |
| GFAP | Polyclonal | GFAP | Cell Signaling Technology | |
| CMKLR1/AF488 | Polyclonal | CMKLR1 | Bioss | |
| MAP2 | Polyclonal | MAP2 | Sigma Millipore | |
| APP | Polyclonal | APP | Cell Signaling Technology | |
| Aβ | Polyclonal | Aβ | Cell Signaling Technology |
Mice and treatments
CMKLR1−/− (CMKLR1 KO) mice in C57BL/6J genetic background were generated by Shanghai Bioray Laboratory. APP/PS1 transgenic mice in C57BL/6J background (APPSWE/PS1ΔE9+/−, stock #005864) were obtained from The Jackson Laboratory. Mouse genotypes were determined by PCR. All mice were housed (≤5 mice per cage) with a 12/12 h light/dark cycle, with ad libitum access to food and water. The housing, breeding, and animal experiments were in accordance with the National Institutes of Health Guide for the care and use of laboratory animals, with procedures approved by the Biological Research Ethics Committee, Shanghai Jiao Tong University.
CMKLR1−/− mice were crossed to APP/PS1 mice to generate APP/PS1-CMKLR1+/− mice. The latter were further crossbred with CMKLR1+/− mice to create the following four groups: WT (APP/PS1−/−-CMKLR1+/+), APP/PS1 (APP/PS1+/−-CMKLR1+/+), CMKLR1−/− (APP/PS1−/−-CMKLR1−/−), and APP/PS1-CMKLR1−/− (APP/PS1+/−-CMKLR1−/−). All four groups of male and female mice at 9 months of age (WT, 4 males and 5 females; APP/PS1, 4 males and 5 females; CMKLR1−/−, 5 males and 5 females; APP/PS1-CMKLR1−/−, 3 males and 3 females) were subjected to a battery of behavioral tests.
Both male and female WT and CMKLR1−/− mice at 6 months of age were randomly divided into control and ICV-STZ groups, respectively. The ICV-STZ mice were produced by ICV injection of a single dose of STZ [2-deoxy-2-(3-(methyl-3-nitrosoureido)-D-glucopyranose)] into both lateral ventricles of the mice brain (Chen et al., 2013). Briefly, mice were anesthetized by intraperitoneal injection of 5 mg/ml pentobarbital sodium and then restrained onto a stereotaxic apparatus. Each mouse received a single ICV injection of 3.0 mg/kg STZ. STZ was freshly prepared in 0.9% saline into both lateral brain ventricles. The bregma coordinates were determined as follows: ±1.0 mm lateral, –0.3 mm posterior, and –2.5 mm below. As controls, ICV-saline mice received an equal volume of 0.9% saline in the same way. After operation, all mice were placed on heating pads (37°C) until recovered from surgery. The body weight of the mice was measured once every 7 d. Six weeks after ICV injection, all mice (WT ICV-saline, 4 males and 4 females; WT ICV-STZ, 3 males and 4 females; CMKLR1−/− ICV-saline, 4 males and 4 females; CMKLR1−/− ICV-STZ, 3 males and 1 female) were subjected to a battery of behavioral tests (see Fig. 2G).
Figure 2.

CMKLR1 deficiency decreases mortality and improves cognition in APP/PS1 mice and/or ICV-STZ mice. A, Survival curves for WT, APP/PS1, CMKLR1−/−, and APP/PS1-CMKLR1−/− mice. Data are mean ± SEM, with 22-29 mice in each group. ***p < 0.001 compared with WT mice. $$p < 0.01 compared with APP/PS1 mice. B–F, Spatial reference learning and memory were determined in Morris water maze. Mice were trained in Morris water maze for consecutive 5 d, four trials per day. The distance traveled (B) and time spent (C) to reach the escape platform are shown. The probe trials were tested on the seventh day after a day off. The percentage of the distance traveled (D) and the time spent (E) within each quadrant are shown. F, Representative swimming paths of a mouse on day 7. Data are mean ± SEM, with 6-10 mice in each group (WT, n = 9; APP/PS1, n = 9; CMKLR1−/−, n = 10; APP/PS1-CMKLR1−/−, n = 6). *p < 0.05; **p < 0.01; compared with WT mice. $p < 0.05 compared with APP/PS1 mice. G, Schematic representation of study design. The ICV-STZ mice and ICV-saline mice were produced by ICV injection of a single dose of 3.0 mg/kg STZ or equal volume of 0.9% saline into both lateral ventricles of the mice brain, respectively. The body weight of these mice was measured every day in the first 7 d, and then once every 7 d. Six weeks after ICV injection, all mice were subjected to a battery of behavioral tests, and then killed for biochemical studies. H, The body weight of WT and CMKLR1−/− mice after receiving STZ or saline was recorded. I–M, Spatial reference learning and memory were examined in Morris water maze. The distance traveled (I) and the time spent (J) to reach the escape platform are shown. The probe trials were tested on seventh day after a day off. The percentage of the distance traveled (K) and the time spent (L) within each quadrant are shown. M, Representative swimming paths of a mouse on day 7. Data are mean ± SEM, with 4-8 mice in each group (WT ICV-saline, n = 8; WT ICV-STZ, n = 7; CMKLR1−/− ICV-saline, n = 8; CMKLR1−/− ICV-STZ, n = 4). *p < 0.05; ***p < 0.001; compared with WT ICV-saline mice. ##p < 0.01 compared with CMKLR1−/− ICV-saline mice. $$p < 0.01 compared with WT ICV-STZ mice.
After behavioral tests, all mice were killed by decapitation and their brains removed immediately. The hippocampi and cerebral cortices of the left hemisphere of the brain were dissected, flash frozen in liquid nitrogen, and stored at −80°C for biochemical analyses later. The right hemispheres of the brain were fixed with 4% PFA in 0.1 m PBS, followed by cryoprotection in 30% sucrose. Sagittal sections (20 μm thickness) were cut using a freezing sliding microtome. The sections were stored in glycol antifreeze solution (12.5 g/L polyvinylpyrrolidone, average MW 40,000, 375 g/L saccharose, 375 ml/L glycol, and 625 ml/L TBS (0.1 M, containing 12.1 g/L Tris base and 40 g/L NaCl)) at −20°C until immunohistochemical staining. In this study, we included 3–6 mice/group for biochemical analyses and immunofluorescence staining.
Behavioral tests
Morris water maze
Spatial reference learning and memory were detected in a water maze (Morris et al., 1982). The test was performed in a white pool of 150 cm in diameter filled with water mixed with nontoxic white coloring emulsifier and maintained at room temperature. During training, an 8-cm-diameter platform was submerged 1 cm below water surface. All mice were given four trials per day for 5 consecutive days. The starting position was randomized among four quadrants of the pool every day. For each trial, the animal was given 90 s to reach the hidden platform. If a mouse failed to find the platform within 90 s, it would be gently guided to it. At the end of each trial, the mouse was left on the platform for 20 s, then dried and returned to its home cage until the next trial. Probe trial was established on the seventh day. Mice were allowed to swim in the pool without the platform for 60 s. The escape latency (s) and swim distance (cm) to reach the platform site were monitored and analyzed using an automated tracking system (Smart video tracking system, Shanghai Mobile Datum).
Accelerating Rotarod test
Motor coordination and balance of mice were detected by using a Rotarod test. Test on an accelerating Rotarod was conducted by three trials on a rotating cylinder. The speed increased steadily from 5 to 40 rpm over a 105 s period. The latency to fall off the Rotarod was calculated. Intertrial intervals were 5-10 min for each mouse. After each trial, the test chambers were cleaned with 75% alcohol.
Open field
Exploratory activities and anxiety were evaluated by allowing mice to freely explore an open field arena for 15 min. The testing device was an open field (a polyvinyl chloride square arena, 100 × 100 cm, with 30 cm high walls), surmounted by a video camera connected to a computer. Each mouse was placed individually in the arena, and the performance was recorded. The distance traveled in the arena, and the time and distance traveled in the center area were recorded and analyzed by Smart video tracking system (Shanghai Mobile Datum).
Elevated plus maze
Elevated plus maze was used to evaluate anxiety/emotionality of the mice. It consisted of four arms (30 × 5 cm) connected by a common 5 × 5 cm center area. Two opposite facing arms were open (open arms), whereas the other two facing arms were enclosed by 20-cm-high walls (closed arms). The entire plus maze was elevated on a pedestal to a height of 80 cm above floor level. The mouse was placed onto the central area facing an open arm and allowed to explore the maze for an 8 min trial. The distance traveled and the time spent in the open arm were recorded and analyzed by Smart video tracking system (Shanghai Mobile Datum). After each trial, the maze floor was cleaned with 75% alcohol.
Total RNA extraction and real-time PCR
Total RNA was extracted from the mouse brain tissue using Trizol reagent (Invitrogen), according to the manufacturer's instructions. Reverse transcription (RT) of RNA was performed with the Toyobo First Strand cDNA Synthesis Kit (Toyobo). The cDNA was subsequently subjected to Real-Time PCR to quantify the transcripts of APP and the housekeeping gene GAPDH, using SYBR Green Real-time PCR Master Mix (Toyobo). The following primers were used: APP (5′-GCC CTG CGG AAT TGA CAA G-3′; 5′-CCA TCT GCA TAG TCT GTG TCT G-3′); GAPDH (5′-CCT TCC GTG TTC CTA CC-3′; 5′-CAA CCT GGT CCT CAG TGT A-3′). The quantitative fold changes in mRNA in each sample were normalized to GAPDH expression and calculated using the 2exp(-ΔΔCt) method.
Aβ ELISA
The hippocampi of WT, APP/PS1, CMKLR1−/−, and APP/PS1-CMKLR1−/− mice were homogenized in lysis buffer containing 0.085 g/ml sucrose, 0.07% β-mercaptoethanol, 2.5 mm Tris-HCl, pH 7.4, 100 mm NaF, 0.016 mm EDTA, 1 × phosphatase inhibitor, and 1 × protease inhibitor. The homogenate was centrifuged at 14,000 rpm for 15 min at 4°C. The pellet was dissolved in 20 volumes of ice-cold 6 m guanidine hydrochloride, and diluted with the lysis buffer as described above containing 1% PMSF. Then insoluble Aβ1-42 was measured using commercially available ELISA kit from Shanghai Jijin Chemistry Technology, according to the manufacturer's instructions.
Western blot
Mouse brain tissues or cultured cells were homogenized in lysis buffer containing 0.085 g/ml sucrose, 0.07% β-mercaptoethanol, 2.5 mm Tris-HCl, pH 7.4, 100 mm NaF, 0.016 mm EDTA, 1 × phosphatase inhibitor, and 1 × protease inhibitor. Protein concentrations were determined by using BCA kits according to the manufacturer's protocol. Quantitative homogenates were added 5 × SDS to heat for 10 min at 99°C and resolved in 8% or 10% SDS-PAGE for separation. After separated samples were transferred onto nitrocellulose membranes (GE Healthcare), blots were incubated with primary antibodies and further treated with corresponding secondary antibodies before scanning using an Odyssey P140-CLx Infrared Imaging (LI-COR Biosciences). Densitometric quantification of protein bands in Western blots was analyzed using the ImageJ software (National Institute of Mental Health).
Immunofluorescence staining
Sections of mouse brain or cultured cells were processed for standard immunofluorescence staining (Peng et al., 2015). Briefly, sections or cells were washed 3 times for 5 min in precold 0.02 M TBS (containing 2.42 g/L Tris-base and 8 g/L NaCl), permeabilized in 3% H2O2 and 50% methanol in TBS for 30 min at room temperature, followed by blocked in TBS with 5% NGS and 0.1% Tween-20 at room temperature for 1 h. Samples were then incubated with anti-CMKLR1/AF488 (1:200) or anti-Aβ (1:200) in TBS with 5% NGS and 0.1% Tween-20 for 1 h at room temperature or overnight at 4°C, respectively. After washing with TBS for 3 times, the samples were further treated with AlexaFluor-488-conjugated anti-rabbit antibody (1:500) for 1 h at room temperature. After three washes in TBS, the samples were stained for nuclei with 100 ng/ml DAPI for 10 min at room temperature and mounted on glass slides.
To identify the expression of CMKLR1 in neuron, microglia, or astrocyte, the brain sections were first stained with anti-microtubule associated protein 2 (MAP2, 1:200) or anti-ionized calcium binding adaptor molecule 1 (Iba1, 1:200) antibody overnight, or anti-GFAP-Cy3TM (1:400) for 1 h at room temperature. After three washes with TBS, the sections were incubated in AlexaFluor-568-conjugated anti-rabbit antibody (1:500) for 1 h. Then the sections were rinsed in TBS and stained with anti-CMKLR1/AF488 (1:200) for 1 h at room temperature. Sections were counterstained with DAPI for 10 min and mounted on glass slides.
To determine whether human tau seeding accelerated tau hyperphosphorylation in primary cultures of mouse neurons, the neurons were stained with anti-pT205 (human and mouse pTau antibody at Thr205 site, 1:200) or anti-pS396 (human and mouse pTau at Ser396 site, 1:200) antibodies overnight at 4°C and incubated with AlexaFluor-488-conjugated anti-rabbit antibody (1:500) for 1 h. Neurons were rinsed in TBS, stained with human tau-specific antibody (Tau13, 1:200) overnight at 4°C, and incubated with AlexaFluor-594-conjugated anti-mouse antibody (1:500) for 1 h. After three washes in TBS, neurons were stained for nuclei with 100 ng/ml DAPI for 3 min at room temperature.
Fluorescent confocal images were taken on a laser-scanning confocal fluorescent microscope (TCS SP8, Leica Microsystems). For quantification of the expression of CMKLR1 and Aβ deposits, the immunofluorescence intensity was quantified using the ImagePro Plus Software (Media Cybernetics). The results were expressed as the mean ± SEM, based on three fields for each region, using at least 3 mice in each group. For quantification of tau proteins, the immunofluorescence area of total human tau (Tau13 antibody, shown in red) and human and mouse phosphorylated tau (Hu and Ms pTau, pT205, and pS396, shown in green) was quantified using the ImagePro Plus Software. The colocalization of total human tau and Hu and Ms pTau (shown in yellow) means the human pTau. The area of mouse pTau (pT205 and pS396) was calculated by subtracting the fluorescence area of human pTau (in yellow) from the fluorescence area of Hu and Ms pTau (shown in green). The results were expressed as the mean ± SEM from three separate experiments, each in duplicate or triplicate.
Neuronal cell cultures
Primary neuronal cultures were prepared from cortices of newborn (postnatal day 0) WT and CMKLR1−/− mouse pups as described previously (Li et al., 2015). Briefly, cerebral cortices were removed from the brains of the mice, the meanings, microvessels, and gray matter were removed. Tissues were minced with a sterile ophthalmic scissor, and then digested with 0.05% trypsin (Sigma Millipore) at 37°C for 6 min. The cell suspension was filtered through a 40 μm sieve; then cells were plated on poly-D-lysine-coated 12-well plates at a density of 2.4 × 106 cells per well. Four hours later, the DMEM medium (containing 10% FBS, 100 U/ml of penicillin, 100 μg/ml streptomycin sulfate, and 2.92 mg/ml L-glutamine) were replaced with neurobasal medium (containing 2% B-27 supplements, 100 U/ml of penicillin, 100 μg/ml streptomycin sulfate, and 2.92 mg/ml L-glutamine) for 2 d. Culture medium was changed to neurobasal with 10% FBS and 2.5 μg/ml cytosine-β-D-arabinofuranoside (Sigma Millipore) in the following 2 d, and then again switched back to neurobasal medium containing 2% B-27 supplements, 100 U/ml of penicillin, 100 μg/ml streptomycin sulfate, and 2.92 mg/ml L-glutamine. Experiments were performed on day 7 after initiation of the culture.
The human neuroblastoma cell line SH-SY5Y (ATCC) was grown in DMEM supplemented with 10% FBS, 100 U/ml of penicillin, and 100 μg/ml streptomycin sulfate.
Treatment with high glucose of SH-SY5Y
When SH-SY5Y cells cultured in plates grew to 60%–70% confluence, the culture medium was replaced with fresh DMEM without FBS for 30 min. Then the cells were incubated with DMEM containing 5, 15, or 30 mm glucose for another 12 and 24 h, with or without a 1 h pretreatment with C15 (1 or 10 μm) at 37°C. At the end of the incubation, conditioned medium (CM) was collected and the cells were lysed in 1 × SDS buffer for immunoblotting analysis.
Tau ELISA
The concentrations of tau in the samples (mouse brain extracts and CM harvested from the cells) were examined by Tau ELISA kits (total tau, #JM-11640M2; pSer199 tau, #JM-11645M2; pThr205 tau, #JM-11661M2; and pSer396 tau, #JM-11636M2) (Jiangsu Jingmei Bio-technology), according to the manufacturer's instructions.
Preparation of tau seeds
The extract with pathologic tau was isolated from the mouse brain tissues as described by Takeda et al. (2015) with minor modification. Briefly, the brain tissues of WT and APP/PS1 mice aged 9 months were homogenized in the lysis buffer containing 0.085 g/ml sucrose, 0.07% β-mercaptoethanol, 2.5 mm Tris-HCl, pH 7.4, 100 mm NaF, 0.016 mm EDTA, 1 × phosphatase inhibitor, and 1 × protease inhibitor. The homogenate was centrifuged at 5,500 rpm for 5 min at 4°C. The supernatants (tau seeds) were collected. The levels of total tau and p-tau in the extract were examined by using tau ELISA. The concentration of total tau, pSer199 tau, pThr205 tau, and pSer396 tau were 8.6, 38.6, 257.5, and 85 ng/L, respectively.
Stereotaxic injection of brain extract
Six-month-old WT and CMKLR1−/− mice in C57BL/6J background were injected with the brain extract (tau seeds) from APP/PS1 mice aged 9 months (ICV-hyper-Tau) (Takeda et al., 2015; Jiang et al., 2019). Briefly, mice were anesthetized with pentobarbital sodium and then restrained onto a stereotaxic apparatus. Each mouse received a single ICV injection of 3 μl of tau seeds (2.6 × 10−2 ng total tau, 1.2 × 10−4 ng pSer199 tau, 7.8 × 10−4 ng pThr205 tau, and 2.6 × 10−4 ng pSer396 tau) into both lateral brain ventricles (±1.0 mm lateral, –0.3 mm posterior, and –2.5 mm below). An equal volume of brain extracts from WT mice of the same age was also injected into the brain of the mice as vehicle controls (ICV-nor-Tau). Postoperatively, all mice (WT ICV-nor-pTau, 2 females; WT ICV-hyper-pTau, 1 male and 2 females; CMKLR1−/− ICV-nor-pTau, 1 male and 2 females; CMKLR1−/− ICV-hyper-pTau, 2 females) were placed on heating pads (37°C) until recovered from surgery. After 3 weeks, the injected mice were killed by decapitation and the brain samples were prepared as the ICV-STZ mice described above.
Trichloroacetic acid/acetone precipitation of tau proteins
Tau proteins secreted from SH-SY5Y cells were precipitated as described by Ivanovova et al. (2008) with minor modification. SH-SY5Y cells were treated with 5 or 30 mm glucose for 24 h. Then the medium was collected and centrifuged at 5,500 rpm for 5 min; 0.11 volume of ice-cold 100% trichloroacetic acid was added to the supernatant, and the mixture was incubated on ice for 10 min. Then 500 μl of 10% trichloroacetic acid was added to the mixture and incubated on ice for 20 min. The mixture was centrifuged at 20,000 rpm for 30 min. The supernatant was discarded, and the precipitate was resuspended in 500 μl of acetone. The mixture was again centrifuged at 20,000 rpm for 10 min. The precipitate was dried at room temperature and resuspended in 1 × SDS loading buffer for immunoblotting analysis.
Treatment with CM of SH-SY5Y
The CM collected from SH-SY5Y cells treated with 5 or 30 mm glucose as described above was purified using dialysis (Schweers et al., 1994). The CM was dialyzed overnight against 500 ml of fresh DMEM with 30 mm glucose at 4°C, using dialysis bags (Union Carbide) with a 0.8-1.4 kDa exclusion limit, which has previously been boiled in EDTA solution, pH 8, 1 mm EDTA, and 2% NaHCO3 (m/v), for 10 min. The dialysis bags were washed 3 times with water before use. After purification, the human tau protein for seeding remained in the dialysis bags, which were also refilled with nutrients needed to culture the neurons. The purified CM was added to the culture of neurons from WT or CMKLR1−/− mice for 72 h. The neurons were examined by immunofluorescence staining as detailed above. The levels of tau protein in CM were examined by using tau ELISA.
Statistical analyses
All data are presented as mean ± SEM. Mean values of paired groups were analyzed using Student's t test. Mean values of multiple groups were analyzed using one-way ANOVA, followed by Tukey's post hoc test. The survival rate and the body weight of the mice were analyzed using Log-rank (Mantel-Cox) test and two-way ANOVA, respectively. All analyses were performed with the statistical software GraphPad Prism 8. p < 0.05 was considered statistically significant.
Results
Increased Aβ accumulation in APP/PS1-CMKLR1−/− mice
Our previous study identified that CMKLR1 is a functional receptor for Aβ. Aβ activates CMKLR1, leading to glia cell migration and clearance of Aβ (Peng et al., 2015). To further verify the scavenging capability of CMKLR1 on Aβ in vivo, we generated APP/PS1-CMKLR1−/− mice and analyzed Aβ levels and accumulation in these mice. We crossed APP/PS1 transgenic mice on a C57BL/6 background to CMKLR1−/− mice (CMKLR1 KO mice also on a C57BL/6 background) to generate APP/PS1-CMKLR1+/− mice, then crossed APP/PS1-CMKLR1+/− mice with CMKLR1−/− mice to generate APP/PS1-CMKLR1−/− mice. We analyzed the brains of APP/PS1-CMKLR1−/− mice aged 6, 9, and 13 months compared with APP/PS1 mice. As shown in Figure 1A-D, CMKLR1 deficiency induced a significant increase in the hippocampus and the cortex of the brain stained for Aβ compared with APP/PS1 mice in all three age groups, especially in 9-month-old mice. WT mice did not show any Aβ staining in their brains at all these age groups (Fig. 1A).
Figure 1.

CMKLR1 deficiency is associated with increased Aβ deposition in APP/PS1 mice. A, Representative immunofluorescence images of Aβ (Aβ deposits, green) frozen in the hippocampus and the cortex of WT, APP/PS1, CMKLR1−/−, and APP/PS1-CMKLR1−/− mice aged 6, 9, and 13 months. Scale bar, 500 μm. Quantification of the fluorescence intensity of Aβ deposits, showing that CMKLR1 deficiency significantly increases Aβ deposition in APP/PS1 mice at the age of 6 months (B), 9 months (C), and 13 months (D). Data are mean ± SEM, based on three fields for each region, using at least 3 mice in each group. *p < 0.05; **p < 0.01; compared with WT mice. ###p < 0.001 compared with CMKLR1−/− mice. $$p < 0.01; $$$p < 0.001; compared with APP/PS1 mice. E, Insoluble Aβ1-42 from the hippocampus of 9-month-old mice were measured using ELISA. Data are mean ± SEM, with at least 3 mice in each group. *p < 0.05 compared with WT mice. ###p < 0.001 compared with CMKLR1−/− mice. $$$p < 0.001 compared with APP/PS1 mice. F, Real-time PCR quantification of the transcripts for APP of the mice at the age of 9 months. Data are mean ± SEM, with at least 3 mice in each group. *p < 0.05 compared with WT mice. #p < 0.05 compared with CMKLR1−/− mice. G, Representative Western blots showing the expression of APP protein in the brain of 9-month-old mice. H, Quantification of the immunoreactivity of the blots, normalized against β-actin. Data are mean ± SEM, with at least 3 mice in each group. *p < 0.05 compared with WT mice. #p < 0.05 compared with CMKLR1−/− mice.
In addition to immunofluorescence staining, ELISA was performed to determine Aβ deposits (insoluble Aβ1-42) in the hippocampus of mice. As shown in Figure 1E, the levels of Aβ deposits were significantly increased in 9-month-old APP/PS1 mice versus age-matched WT mice. Consistent with the results of Aβ immunofluorescence staining, CMKLR1 deficiency induced a pronounced increase of Aβ deposits in APP/PS1-CMKLR1−/− mice compared with APP/PS1 mice (Fig. 1E). These results indicate that CMKLR1 affects the content and accumulation of Aβ in vivo.
Since Aβ is derived by proteolytic processing from the APP, we further investigate whether the deletion of CMKLR1 affects the expression of APP. As expected, a significant increase in APP mRNA and protein expression was observed in the brain of APP/PS1 mice compared with the WT mice; however, no significant difference was found in APP/PS1-CMKLR1−/− mice compared with the APP/PS1 mice (Fig. 1F-H). These results suggest that increased Aβ deposition in APP/PS1-CMKLR1−/− mouse brain may be because of decreased CMKLR1-induced Aβ clearance and not because of the regulation of APP expression.
Decreased mortality and improved cognition in APP/PS1-CMKLR1−/− mice
Since the scavenging capability of CMKLR1 on Aβ in vivo and in vitro has been confirmed, we further validate the effect of CMKLR1 on other pathologies of AD. We first observed the mortality in AD mice. All four groups of mice (WT, APP/PS1, CMKLR1−/−, and APP/PS1-CMKLR1−/− mice) showed healthy initially. However, at the age of 8 months, there was a significant increase in the mortality rate of APP/PS1 mice compared with the WT mice (Fig. 2A). Interestingly, the mortality rate of APP/PS1-CMKLR1−/− mice was markedly decreased compared with APP/PS1 mice. By the age of 10 months, 20% of APP/PS1-CMKLR1−/− mice died compared with 60% of APP/PS1 mice (Fig. 2A). These data show that CMKLR1 deficiency decreases the mortality in APP/PS1 AD mice.
To investigate the effect of CMKLR1 on cognitive performance, we tested 9-month-old APP/PS1 mice in Morris water maze. It was found that APP/PS1 mice swam a longer distance (Fig. 2B) and took a longer time (Fig. 2C) than the WT mice to find the escape platform, suggesting a learning impairment in APP/PS1 mice. However, in APP/PS1-CMKLR1−/− mice, a significant shorter swimming distance and escape latency on day 5 to reach the escape platform were observed compared with the APP/PS1 mice (Fig. 2B,C). Interestingly, APP/PS1-CMKLR1−/− mice showed longer swimming distance on training days 1 and 2 to reach the escape platform than the APP/PS1 mice, but this change was not statistically significant (Fig. 2B). Together, these results indicate that CMKLR1 deficiency improves learning of the APP/PS1 mice. After 1 d off, probe trials were performed on day 7 to evaluate the spatial reference memory of the mice. As expected, APP/PS1 mice traveled less distance and spent less time in the target quadrant than the WT mice, suggesting an impairment of spatial reference memory (Fig. 2D–F). In addition, although CMKLR1−/− mice showed a trend of decrease in swimming distance and time in the target quadrant compared with the WT mice, this trend was not statistically significant. APP/PS1-CMKLR1−/− mice showed no significant difference in distance traveled or time spent in the target quadrant compared with CMKLR1−/− mice (Fig. 2D–F). This result indicates that, in the absence of CMKLR1, overexpression of human mutant APP and PS1 does not induce the spatial reference memory impairment in mice.
In addition, we established another mouse model of AD, ICV-STZ injection mice, to confirm the effect of CMKLR1 on cognitive function. This model was generated by ICV injection of STZ in CMKLR1−/− mice and WT littermates, using normal saline as a control (Fig. 2G). Since STZ-induced animal models are characterized by insulin deficiency and lower body weight, we monitored body weight changes after ICV injection. ICV-STZ mice showed lower body weight compared with ICV-saline mice (Fig. 2H), as reported previously (Chen et al., 2013). CMKRL1 deficiency had no effect on the decrease in body weight of ICV-STZ mice (Fig. 2H). For the Morris water maze test, consistent with the results of the APP/PS1 mouse model, CMKLR1 deficiency induced shorter distance swum to reach the escape platform on days 3, 4, and 5 in the mice receiving ICV-STZ (Fig. 2I). CMKLR1 deficiency did not affect the time spent to find the escape platform (Fig. 2J), maybe because the swimming speed of mice in this group was decreased compared with other groups. Probe trials test found that the distance swum and time spent of CMKLR1−/− ICV-STZ mice in the target quadrant were not different from that of CMKLR1−/− ICV-saline mice but displayed a markedly increasing trend compared with APP/PS1 mice, although the trend was not statistically significant (Fig. 2K–M). These data suggest that CMKLR1 deficiency could also improve the learning and memory of ICV-STZ AD mice.
In addition to testing cognitive functions, we also evaluated the general behaviors of the mice. Motor coordination and balance ability were measured using Rotarod test. APP/PS1 mice displayed a decreased fall latency compared with WT mice (Fig. 3A). CMKLR1 deficiency showed a trend of attenuating the decrease in fall latency of APP/PS1 mice (Fig. 3A). This result indicates that CMKLR1 deficiency may improve motor coordination and balance of AD mice. Next, we evaluated spontaneous exploratory activity of mice. We found no significant difference in the total distance explored between four groups in the open field test, indicating that CMKLR1 has no effect on spontaneous exploratory ability in mice (Fig. 3B). We further measured anxiety in mice using elevated plus maze and open field test. Compared with WT mice, APP/PS1 mice traveled shorter distance and spent less time in the open arms of the elevated plus maze (Fig. 3C–E) and in the center area of open field (Fig. 3F–H). CMKLR1 deficiency did not significantly alter the distance traveled and the time spent in the open arms of the elevated plus maze or in the center of the open field, compared with the APP/PS1 mice (Fig. 3C–H). These results suggest that CMKLR1 does not affect the anxiety of the mice.
Figure 3.

CMKLR1 deficiency has no effect on the locomotion, spontaneous exploratory, and anxiety of APP/PS1 mice aged 9 months. A, The locomotion of mice was tested on Rotarod, and the fall latency was recorded. B, The spontaneous exploratory activity of mice was examined in an open field, and the distance traveled during 15 min was recorded. The anxiety of mice was tested in an elevated plus maze (C–E) and an open field (F–H). The distance traveled (C) and the time spent (D) in the open arm were recorded. E, Representative traces of a mouse's movements during the elevated plus maze. The distance traveled (F) and the time spent (G) in the center were recorded. H, Representative traces of a mouse's movements during the open field test. Data are mean ± SEM, with 6-10 mice in each group (WT, n = 9; APP/PS1, n = 7; CMKLR1−/−, n = 10; APP/PS1-CMKLR1−/−, n = 6). *p < 0.05; **p < 0.01; compared with WT mice. #p < 0.05 compared with CMKLR1−/− mice.
CMKLR1 deficiency attenuates hyperphosphorylation of tau in AD mouse models
The above results showed that CMKLR1 deficiency aggravated the accumulation of Aβ in the brain of AD mice but reduced the mortality and alleviated the learning and memory impairment of AD mice. Increasing evidence demonstrates that the cognitive dysfunction is closely related to the excessive phosphorylation of tau (Tracy et al., 2016; DeVos et al., 2017). Therefore, we further explored the effect of CMKLR1 on tau phosphorylation in AD mice. We detected the levels of total tau (Tau5 antibody), nonphosphorylated tau (Tau1 antibody), and phosphorylated tau at several AD-related amino acid positions, including Ser199, Thr205, Ser396, and Ser404 in mice. As expected, in the hippocampus of APP/PS1 mice, there was an increase in tau phosphorylation at Ser199, Thr205, Ser396, and Ser404 compared with the WT mice (Fig. 4A–C). CMKLR1 deficiency attenuated hyperphosphorylation of tau at these sites in APP/PS1 mice. Consistent with this result, in the brain of ICV-STZ mice, a marked increase in tau phosphorylation at Ser199, Thr205, Ser396, and Ser404 was observed as compared with the ICV-saline mice (Fig. 4D–F). CMKLR1 deficiency attenuated hyperphosphorylation of tau in ICV-STZ mice. These results confirmed that CMKLR1 is involved in tau pathology in AD.
Figure 4.

CMKLR1 deficiency attenuates hyperphosphorylation of tau in the brain of APP/PS1 mice and ICV-STZ mice. A, Representative Western blots showing tau phosphorylation at Ser199, Thr205, Ser396, and Ser404 in the hippocampus of WT, APP/PS1, CMKLR1−/−, and APP/PS1-CMKLR1−/− mice at the age of 9 months. The levels of nonphosphorylated tau (Tau1) and total tau (Tau5) were also measured. Quantification of the immunoreactivity of the blots, normalized against total tau (B) and β-actin (C). Data are mean ± SEM, with 6 mice in each group. *p < 0.05 compared with WT mice. $p < 0.05 compared with APP/PS1 mice. D, Representative Western blots showing tau phosphorylation in the brain of WT and CMKLR1−/− mice receiving ICV injection of STZ or saline. Quantification of the immunoreactivity of the blots, normalized against total tau (E) and β-actin (F). Data are mean ± SEM, with 6 mice in each group. *p < 0.05; **p < 0.01; ***p < 0.001; compared with WT ICV-saline mice. $p < 0.05; $$p < 0.01; compared with WT ICV-STZ mice.
Our previous study has found that CMKLR1 expressed in primary cultures of glial cells, including microglia and astrocytes (Peng et al., 2015). To verify the mechanism of CMKLR1 involved in tau phosphorylation, we first investigated the expression and distribution of CMKLR1 in AD mouse brain. Immunofluorescence staining of serial slices from mouse brain with an anti-CMKLR1 antibody identified a significant upregulation of CMKLR1 in CA and dentate gyrus (DG) regions of the hippocampus as well as in the cortex of APP/PS1 mice aged 6 and 9 months, compared with the WT mice (Fig. 5A–C). To identify the cellular distribution of CMKLR1, double immunostaining was conducted for CMKLR1 and the cell-specific markers MAP2 (neurons), Iba1 (microglia), and GFAP (astrocytes). CMKLR1 was colocalized with neurons, microglia, and astrocytes in the hippocampus and the cortex of mice aged 9 months (Fig. 5D–F). The results of the first part in this study and our previous data (Peng et al., 2015) confirmed that the expression of CMKLR1 on glial cells is involved in Aβ clearance. Next, we further verified whether CMKLR1 expressed on neurons is directly involved in tau pathology.
Figure 5.

Upregulation of CMKLR1 and colocalization with MAP2, Iba1, and GFAP in the brain of APP/PS1 mice. A, Representative immunofluorescence images of CMKLR1 (green) in CA1, CA3, and DG regions of the hippocampus and in the cortex of WT and APP/PS1 mice aged 6 months and 9 months. Scale bar, 50 µm. Quantification of the fluorescence intensity of CMKLR1 in each of the regions, showing that CMKLR1 was significantly increased in the hippocampus and the cortex of APP/PS1 mice at the age of 6 months (B) and 9 months (C). Data are mean ± SEM, with at least 3 mice in each group. *p < 0.05; **p < 0.01; compared with WT mice. D–F, Colocalization of CMKLR1 (green) with neurons (anti-MAP2, red, D), microglia (anti-Iba1, red, E), and astrocytes (anti-GFAP, red, F) in the hippocampus and the cortex of APP/PS1 mice aged 9 months. Scale bar, 50 μm.
Neuronal CMKLR1 affects tau phosphorylation
Since impaired glucose utilization in the brain is one of the common pathologic features of AD (Chen et al., 2016), we established an AD cell model by using high glucose to induce tau hyperphosphorylation in human neuroblastoma SH-SY5Y cells. We found that endogenous CMKLR1 was expressed in the cell membrane, cytoplasm, and nucleus of SH-SY5Y cells (Fig. 6A), consistent with a previous study (Zhang et al., 2019b). High glucose (15 and 30 mm) treatment for 24 h induced a significant concentration-dependent increase in tau phosphorylation at Ser199, Thr205, and Ser396 in SH-SY5Y cells (Fig. 6B–D). The role of CMKLR1 in high glucose-induced tau hyperphosphorylation was further examined using C15, a chemerin-derived 15-residue peptide lacking the agonistic activity of chemerin (Cash et al., 2008). As expected, in the presence of C15, high glucose treatment could no longer stimulate tau hyperphosphorylation in SH-SY5Y cells (Fig. 6E–G). These results indicate that the neuronal CMKLR1 is involved in the regulation of tau phosphorylation.
Figure 6.

Inhibition of neuronal CMKLR1 attenuates tau hyperphosphorylation induced by high glucose treatment in SH-SY5Y cells. A, Representative immunofluorescence images of CMKLR1 (green) in SH-SY5Y cells. Scale bar, 50 μm. B, Representative Western blots showing tau hyperphosphorylation at Ser199, Thr205, and Ser396 from SH-SY5Y treated with 5, 15, or 30 mm glucose for 12 or 24 h. Quantification of the immunoreactivity of the blots, normalized against total tau (C) and β-actin (D). E, Representative Western blots showing tau hyperphosphorylation at Ser199 and Ser396 from SH-SY5Y incubated with 5 or 30 mm glucose for 24 h, with or without a 1 h pretreatment with C15 (1 or 10 µm). Quantification of the immunoreactivity of the blots, normalized against total tau (F) and β-actin (G). Data are mean ± SEM from three separate experiments, each in duplicate or triplicate. *p < 0.05; **p < 0.01; ***p < 0.001; compared with 5 mM glucose treatment group. $p < 0.05; $$p < 0.01; compared with 30 mm glucose treatment group. Glu: glucose.
Neuronal CMKLR1 correlates with hyperphosphorylated tau seeding
Recent research has shown that tau pathology may be driven by tau seeding. The abnormally hyperphosphorylated or oligomeric tau can be secreted from neurons into the extracellular space and then be taken up by other normal neurons (Yanamandra et al., 2013; Takeda et al., 2015; Dai et al., 2018; DeVos et al., 2018). Therefore, we explore whether neuronal CMKLR1 affects tau phosphorylation by regulating tau seeding. The above results have shown that tau phosphorylation at Ser199, Thr205, Ser396, and Ser404 sites was significantly increased in the hippocampus of 9-month-old APP/PS1 mice compared with age-matched WT mice (Fig. 4A–C). Then we prepared the brain extracts containing hyperphosphorylated tau (from 9-month-old APP/PS1 mice) or normal phosphorylated tau (from age-matched WT mice), by centrifugation at 5,500 rpm, and injected the supernatant to the ventricles of 6-month-old WT mice and age-matched CMKLR1−/− mice to establish the ICV-hyper-pTau (ICV hyperphosphorylated tau) mice and ICV-nor-pTau (ICV normal phosphorylated tau) mice (Fig. 7A). As expected, 3 weeks after the injection, a significant increase of tau phosphorylation at T205 and S396 was observed in the hippocampus and cortex of WT ICV-hyper-pTau mice, compared with WT ICV-nor-pTau mice (Fig. 7B–G). However, for CMKLR1−/− mice, the injection of brain extracts containing hyperphosphorylated tau did not induce the hyperphosphorylation of tau at T205 and S396 sites in the brain of mice (Fig. 7B–G).
Figure 7.

CMKLR1 deficiency is associated with tau spreading in ICV-Tau model mice. A, Schematic representation of ICV-Tau model mouse construction. The hippocampus extracts containing hyperphosphorylated tau (from 9-month-old APP/PS1 mice) or normal phosphorylated tau (from age-matched WT mice) were centrifuged at 5,500 rpm for 5 min in 4°C, and then the supernatant was injected into the ventricles of 6-month-old WT mice and age-matched CMKLR1−/− mice to establish the ICV-hyper-pTau mice and ICV-nor-pTau mice. Mice were killed 3 weeks after the injection. B, Representative Western blots showing tau hyperphosphorylation at Ser199, Thr205, and Ser396 in the hippocampus of WT and CMKLR1−/− mice receiving ICV injection of the extracts containing hyper-pTau or nor-pTau. Quantification of the immunoreactivity of the blots, normalized against total tau (C) and β-actin (D). E, Representative Western blots showing tau hyperphosphorylation in the cortex of WT and CMKLR1−/− mice receiving ICV injection of the extracts containing hyper-pTau or nor-pTau. Quantification of the immunoreactivity of the blots, normalized against total tau (F) and β-actin (G). Data are mean ± SEM, with at least 2 mice in each group (WT ICV-nor-pTau, n = 2; WT ICV-hyper-pTau, n = 3; CMKLR1−/− ICV-nor-pTau, n = 3; CMKLR1−/− ICV-hyper-pTau, n = 2). *p < 0.05; **p < 0.01; compared with WT ICV-nor-pTau mice. $p < 0.05; $$p < 0.01; compared with WT ICV-hyper-pTau mice.
To further confirm the involvement of neuronal CMKLR1 in tau seeding, we used primary cultures of neurons from the brain of 1-day-old CMKLR1−/− and WT mice. The above result has shown that high glucose (15 and 30 mm) treatment induced a significant concentration-dependent increase in tau phosphorylation in SH-SY5Y cells (Fig. 6B–D). ELISA assay and Western blot detected that the supernatant from SH-SY5Y cells stimulated with high glucose (30 mm) contained human specific tau protein (Fig. 8A,B). Then we collected the supernatant, purified the supernatant with dialysis bags, and then applied it to the mouse primary cultures of neurons. As shown in Figure 8C, we detected the endogenous expression of CMKLR1 in primary cultures of neurons. After 3 d of treatment, human tau uptake was observed in WT mouse primary neurons by immunofluorescence labeling of intracellular human tau (Fig. 8D,E; anti-Tau13 antibody, shown in red). However, no human tau was detected in CMKLR1−/− mouse primary neurons (Fig. 8D,E). These results suggest that CMKLR1 expressed on mouse primary neurons is involved in the uptake of extracellular human tau. Then we further explored the effect of human tau taken up by neurons on the phosphorylation of mouse tau. For quantification of tau proteins (Fig. 8F,G), the immunofluorescence area of human tau (Tau13 antibody, shown in red) and human and mouse tau (Hu and Ms tau, pT205 and pS396; shown in green) was quantified. The colocalization of human tau and Hu and Ms tau (shown in yellow) means the human tau. The area of mouse tau (pT205 and pS396) was calculated by subtracting the fluorescence area of human tau (shown in yellow) from the fluorescence area of Hu and Ms tau (shown in green). As shown in Figure 8F, G, human tau taken up by mouse neurons significantly accelerated the hyperphosphorylation of mouse tau in primary cultures of mouse neurons. All these results suggest that CMKLR1 affects tau phosphorylation through regulating tau seeding.
Figure 8.
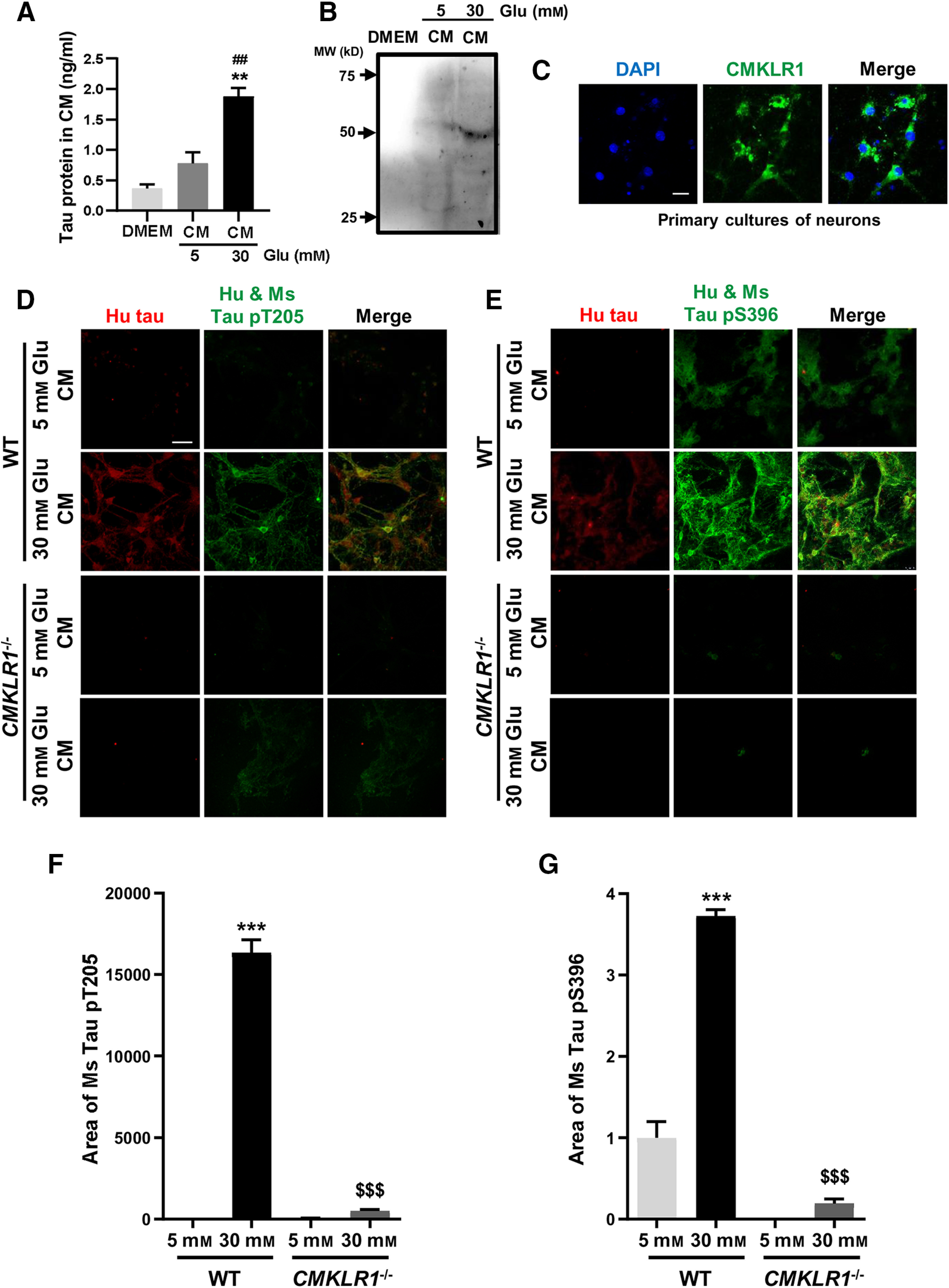
Neuronal CMKLR1 affects tau phosphorylation through regulating tau seeding. A, Tau protein in the supernatant from SH-SY5Y stimulated with 5 or 30 mm glucose was examined using ELISA. Data are mean ± SEM from three separate experiments, each in duplicate or triplicate. **p < 0.01 compared with DMEM medium without glucose treatment. ##p < 0.01 compared with 5 mm glucose treatment group. B, Representative Western blots showing the human specific tau protein in the supernatant from SH-SY5Y stimulated with 5 or 30 mm glucose. C, Representative immunofluorescence images of CMKLR1 (green) in primary cultures of mouse neurons. Scale bar, 50 μm. D–G, The supernatant from SH-SY5Y treated with 5 or 30 mm glucose was collected, purified, and then applied to primary cultures of neurons from WT mice and CMKLR1−/− mice for 3 d. Representative immunofluorescence images of human specific tau protein (red) and human and mouse phosphorylated tau at Thr205 site (green, D) and Ser396 site (green, E) in primary cultures of neurons from WT mice and CMKLR1−/− mice. The primary cultures of neurons were incubated with the purified supernatant form SH-SY5Y treated with 5 or 30 mm glucose. Scale bar, 50 μm. Quantification of the area of mouse phosphorylated tau at pT205 (F) and pS396 (G), calculated by subtracting the fluorescence area of human tau (yellow) from the fluorescence area of Hu and Ms tau (green), showing that human tau taken up by mouse neurons significantly accelerated the hyperphosphorylation of mouse tau in primary cultures of mouse neurons. Data are mean ± SEM from three separate experiments, each in duplicate or triplicate. ***p < 0.001 compared with primary cultures of neurons from WT mice treated with the purified supernatant stimulated with 5 mm glucose. $$$p < 0.001 compared with primary cultures of neurons from WT mice treated with the purified supernatant stimulated with 30 mm glucose. Glu: glucose.
Discussion
We recently identified that CMKLR1 is a new functional receptor for Aβ. The expression of CMKLR1 is increased in the brain of AD patients, and it is colocalized with Aβ in the hippocampus and the cortex of APP/PS1 transgenic mice. Furthermore, Aβ activation of CMKLR1 induces microglial cell migration and internalization of the receptor-Aβ complex in primary cultures of microglia and astrocytes (Peng et al., 2015). All these data suggest that CMKLR1 contributes to the uptake of Aβ. However, it is unclear whether CMKLR1 ameliorates or aggravates the process of AD. In this manuscript, we found that genetic deficiency in CMKLR1 led to a significant increase of Aβ deposition in the brain of APP/PS1 mice, providing the evidence in vivo that CMKLR1 is a key receptor involved in the clearance of Aβ. However, interestingly, we also found that the absence of CMKLR1 markedly decreased the mortality of APP/PS1 mice, improved the cognitive impairment, and attenuated the hyperphosphorylation of tau in the brain of both APP/PS1 mice and ICV-STZ mice. Several receptors have been identified in glial cells as endogenous binding sites for Aβ, and the activation of these receptors may induce phagocytosis of Aβ and affect the progression of AD (El Khoury et al., 2007; Bruban et al., 2011; Frenkel et al., 2013). Frenkel et al. (2013) reported that Scara1, the scavenger receptor for sAβ, is also an Aβ clearance receptor. Genetic deficiency in Scara1 markedly accelerated Aβ accumulation and increased the mortality of APP/PS1 mice (Frenkel et al., 2013). Although both the CMKLR1 and Scara1 have the ability to clear Aβ and the deficiency of these two receptors significantly increased Aβ deposit in the AD mouse brain, the effect on the survival rate of AD mice is reversed. There is currently no report about the effect of Scara1 on cognitive function and tau phosphorylation in the brain of AD mice. Accumulating evidence suggests that the cognitive dysfunction of AD is more closely related to tau pathology (Tracy et al., 2016; DeVos et al., 2017). Therefore, we propose that absence of CMKLR1 improves the cognitive ability and increases the survival rate of AD mice, which may be because of its involvement in the pathologic progress of tau. In addition, formyl peptide receptor 2 (FPR2), another functional receptor for Aβ, could externalize and clear Aβ in macrophages (Le et al., 2001). It has also been reported that the deletion of FPR2 alleviated the spatial learning and memory of ICV-STZ mice, and attenuated the hyperphosphorylation of tau in the hippocampus and cortex of ICV-STZ mice (Zhang et al., 2019a). Therefore, to date, all these studies suggest that, in the middle and late stages of AD progression, cognitive function and survival rate may be affected by many factors, and the content of Aβ deposit in the brain may not play a leading role. This may be one of the reasons for the failure of clinical trials of Aβ antibody treatment for patients with advanced AD in recent years. Our results support the notion that preventing or slowing the progression of tau pathology, such as inhibition of CMKLR1 expression or activation, may benefit patients with advanced AD.
Recent studies have shown that abnormally hyperphosphorylated tau can be secreted from intact neurons into the extracellular space, and then the extracellular tau can be taken up by normal neurons, suggesting that tau pathology may be driven by a prion-like spread mechanism (Yanamandra et al., 2013; Takeda et al., 2015; Dai et al., 2018; DeVos et al., 2018). In this manuscript, we found that CMKLR1 deficiency or inhibition attenuated the hyperphosphorylation of tau in the brains of AD mouse models in vivo and in the neuronal cells in vitro. Moreover, the expression of CMKLR1 on the neurons affected tau phosphorylation by participating in tau spreading. It has been proposed that intracellular tau is released from neurons by different mechanisms of exocytosis, including secretion (Mohamed et al., 2017), exosome (Asai et al., 2015), and neuron death (Shafiei et al., 2017). Also, the extracellular tau can be taken up by neighboring cells, both neurons and glial cells (Santello et al., 2019), by various mechanisms, such as endocytosis (Christianson and Belting, 2014), pinocytosis (Calafate et al., 2016), phagocytosis (McInnes et al., 2018), and mediated by receptors (Bolós et al., 2017; Leyns et al., 2019; Perez et al., 2019). Bolós et al. (2017) reported that extracellular tau binding to CX3CR1, the microglial fractalkine receptor, triggered the internalization of tau by microglia. This result suggests that the interaction of tau with the receptors in microglia may contribute to tau spreading (Asai et al., 2015; Maphis et al., 2015). Our results have shown that CMKLR1 receptor is expressed on neurons, microglia, and astrocytes. Also, CMKLR1 expressed on neurons participates in tau pathology via regulating tau seeding. Therefore, whether CMKLR1 on glial cells contributes to tau seeding needs additional investigation in the future.
In addition, increasing evidence indicates that the inflammatory response induced by sustained activated glial cells is related to tau hyperphosphorylation and cognitive impairment in AD (Fakhoury, 2018; Reichenbach et al., 2019). In this study, we observed that CMKLR1 markedly colocalized with microglia, and the fluorescence intensity of CMKLR1 in microglia was even stronger than in neurons, suggesting that CMKLR1 is abundantly expressed in microglia. This result is consistent with that shown in RNA-seq database provided by Zhang et al. (2014, 2016). Therefore, we verified whether CMKLR1 deficiency affects the activation of microglia. It was found that CMKLR1 deficiency does not reduce microglial activation, and even further increases its activation in the cortex of APP/PS1 mice (data not shown). Our previous findings showed that CMKLR1 activation does not induce the production of inflammatory mediators, such as IL-6, IL-12, and iNOS in microglia (Yu and Ye, 2015), suggesting that CMKLR1 has no effect on microglial activation. Also, in addition to CMKLR1, Aβ may bind to other receptors on microglia, such as the receptor for advanced glycation end product, CD36, CD36/CD47/α6β1-intergrin, CD14/TLR2/TLR4, and FPR2. These receptors are responsible for the activation of microglia and/or production of proinflammatory mediators following Aβ binding (Yu and Ye, 2015). Therefore, CMKLR1 deficiency, leading to the further activation of microglia induced by increased Aβ deposition, may be because of the participation of these receptors.
Impaired glucose utilization in the brain is one of the common pathologic features of AD (Chen et al., 2016). In this study, we established a cellular model of AD with high glucose treatment to induce tau hyperphosphorylation in SH-SY5Y cells. We found that high glucose (15 and 30 mm) treatment for 24 h induced a significant concentration-dependent increase in tau phosphorylation at Ser199, Thr205, and Ser396 sites in SH-SY5Y cells. Lee et al. (2016) reported that high glucose treatment (25 mm) for 24 h increased Aβ secretion and tau phosphorylation at Ser396 site in human neuroblastoma SK-N-MC cells. Their further mechanism experiments found that high glucose treatment induced beta-site APP cleaving enzyme 1 expression, which led to the phosphorylation of AKT and GSK-3β, a major tau kinase leading to tau hyperphosphorylation in AD (Hernandez et al., 2010; Lee et al., 2016). High glucose also induced oxidative stress, which could increase the phosphorylation of GSK-3β by activating calpain (calcium-activated neutral proteinase) (Saito et al., 1993; Liu et al., 2005; Feng et al., 2013). Studies have also found that TLR9/p38 MAPK and ErbB4/mTOR/S6K signaling pathways are related to chronic high glucose treatment-induced tau hyperphosphorylation in primary hippocampal neurons and the differentiated SH-SY5Y cells, respectively (Sun et al., 2017; Nie et al., 2018). Our and previous studies have found that Aβ and chemerin are endogenous ligands of CMKLR1 (Meder et al., 2003; Wittamer et al., 2003; Peng et al., 2015). Lee et al. (2016) reported that high glucose treatment induced Aβ secretion in SK-N-MC cells. Several studies have reported that Aβ can lead to tau hyperphosphorylation in neurons by activating the protein phosphatases or kinases (Takashima et al., 1998; Town et al., 2002; Blurton-Jones and Laferla, 2006; Oliveira et al., 2015). These data suggest that Aβ may induce tau hyperphosphorylation by interacting with neuronal CMKLR1, and CMKLR1 deficiency may alleviate this effect. Therefore, a possible alternative mechanism by which CMKLR1 involved in tau hyperphosphorylation is interaction with Aβ. Additionally, chemerin, also known as retinoic acid receptor responder protein 2, is another endogenous ligand of CMKLR1. RNA-seq database shows the expression of chemerin/retinoic acid receptor responder protein 2 in neurons, microglia, and astrocytes, and it is most expressed in astrocytes (Zhang et al., 2014, 2016). Studies have found that chemerin could decrease insulin-induced AKT phosphorylation and glucose uptake in vascular smooth muscle cells via CMKLR1-dependent mechanism (Neves et al., 2018). Zhang et al. (2019b) demonstrated that chemerin ameliorated neuronal apoptosis partially via activating CMKLR1/CAMKK2/AMPK signaling pathway. However, there are few reports on the role of chemerin in neurons. More experiments are needed to verify how chemerin affects tau phosphorylation in neurons through CMKLR1.
Footnotes
The authors declare no competing financial interests.
This work was supported by National Natural Science Foundation of China Grants 81571027 and 81870835.
References
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. 10.1038/nn.4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blurton-Jones M, Laferla FM (2006) Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res 3:437–448. 10.2174/156720506779025242 [DOI] [PubMed] [Google Scholar]
- Bolós M, Llorens-Martín M, Perea JR, Jurado-Arjona J, Rábano A, Hernández F, Avila J (2017) Absence of CX3CR1 impairs the internalization of tau by microglia. Mol Neurodegener 12:59. 10.1186/s13024-017-0200-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruban J, Maoui A, Chalour N, An N, Jonet L, Feumi C, Treton J, Sennlaub F, Behar-Cohen F, Mascarelli F, Dinet V (2011) CCR2/CCL2-mediated inflammation protects photoreceptor cells from amyloid-beta-induced apoptosis. Neurobiol Dis 42:55–72. 10.1016/j.nbd.2011.01.004 [DOI] [PubMed] [Google Scholar]
- Calafate S, Flavin W, Verstreken P, Moechars D (2016) Loss of bin1 promotes the propagation of tau pathology. Cell Rep 17:931–940. 10.1016/j.celrep.2016.09.063 [DOI] [PubMed] [Google Scholar]
- Cash JL, Hart R, Russ A, Dixon JP, Colledge WH, Doran J, Hendrick AG, Carlton MB, Greaves DR (2008) Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J Exp Med 205:767–775. 10.1084/jem.20071601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liang Z, Blanchard J, Dai CL, Sun S, Lee MH, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX (2013) A non-transgenic mouse model (icv-STZ mouse) of Alzheimer's disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol Neurobiol 47:711–725. 10.1007/s12035-012-8375-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang J, Zhang B, Gong CX (2016) Targeting insulin signaling for the treatment of Alzheimer's disease. Curr Top Med Chem 16:485–492. 10.2174/1568026615666150813142423 [DOI] [PubMed] [Google Scholar]
- Christianson HC, Belting M (2014) Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol 35:51–55. 10.1016/j.matbio.2013.10.004 [DOI] [PubMed] [Google Scholar]
- Dai CL, Hu W, Tung YC, Liu F, Gong CX, Iqbal K (2018) Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 × Tg-AD mice. Alzheimers Res Ther 10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, Zanardi TA, Kordasiewicz HB, Swayze EE, Bennett CF, Diamond MI, Miller TM (2017) Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9:eaag0481 10.1126/scitranslmed.aag0481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, Commins C, Gonzalez JA, Dooley PM, Frosch MP, Hyman BT (2018) Synaptic tau seeding precedes tau pathology in human Alzheimer's disease brain. Front Neurosci 12:267. 10.3389/fnins.2018.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 13:432–438. 10.1038/nm1555 [DOI] [PubMed] [Google Scholar]
- Fakhoury M. (2018) Microglia and astrocytes in Alzheimer's disease: implications for therapy. Curr Neuropharmacol 16:508–518. 10.2174/1570159X15666170720095240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Xia Y, Yu G, Shu X, Ge H, Zeng K, Wang J, Wang X (2013) Cleavage of GSK-3β by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK-3β activity induced by H2O2. J Neurochem 126:234–242. 10.1111/jnc.12285 [DOI] [PubMed] [Google Scholar]
- Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, Farfara D, Kingery ND, Weiner HL, El Khoury J (2013) Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat Commun 4:2030. 10.1038/ncomms3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, et al. (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14:388–405. 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez F, Gomez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J (2010) GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol 223:322–325. 10.1016/j.expneurol.2009.09.011 [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I (2010) Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res 7:656–664. 10.2174/156720510793611592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovova N, Handzusova M, Hanes J, Kontsekova E, Novak M (2008) High-yield purification of fetal tau preserving its structure and phosphorylation pattern. J Immunol Methods 339:17–22. 10.1016/j.jim.2008.07.014 [DOI] [PubMed] [Google Scholar]
- Jiang L, Ash PE, Maziuk BF, Ballance HI, Boudeau S, Abdullatif AA, Orlando M, Petrucelli L, Ikezu T, Wolozin B (2019) TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol 137:259–277. 10.1007/s00401-018-1937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AJ, Davenport AP (2018) International Union of Basic and Clinical Pharmacology CIII: chemerin receptors CMKLR1 (Chemerin1) and GPR1 (Chemerin2) nomenclature, pharmacology, and function. Pharmacol Rev 70:174–196. 10.1124/pr.116.013177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Y, Gong W, Tiffany HL, Tumanov A, Nedospasov S, Shen W, Dunlop NM, Gao JL, Murphy PM, Oppenheim JJ, Wang JM (2001) Amyloid β42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci 21:RC123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Ryu JM, Jung YH, Lee SJ, Kim JY, Lee SH, Hwang IK, Seong JK, Han HJ (2016) High glucose upregulates BACE1-mediated Aβ production through ROS-dependent HIF-1α and LXRα/ABCA1-regulated lipid raft reorganization in SK-N-MC cells. Sci Rep 6:36746. 10.1038/srep36746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns CE, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VM, Ulrich JD, Holtzman DM (2019) TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci 22:1217–1222. 10.1038/s41593-019-0433-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SQ, Yu Y, Han JZ, Wang D, Liu J, Qian F, Fan GH, Bucala R, Ye RD (2015) Deficiency of macrophage migration inhibitory factor attenuates tau hyperphosphorylation in mouse models of Alzheimer's disease. J Neuroinflammation 12:177. 10.1186/s12974-015-0396-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX (2005) Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem 280:37755–37762. 10.1074/jbc.M507475200 [DOI] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138:1738–1755. 10.1093/brain/awv081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes J, Wierda K, Snellinx A, Bounti L, Wang YC, Stancu IC, Apostolo N, Gevaert K, Dewachter I, Spires-Jones TL, De Strooper B, De Wit J, Zhou LJ, Verstreken P (2018) Synaptogyrin-3 mediates presynaptic dysfunction induced by tau. Neuron 97:823–835.e8. 10.1016/j.neuron.2018.01.022 [DOI] [PubMed] [Google Scholar]
- Meder W, Wendland M, Busmann A, Kutzleb C, Spodsberg N, John H, Richter R, Schleuder D, Meyer M, Forssmann WG (2003) Characterization of human circulating TIG2 as a ligand for the orphan receptor ChemR23. FEBS Lett 555:495–499. 10.1016/s0014-5793(03)01312-7 [DOI] [PubMed] [Google Scholar]
- Mohamed NV, Desjardins A, Leclerc N (2017) Tau secretion is correlated to an increase of Golgi dynamics. PLoS One 12:e0178288. 10.1371/journal.pone.0178288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J (1982) Place navigation impaired in rats with hippocampal lesions. Nature 297:681–683. 10.1038/297681a0 [DOI] [PubMed] [Google Scholar]
- Neves KB, Nguyen Dinh Cat A, Alves-Lopes R, Harvey KY, Costa RM, Lobato NS, Montezano AC, Oliveira AM, Touyz RM, Tostes RC (2018) Chemerin receptor blockade improves vascular function in diabetic obese mice via redox-sensitive and Akt-dependent pathways. Am J Physiol Heart Circ Physiol 315:H1851–H1860. 10.1152/ajpheart.00285.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie SD, Li X, Tang CE, Min FY, Shi XJ, Wu LY, Zhou SL, Chen Z, Wu J, Song T, Dai ZJ, Zheng J, Liu JJ, Wang S (2018) High glucose forces a positive feedback loop connecting ErbB4 expression and mTOR/S6K pathway to aggravate the formation of tau hyperphosphorylation in differentiated SH-SY5Y cells. Neurobiol Aging 67:171–180. 10.1016/j.neurobiolaging.2018.03.023 [DOI] [PubMed] [Google Scholar]
- Schweers O, Man A, Mandelkow E (1994) Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for p-structure. J Biol Chem 269:24290–24297. [PubMed] [Google Scholar]
- Oliveira JM, Henriques AG, Martins F, Rebelo S, da Cruz e Silva OA (2015) Amyloid-β modulates both AβPP and tau phosphorylation. J Alzheimers Dis 45:495–507. 10.3233/JAD-142664 [DOI] [PubMed] [Google Scholar]
- Peng L, Yu Y, Liu J, Li S, He H, Cheng N, Ye RD (2015) The chemerin receptor CMKLR1 is a functional receptor for amyloid-β peptide. J Alzheimers Dis 43:227–242. 10.3233/JAD-141227 [DOI] [PubMed] [Google Scholar]
- Perez M, Avila J, Hernandez F (2019) Propagation of tau via extracellular vesicles. Front Neurosci 13:698. 10.3389/fnins.2019.00698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362:329–344. 10.1056/NEJMra0909142 [DOI] [PubMed] [Google Scholar]
- Reichenbach N, Delekate A, Plescher M, Schmitt F, Krauss S, Blank N, Halle A, Petzold GC (2019) Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer's disease model. EMBO Mol Med 11:e9665 10.15252/emmm.201809665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries M, Sastre M (2016) Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci 8:160. 10.3389/fnagi.2016.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA (1993) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA 90:2628–2632. 10.1073/pnas.90.7.2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Toni N, Volterra A (2019) Astrocyte function from information processing to cognition and cognitive impairment. Nat Neurosci 22:154–166. 10.1038/s41593-018-0325-8 [DOI] [PubMed] [Google Scholar]
- Shafiei SS, Guerrero-Muñoz MJ, Castillo-Carranza DL (2017) Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci 9:83. 10.3389/fnagi.2017.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Xiao Q, Luo C, Zhao Y, Pu D, Zhao K, Chen J, Wang M, Liao Z (2017) High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp Cell Res 359:312–318. 10.1016/j.yexcr.2017.07.032 [DOI] [PubMed] [Google Scholar]
- Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, Ishiguro K, Yamaguchi H (1998) Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res 31:317–323. 10.1016/S0168-0102(98)00061-3 [DOI] [PubMed] [Google Scholar]
- Takeda S, Wegmann S, Cho HS, Devos SL, Commins C, Roe AD, Nicholls SB, Carlson GA, Pitstick R, Nobuhara CK, Costantino I, Frosch MP, Muller DJ, Irimia D, Hyman BT (2015) Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer's disease brain. Nat Commun 6:8490. 10.1038/ncomms9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Zolton J, Shaffner R, Schnell B, Crescentini R, Wu Y, Zeng J, DelleDonne A, Obregon D, Tan J, Mullan M (2002) p35/Cdk5 pathway mediates soluble amyloid-beta peptide-induced tau phosphorylation in vitro. J Neurosci Res 69:362–372. 10.1002/jnr.10299 [DOI] [PubMed] [Google Scholar]
- Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li YQ, Zhou YG, Le D, Lo I, Ponnusamy R, Cong X, Schilling B, Ellerby LM, Huganir RL, Gan L (2016) Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90:245–260. 10.1016/j.neuron.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I, Brezillon S, Tyldesley R, Blanpain C, Detheux M, Mantovani A, Sozzani S, Vassart G, Parmentier M, Communi D (2003) Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med 198:977–985. 10.1084/jem.20030382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. (2006) Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 12:1005–1015. 10.1038/nm1484 [DOI] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma SM, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80:402–414. 10.1016/j.neuron.2013.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Li S, Xu H, Walsh DM, Selkoe DJ (2017) Large soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 37:152–163. 10.1523/JNEUROSCI.1698-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Ye RD (2015) Microglial Aβ receptors in Alzheimer's disease. Cell Mol Neurobiol 35:71–83. 10.1007/s10571-014-0101-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wang D, Gong P, Lin A, Zhang Y, Ye RD, Yu Y (2019a) Formyl peptide receptor 2 deficiency improves cognition and attenuates tau hyperphosphorylation and astrogliosis in a mouse model of Alzheimer's disease. J Alzheimers Dis 67:169–179. 10.3233/JAD-180823 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu N, Ding Y, Doycheva DM, Zhang Y, Li Q, Flores J, Haghighiabyaneh M, Tang J, Zhang JH (2019b) Chemerin reverses neurological impairments and ameliorates neuronal apoptosis through ChemR23/CAMKK2/AMPK pathway in neonatal hypoxic-ischemic encephalopathy. Cell Death Dis 10:97. 10.1038/s41419-019-1374-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89:37–53. 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]