


Abstract
Myocyte enhancer factor-2B (MEF2B) has the unique capability of binding to its DNA target sites with a degenerate motif, while still functioning as a gene-specific transcriptional regulator. Identifying its DNA targets is crucial given regulatory roles exerted by members of the MEF2 family and MEF2B’s involvement in B-cell lymphoma. Analyzing structural data and SELEX-seq experimental results, we deduced the DNA sequence and shape determinants of MEF2B target sites on a high-throughput basis in vitro for wild-type and mutant proteins. Quantitative modeling of MEF2B binding affinities and computational simulations exposed the DNA readout mechanisms of MEF2B. The resulting binding signature of MEF2B revealed distinct intricacies of DNA recognition compared to other transcription factors. MEF2B uses base readout at its half-sites combined with shape readout at the center of its degenerate motif, where A-tract polarity dictates nuances of binding. The predominant role of shape readout at the center of the core motif, with most contacts formed in the minor groove, differs from previously observed protein–DNA readout modes. MEF2B, therefore, represents a unique protein for studies of the role of DNA shape in achieving binding specificity. MEF2B–DNA recognition mechanisms are likely representative for other members of the MEF2 family.
INTRODUCTION
Interactions between proteins and their DNA target sites are essential components in the regulation of gene expression (1). Investigating protein–DNA binding recognition mechanisms is an important step in understanding how proteins select their in vivo target sites (2). This intricate recognition process could be affected by nucleotide variations that influence protein coding or DNA regulatory regions (3). One way that variations within regulatory regions affect gene regulation is by altering how transcription factors (TFs) recognize their DNA target sites (4). Structural mechanisms of binding specificity for protein–DNA interactions have been extensively studied (5–10), with base and shape readout mechanisms comprising the two primary categories of recognition modes (11). Yet, the lack of a universal code to explain how proteins select their binding sites (1) highlights the need for further studies aimed at disentangling steps in the protein–DNA recognition process and decoding their interplay.
Members of the myocyte enhancer factor-2 (MEF2) family of TFs display a remarkable DNA binding function: while acting as gene-specific transcriptional regulators (12,13), they are capable of binding degenerate sequences (14,15). The four human MEF2 family members (MEF2A, B, C and D) play important roles in different biological processes (16–18). Mutations in these proteins have been associated with distinct pathologies, including cardiac disease (19), neuronal disorders (20), and cancer (21,22). Establishing a thorough and detailed understanding of the intricate DNA binding properties of the MEF2 family is one of the first critical steps towards unraveling how MEF2 proteins exert their functions in vivo.
One of the main DNA recognition elements of MEF2 is the MADS (MCM1, Agamous, Deficiens and SRF)-box domain (12). Together with the MEF2 domain, the MADS-box domain forms the DNA binding domain (DBD) and mediates DNA binding, dimerization and cofactor recruitment (12,23,24). Members of the MADS-box family of proteins recognize CArG (C-A/T-rich-G)-box sequence elements that comprise variable consensus motifs (25). MEF2 TFs recognize their DNA consensus motif 5′-YTAW4TAR-3′ (Y = C or T; W = A or T; R = A or G) (14,15,26) as homo- or heterodimers (24,27).
The DNA binding mode of the MADS-box family of TFs is fundamentally different from that observed for other homo- or heterodimeric proteins. For example, basic helix-loop-helix (bHLH) TFs bind as dimers through extensive base contacts in the major groove of the E-box core motif (28). Glucocorticoid receptors (GRs) bind to adjacent half-sites separated by a spacer, using a base and shape readout mechanism that is similar to the DNA readout mechanism employed by Hox proteins (29,30). In contrast, MEF2 homodimers bind through a few base-specific major groove contacts to only the peripheral YTA and TAR half-sites of the 10-base pair (bp) core motif, using basic amino acids to form extensive contacts in the minor groove of the central W4 region of the core motif (24,27,31). The ability of MEF2 TFs to bind degenerate sequences in the W4 region suggests that their DNA recognition mechanisms go beyond recognizing the nucleotide identity of each individual position within the binding site. This mechanism may account for the DNA binding specificities that are observed in a wide range of plant MADS-box proteins that recognize CArG boxes, such as SEPALLATA3 (32). In this context, the low percentage of CArG boxes that are bound in vivo cannot be explained by the number of CArG boxes that are observed in the genome (25,33).
One question raised by our current knowledge of MEF2 binding is how a relatively small number of base-specific contacts in the major groove, coupled with a degenerate sequence in the central AT-rich region, can dictate DNA recognition. It is particularly unclear how variations of A and T nucleotides within the central degenerate 4-bp AT-rich region of the core motif interfere with protein–DNA binding and contribute to binding specificity. To answer these questions in the context of MEF2 binding, we comprehensively investigated the DNA binding mechanisms of MEF2B. Recent evidence suggests that MEF2B takes on regulatory roles across numerous tissues (34), and is involved in the development of B-cell lymphoma (21,22,35). Additionally, MEF2B exhibits a more distinct protein sequence compared to other MEF2 family members (12).
To investigate the mechanisms governing MEF2B–DNA binding specificity, we interrogated DNA binding preferences of MEF2B using a high-throughput binding assay. Initial analysis of co-crystal structures suggested that MEF2B TFs employ both base and shape readout modes, which prompted us to perform a high-throughput study of the binding sites of MEF2B using systematic evolution of ligands by exponential enrichment combined with massively parallel sequencing (SELEX-seq) (36,37). We quantitatively modeled MEF2B binding by analyzing thousands of sequences and further investigated the unique signatures of binding sites that revealed base and shape readout preferences. Interestingly, our analysis indicates that A-tract polarity is a source of binding specificity. Some of these signatures are likely to be TF family-specific, and therefore shared with MEF2A, C and D, and MADS proteins more broadly.
MATERIALS AND METHODS
Structural and computational analysis of DNA derived from co-crystal structures of protein–DNA complexes
To obtain DNA structural features from crystal structures of MEF2–DNA complexes (24,27,31), DNA was analyzed with the Curves 5.3 algorithm (38). Minor groove width at each nucleotide position was calculated according to a previously reported protocol (39). Electrostatic potential at the center of the DNA minor groove was calculated with DelPhi (40), which solves the nonlinear Poisson-Boltzmann equation at physiological ionic strength of 0.145 M. The AMBER force field was used to assign partial charges and atomic radii (41), as previously described (39). Protein–DNA interactions were analyzed and visualized with DNAproDB (https://dnaprodb.usc.edu/) (42,43).
Protein expression and purification
The DBD of wild-type human MEF2B (residues 1–93) containing a C-terminal His-tag (LVPRGSKLAAALEHHHHHH) was cloned into pET30-b(+). Protein was expressed in Escherichia coli Rosetta™(DE3) pLysS (MilliporeSigma, Burlington, MA, USA) as follows. Briefly, culture was grown at 37°C in 2× YT media in the presence of kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml) to an OD600 of ∼0.6. Protein expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside at 23°C overnight. Cell pellets were stored at −20°C. For protein purification, cells were lysed by sonication in lysis buffer (250 mM NaCl, 50 mM HEPES at pH 7.6, 1 mM EDTA, 1 mM DTT and 5% glycerol) containing protease inhibitors, followed by ultracentrifugation to separate soluble and insoluble fractions. Purification by chromatography was performed by using SP Sepharose (GE Healthcare, Chicago, IL, USA) and Ni-NTA (Qiagen, Germantown, MD, USA), according to the manufacturer's instructions. Amicon filter units (3K device; MilliporeSigma) were used for buffer exchange and protein concentration. Purified MEF2B was stored in buffer containing 250 mM NaCl, 10 mM HEPES (pH 7.6), 1 mM EDTA, 1 mM DTT and 5% glycerol. Protein concentration was estimated by NanoDrop™.
Oligonucleotides
All oligonucleotides were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA). A complete list of oligonucleotides is available in Supplementary Table S1.
SELEX-seq protocol for MEF2B
The DNA library used for SELEX-seq (36,37) was designed to contain a 16-bp random region and allowed for multiple indexes to be used in the same sequencing run (for sequences, refer to Supplementary Table S1). The initial oligonucleotide library containing a 16-bp random region was commercially obtained from IDT using the hand-mix option as described elsewhere (36). An SR1 primer and a 60-bp oligonucleotide (Selex-Lib), which contains regions compatible with Illumina sequencing, were annealed to allow for dsDNA library generation (SELEX-library) through a Klenow reaction, using DNA Polymerase I, Large (Klenow) fragment (New England Biolabs [NEB], Ipswich, MA, USA), according to an established protocol (36). The library was gel-purified by using the minElute kit (Qiagen). A similar procedure was carried out by using a 5′ 6-FAM (fluorescein) SR1 (SR1-FAM) to generate a 5′ 6-FAM-labeled library and positive or negative control probes (Supplementary Table S1) for the electrophoretic mobility shift assay (EMSA). Additional unlabeled EMSA probes were generated by annealing oligonucleotides (Supplementary Table S1). A schematic representation of the SELEX-seq protocol is shown in Supplementary Figure S1.
Binding reactions were carried out in 150 mM NaCl, 10 mM HEPES (pH 7.6), 0.5 mM EDTA, 0.5 mM DTT, 0.05 mg/ml BSA and 5% glycerol in a 30-μl binding reaction with 200 nM of SELEX-library DNA and 20 nM of MEF2B DBD dimer. EMSA was performed on an 8% polyacrylamide gel. To isolate bound fragments, a reaction was run in parallel using the 5′ 6-FAM library in a PharosFX™ imager (Bio-Rad, Hercules, CA, USA). Bound fractions were gel-purified and isolated.
Isolated DNA was subjected to a 15-cycle polymerase chain reaction (PCR), according to a published protocol (36,37), using primers SF1 and SR1 (Supplementary Table S1), followed by PCR-purification with the minElute PCR purification kit (Qiagen) and quantification by NanoDrop™. Product obtained in round 1 (R1) of selection was used as the template for round 2 (R2) of selection (in a binding reaction following the same steps described above) or as the PCR template for preparing the final sequencing library. For the latter, Phusion polymerase (NEB) was used in a four-cycle PCR as described elsewhere (36,37), with RP1 and a variable ‘RPI#’ indexing primer (Supplementary Table S1). Sequencing was performed on a NextSeq 500 platform (Illumina, San Diego, CA, USA) at the USC Norris Molecular Genomics Core.
SELEX-seq data processing and analysis of DNA features
Sequencing data were pre-processed to trim the 3′ ends of reads containing the adapters and indexing regions. Data were analyzed with the SELEX R-package version 1.8.0 (36) available at Bioconductor (https://bioconductor.org/packages/SELEX). A fifth-order Markov model was generated based on round zero (R0) data. Relative binding affinities for oligomers of length k = 10 (10-mers) with counts >100 were estimated based on the SELEX-seq method (36). Refined dataset tables containing relative affinities for respective 10-mers after R2 were used to perform DNA sequence and shape analysis, as described below. Whenever the MEF2B motif was required for binding site alignment and filtering, the known consensus DNA sequence motif for the MEF2 family YTAW4TAR was used, unless otherwise stated.
We considered two main classes of DNA features: DNA sequence and shape. We refer to DNA shape as a set of four sequence-dependent local DNA structural features per bp (minor groove width and propeller twist) or bp step (helix twist and roll) (44). All data analysis was performed with R version 3.4. For the initial analysis of base and shape readout signatures, 10-mers with relative affinity >0.7 were selected and aligned based on the consensus motif YTAW4TAR. We excluded 10-mers containing shifted motifs (additional nucleotides at the 5′ or 3′ flanks). A position weight matrix (PWM) was generated by using the MEME Suite platform (45) assuming palindromic sites. Averages of DNA shape features were calculated based on DNA shape predictions obtained with the DNAshape method (46), as described below. DNA sequence and shape signatures were comprehensively analyzed for sequences based on alignment with the consensus motif, allowing for a variable number of mismatches within the core binding site (for details, refer to Results). DNA sequence analysis was performed to characterize k-mer signatures of the binding sites. For enrichment analysis, the highest affinity 10-mer CTAAAAATAG was used. Point mutations at every position within the binding site were considered to generate a position-specific affinity matrix (47). Binding free energies for each nucleotide at each position were computed as ΔΔG/RT (47) and used as the energy logo representation. Code used for data analysis is available in the GitHub repository https://github.com/acdantas/mef2-selexseq.
ChIP-seq analysis
ChIP-seq raw sequencing reads from publicly available data (48) targeting MEF2B were used for additional analysis. Two replicates and their inputs were aligned to hg38 using the BWA-MEM algorithm implemented in bwa version 0.7.17 (49). Homer (50) version 4.9.1 was used to call differentially bound peaks, and bedtools (51) version 2.27.1 was used to extract regions from the two replicates that overlapped by at least 1 bp. For overlapping regions, the start point was considered as the minimum starting point between two replicates, and the end point was considered as the maximum. Resulting sequences were scanned for DNA sequences of interest. Scripts used to automate the process are available on the GitHub repository https://github.com/bhcooper/ChIP-seq_analysis.
High-throughput DNA shape prediction
DNA shape features were predicted with the R-package DNAshapeR version 1.4.0 available at Bioconductor (https://bioconductor.org/packages/DNAshapeR) (52), which is based on the DNAshape method (46). This approach allows for prediction of sequence-dependent DNA shape features based on a sliding pentamer window (53). For heat map representations of DNA shape features, filtered sequences were ordered based on relative binding affinity, and predicted shape feature values were binned. The resulting heat map represented the mean average of shape features of each bin (rows) for each position (columns) of the 10-mer. Significance levels of differences in shape features for high- and low-affinity binding sites were given by the P value, calculated with a one-sided Mann–Whitney U test.
The Monte Carlo (MC)-based DNA shape predictions are a high-throughput approach (46,52). Alternative approaches with lower throughput and lesser statistical coverage of the sequence space include molecular dynamics (MD) simulations of unbound DNA fragments (54) and experimentally solved 3D structures of oligonucleotides using X-ray crystallography and NMR spectroscopy as curated in the Nucleic Acid Database (55). A previous study using these DNA features derived from MD simulations and experimental structures confirmed models derived from MC-derived DNA shape features (53). The MC-based DNA shape predictions were experimentally validated in hydroxyl radical cleavage measurements (56).
Molecular dynamics simulations
To perform MD simulations with Gromacs 5.1.4 (57), we selected a starting co-crystal structure of a MEF2B–DNA complex (PDB ID: 1TQE) (58), which is the available co-crystal structure for bound MEF2B DBD with the largest protein–DNA interface. The co-crystal structure was solvated with explicit water molecules in a cubic box in which the solute was ≥1.5 Å from its boundaries, and charge neutrality of the system was obtained by replacing some water molecules with sodium ions. The solute and ions were modeled with the AMBER99-parmbsc1 force field (59,60), while the TIP3P model was adopted for the solvent. The solvated system was equilibrated in the NPT ensemble at a temperature of 300 K and pressure of 1 bar following a standard minimization-equilibration protocol. A time step of 2 fs was used to integrate Newton's equation of motion for a production run of 1 μs. Trajectories were obtained with the same approach for MEF2B mutants. All MD parameter files used are provided in the GitHub repository https://github.com/bhcooper/MDAnalysis.
For the analysis presented here, dynamical frames were retained every 1 ns. Clustering was performed on each trajectory based on solute heavy atoms, with an RMSD cut-off of 0.18 nm, yielding the most representative structure as the centroid of the most populated cluster. For the most representative structure of each trajectory, the average minimum distance between every pair of interface residues (amino acids and nucleotides) was calculated based on the two closest atoms in each frame. Final distances at the MEF2B–DNA interface were discretized into 40 bins and provided in xpm format. Residue distances from different MD simulations were compared by subtracting their corresponding matrices and are shown as difference contact maps. As an additional analysis, principal component analysis on heavy atoms was applied with Gromacs tools, to characterize the mobility of the complex throughout the simulation.
L2-regularized multiple linear regression
An L2-regularized multiple linear regression (MLR) model with 10-fold cross-validation (61,62) was trained to predict binding affinities based on experimentally obtained relative binding affinities from the SELEX-seq data for MEF2B-bound sequences. Datasets utilized for MLR included sequences (10-mers) with counts > 100 from R2 of selection. These sequences were aligned based on the consensus motif and allowed a variable number of mismatches (see Results). Trained models encoded features based on sequence and/or shape parameters. DNA sequence features included k-mers (1-mer, 2-mer, and 3-mer). DNA shape features included helix twist, minor groove width, propeller twist, and roll. Model performance was specified by the coefficient of determination (R2). To determine which binding site positions had the largest contributions to model performance, we further applied a feature selection approach (61,63), whereby trained models had shape features added or removed one position at a time.
RESULTS AND DISCUSSION
Characterization of sequence-specific variations of MEF2B binding sites reveals preferred DNA signatures in and outside the core binding site
Analysis of available MEF2–DNA co-crystal structures (23,24,27,31) showed that the major DNA binding elements of MEF2—the N-terminal tail and DNA binding helix H1—interacted extensively with the minor groove and phosphodiester backbone of DNA. Moreover, these interactions accounted for the majority of protein–DNA interactions (Figure 1A and Supplementary Figure S2). Major groove contacts accounted for relatively few base-specific contacts (Supplementary Figure S2). Despite this, MEF2 proteins had a strong preference for AT-rich regions. Throughout the remainder of the manuscript, we use a simplified description of the DNA binding site, referring to different regions as (i) the peripheral 3-bp half-sites (underlined outer regions of the core binding site, based on consensus motif YTAW4TAR), also denoted as ‘half-sites’ and (ii) the central 4-bp core region (underlined central W4 region of the core binding site, based on consensus motif YTAW4TAR), denoted as the ‘central core’.
Figure 1.

Sequence variations for MEF2B binding sites. (A) Major MEF2B–DNA interactions based on co-crystal structure of MEF2B in complex with DNA (PDB ID: 1N6J) (27) are observed at the DNA minor groove and backbone. Amino acids that interact with DNA, including positively charged residues in the vicinity of DNA, are shown in stick representation. Different views are shown to depict binding recognition by helix H1 and N-terminal tail regions. (B) PWM obtained from SELEX-seq data using MEME Suite (45). Analyzed sequences were obtained after two rounds of selection for top 10-mers with relative affinity >0.7. (C) Strip chart showing relative binding affinities for 10-mers displaying full or partial matches to MEF2B consensus motif, represented by YTAW4TAR (Y = C or T; W = A or T; R = A or G). Variations in core motif are represented by underlined regions, with W4, YTA and TAR denoting regions that deviate from W4, YTA and TAR, respectively. (D) Heat map of relative affinities for triplet variations at 5′ peripheral half-site (YTAW4TAR) based on central core (W4) preferences.
We obtained relative binding affinities from successive rounds of SELEX-seq experiments (Supplementary Figure S1). Validation of library design was performed by EMSA. Data from R2 of selection with a k-mer length of 10-bp were chosen for analysis because they maximized information gain (Supplementary Figure S3). The 10-mer with highest relative binding affinity was CTAAAAATAG, in agreement with the consensus motif YTAW4TAR for MEF2 family members (14,26). The PWM generated from the most enriched k-mers (relative affinity > 0.7) revealed that some positions within the binding site displayed greater sequence conservation than others (Figure 1B). A PWM inherently assumes that every position contributes independently to binding affinity. However, promiscuity of the central W4 region raised the question of whether, for example, the T at position −2 favored an A or a T at position −1.
Binding sites conforming to this consensus sequence exhibited some of the highest observed affinities (Figure 1C). However, the presence of several ambiguous nucleotides still allowed for substantial variability within the set of consensus-conforming binding sites. Variations at the 3-bp half-sites of consensus motif YTAW4TAR had different effects on binding affinity (Figure 1C, D). Certain 10-mers that deviated from the consensus motif and displayed variations at the 3-bp half-sites (i.e. lack of the CTA) were within the highest affinity sites (Figure 1D). Although these sites varied mainly in only one nucleotide with respect to the consensus motif, comparison of the relative binding affinity values suggested preferences for optimal base compositions at specific nucleotide positions that were more important for high-affinity sites (Figure 1D). For example, a variation at position −5 was less detrimental to binding as long as a TpA bp step at positions −4 and −3 was present (Figure 1D). Interestingly, sequences with a CpC bp step at positions −5 and −4 exhibited high enrichment relative to other sequences with a mismatch at position −4 (Figure 1D). Although most optimal sites presented a T at position −4, we observed some C substitutions that did not drastically reduce binding affinity (Figure 1D). Notably, the CpC dinucleotide observed in our studies was previously reported at in vivo target sites of MEF2B (14,48) and was part of the consensus motif of binding sites of serum response factor (SRF) (64,65), another MADS-box protein.
In addition to exploring variations at peripheral half-sites, we analyzed variations within the central W4 region. The highest affinity site contained the longest possible A-tract (a run of at least three consecutive As and Ts without a TpA step) in this region, and most high-affinity sequences displayed an A-tract within the core-binding site (Figure 1D). Based on interrogation of AT-rich regions at the central 4-bp region, diverse combinations of As and Ts in the W4 core had different effects on relative binding affinity (Figure 1C, D). Due to the greater flexibility introduced by TpA steps (called ‘hinge’ steps due to weak stacking interactions (66)), the effect of the central core on MEF2B binding affinity is unclear.
Along with the DBD, MEF2 family members display a transcriptional activation domain (TAD) (12). The TAD is the most divergent region across MEF2 family members (12) and may contribute to the activation of different sets of genes across the family. For example, studies have shown that post-translational modification of the TAD modulates MEF2D binding to the promoter of myogenin (67). The presence of cofactors that interact with the DBD can also mediate DNA binding (68).
By uncovering the interplay between individual nucleotides of the binding site, our study provides support for the overall DNA recognition mode by MEF2 TFs. Results from our selection experiment corroborate the reported consensus sequence motif (15,48) and highlight the potential importance of higher order features such as DNA shape, as is suggested by crystallographic studies (24). Whereas variations at peripheral half-sites can be associated with position-specific nucleotide preferences, the central core displays a degenerate recognition mode that likely involves recognition of intrinsic DNA shape characteristics and conformational flexibility, which is usually increased at AT-rich regions.
Position-specific variations at MEF2 target sites suggest that A-tract polarity is a crucial component that affects binding
To examine effects of any single-nucleotide variation on binding affinity, we first determined the extent to which any substitution at the core affected binding based on the highest affinity sequence, CTAAAAATAG. Variations toward the 3′ end of the binding site, at nucleotide positions +2, +3 and +4, had the greatest effect on binding energy, as visualized by an energy logo (Figure 2A, Supplementary Figure S4A). This observation was surprising because the peripheral half-sites of MEF2B are palindromic to each other and not expected to exert a dissimilar effect on recognition, especially considering that MEF2B binds as a homodimer. The most important positions were located 3′ of A-tracts, which influence the structural characteristics of flanking sequences differently at their 5′ versus 3′ ends. This phenomenon, known as A-tract polarity (69,70), may explain why we see this difference in selectivity at peripheral half-sites.
Figure 2.

Effect of nucleotide variations on MEF2B binding. (A, B) Comparison of relative affinities for each of the central four nucleotide positions, considering all sequences based on the CTAW4TAG reference motif. (A) Affinity logos generated based on the reference sequence show effects of single-nucleotide substitutions on a MEF2B target site. (B) Mean average relative affinity based on each nucleotide substitution. (C, D) Box plots showing changes in relative binding affinities for (C) A→ G and A→C substitutions at each position (−2, −1, +1, +2) or (D) for AAAA to alternative indicated 4-mers. (E, F) Bar plots showing numbers of specific sequence hits (as indicated) in ChIP-seq peaks.
Interestingly, the polarity effect was not observed for palindromic sequences (Supplementary Figure S4A). In this case, positions where single-nucleotide substitutions had the largest effect on binding were those with the greatest number of base-specific contacts, although mainly in the minor groove (Supplementary Figure S2). Largest changes in binding affinity for variations in AT-rich regions were seen for positions −4/+4, −3/+3 and −2/+2, but these changes were dependent on the A-tract polarity. Nucleotide substitutions at these positions could affect base-specific contacts with G2, R3 and K23, which participate in base readout according to MEF2A crystallographic studies (24). With the exception of the major-groove–contacting residue K23, most residues interacted with the sequence-degenerate DNA minor groove (24). Surprisingly, substitutions at the first or last position of the binding site (positions −5/+5), which represent the few major groove recognition nucleotides, exhibited one of the smallest effects on binding energy (Figure 2A, Supplementary Figure S4A).
To examine if the effects of position-specific nucleotide variations can be generalized, we analyzed relative affinities of all sequences conforming to CTAW4TAG (Figure 2B and C, Supplementary Figure S4B). We considered relative binding affinities of each reference k-mer and all possible single-nucleotide substitutions at each of the four central positions for every sequence with alternative AT-rich central 4-mers (Figure 2B). We also investigated affinity changes for substitutions of individual nucleotide pairs, wherein a reference higher affinity nucleotide was substituted for an alternative lower affinity one (Figure 2C). Although the highest-affinity sequence could tolerate C or G substitutions at the central AT-rich region (W4) (Figure 2B), these substitutions were substantially more detrimental to the relative binding affinity when other consensus-conforming sequences were considered (Figure 2A–C, Supplementary Figure S4). The effect on relative binding affinity of such substitutions at positions −2 and +2 was stronger for GpT and ApC bp steps, respectively (Supplementary Figure S4B). Although this analysis demonstrated overall effects based on a pool of sequences, there was some evidence that the sequence-dependent context also influenced MEF2 binding due to polarity of the binding site (Figure 2A–D, Supplementary Figure S4).
MEF2B preferentially binds sequences with an AT-rich region at its central core (14,71), despite an apparent lack of base-specific contacts based on co-crystal structure analysis (24,27) (Supplementary Figure S2). Our study suggests that the position and polarity of the A-tract each exert distinct effects on the observed relative binding affinity. This pattern could be important for in vivo function, given that the A-tract motif composition and polarity were not uniform across MEF2B ChIP-seq peaks (Figure 2E and F). In agreement with our in vitro data, ChIP-seq data revealed an increased number of sites with a conserved A-tract towards the 3′ peripheral 3-bp region of the binding site (Figure 2D). This finding was supported by EMSAs showing that mutations located 3′ of the central A-tract resulted in decreased binding relative to the wild-type sequence (72). In addition, motif matches containing A-tracts had an increased number of conserved peripheral half-sites at the 3′ region compared to the 5′ region (Figure 2F). This finding is particularly intriguing because it opens new questions regarding the sources of specificity of MEF2B and potentially other MADS proteins.
The observed changes in relative affinity based on nucleotide substitutions are indicative of a dual-recognition mode in which base and shape readout mechanisms (11) could be intertwined. Although some nucleotide substitutions were clearly more detrimental than others, further studies were needed to determine how these changes affected the intrinsic shape of DNA.
DNA shape signatures of MEF2B binding sites suggest distinct structural preferences
When analyzing DNA shape features of MEF2B binding sites, we considered four parameters: helix twist, minor groove width, propeller twist, and roll. Initial analysis of shape parameters predicted for 10-mers obtained from SELEX-seq experiments revealed that high-affinity sites exhibited enhanced negative propeller twist, increased helix twist, and narrow minor groove within the central region of the binding site (Figure 3A). Helix twist and propeller twist showed the most significant differences between high- and low-affinity binding sites, although minor groove width values also differed significantly (Figure 3A and B).
Figure 3.

Conservation of DNA shape features across MEF2B target sites. (A) DNA shape profiles across MEF2 target sites suggest that some DNA shape features can be used to discriminate high-affinity sites. Each column represents a position within the binding site. Each row represents a bin of 200 sequences. Rows are ordered from the top to bottom of the heat map by descending binding affinity. Shape parameters at central positions display the greatest differences between high- and low-affinity sites. (B) Box plots comparing mean shape values for central two positions of sites analyzed in panel (A) between high- and low-affinity sites (n = 2000). (C) Comparison between high- and low-affinity sites for sequences showing variations at the central W4 region or deviations from it (non-W4) (mean of shape parameters, n = 100). (A–C) Sequences were aligned based on the DNA motif YTAN4TAR with up to two mismatches allowed. Four DNA shape parameters were considered: helix twist (HelT), minor groove width (MGW), propeller twist (ProT) and Roll. Only sites with relative affinity >0.2 were included. P-values were calculated by Mann–Whitney U test.
Investigation of DNA shape features from MEF2B binding sites with variable sequence context (W4 versus non-W4) suggested that some shape features might aid in discriminating such binding sites (Figure 3C). Helix twist was increased at central positions in high-affinity sites that displayed a conserved central W4 region of the binding site (Figure 3C). Deviations from the W4 sequence at the central region (non-W4) generated sites that had conserved helix twist patterns for higher- but not for lower-affinity binding sites (Figure 3C). Some DNA shape features (e.g. roll angle between adjacent bp) displayed indistinguishable overall shape patterns (Figure 3B). AT-rich regions showed enhanced negative values for propeller twist (Figure 3C). For most cases where distinguishable shape patterns were noted, these differences were mainly observed at the central 2–3 positions of the binding site.
Overall, the DNA shape characteristics highlighted here suggest unique structural signatures of the MEF2 target sites. These findings are in agreement with previously reported individual structural features of MEF2–DNA complexes, including the narrow minor groove and enhanced negative propeller twist observed from co-crystal structural analysis (24). Increased DNA bending has also been associated with high-affinity binding to MEF2C target sites (73). Although our analysis was based on MEF2B binding data, we expect to observe most of the same characteristics with other MEF2 family members, as they share the same DNA recognition motif (14). However, specific differences in the DBD among MEF2 homologs could affect variations in binding specificity.
For example, a specific MEF2B residue, Q14, has a lower affinity and transcriptional activity than the alternative E14 residue found in other MEF2 proteins (18). In MEF2A, mutating E14 to the SRF-like residue K14 (E14K) increased bending and induced binding to the SRF target site (74). Whereas co-crystal structures of MEF2–DNA complexes did not show substantial bending (24,27), in vitro binding studies associated E14 of MEF2A with bending. Thus, Q14 at this position in MEF2B (Supplementary Figure S5) could lead to differential sensitivity to the intrinsic DNA bending propensity, and may distinguish MEF2B from other family members. MEF2 homologs are involved in various biological processes (12,16–18), and their ultimate functions depend on a complex network of events that are influenced by transcriptional regulation, post-translational modifications, and cofactor recruitment, among others.
By analyzing the specific shape profiles of high affinity sequences, we have been able to identify the key structural features contributing to target site recognition. MEF2 is unique among proteins in the importance of DNA shape for protein recognition (29,75); although MEF2 is capable of recognizing seemingly degenerate sites, it is still drawn to gene-specific targets in vivo. Through the analysis of thousands of target sites, we were able to infer the conserved DNA shape patterns that are critical for MEF2B binding.
Quantitative modeling of MEF2B DNA binding affinities
As SELEX-seq experiments generate relative binding affinity data from a random pool of sequences, we next investigated how accurately a trained model could predict relative binding affinity for any given sequence. To model MEF2B binding to its target sites quantitatively, we used L2-regularized MLR to train models using DNA sequence and shape features (61,63) for 10-mer sequences obtained from SELEX-seq experiments with their respective relative binding affinities. We trained models using 10-fold cross-validation and assessed model accuracy using the coefficient of determination (R2) (Figure 4 and Supplementary Figure S6A). In the initial step of data processing, we selected 10-mers based on the consensus binding sequence YTAW4TAR, allowing up to one mismatch to obtain a sufficient number of sequences for MLR (63). Models considering DNA shape (‘1-mer+shape models’) or DNA shape combined with interdependencies between nucleotide positions (‘3-mer+shape models’) performed better than those considering sequence alone when encoded in mono-nucleotide form (‘1-mer models’), with R2 values of about 0.82, 0.86 and 0.74, respectively (Supplementary Figure S6A).
Figure 4.

Comparison of model performance when considering different DNA features. Comparison of model performance (R2) between (A) sequence-only mono-nucleotide (1-mer) and shape-augmented (1-mer+shape) models, or (B) sequence-only (1-mer) and shape models. Adding shape features to a sequence-only model improved performance regardless of the DNA sequence motif used for alignment. Differently colored models represent different data filtering schemes employed. Sequences were selected based on motif shown. Number of permitted mismatches is indicated. (C) Contribution to model performance is shown as the ΔR2 when adding shape information one position at a time to a sequence-only model (top panel) or removing shape information one position at a time from a shape-only model (bottom panel).
Filtering sequences based on a known consensus motif will inherently exclude non-canonical sequences from analysis (61,63). Therefore, to include additional sequences into our modeling approach, we considered a more ambiguous consensus sequence by allowing for more mismatches. Resulting model performances are illustrated in Figure 4A, B and Supplementary Figure S6B. Not surprisingly, we found lower R2 values when training MLR models with this less stringent filtering scheme (Supplementary Figure S6C). Yet, this filtering scheme can be advantageous for the investigation and rationalization of binding mechanisms because it allows for analysis of a greater number of sequences.
Regardless of MLR filtering parameters, models that included shape features always performed better than sequence-only mono-nucleotide (‘1-mer’) models (Figure 4A and Supplementary Figure S6B). Interestingly, in cases where dataset filtering allowed for any nucleotide variation within the central region (N4vs. W4), sequence-only models had comparable performance to shape-only models (Figure 4B). Thus, the four shape parameters (helix twist, minor groove width, propeller twist, and roll) and the sequence features that considered interdependencies between positions (2-mers, 3-mers) generally increased model performance in an MLR analysis.
The observed performance increase with inclusion of shape features points to the importance of DNA shape for the binding affinity of the sequence pool. However, this performance increase does not reveal which features and, most importantly, at which nucleotide positions these features are important for binding. To address these issues, we used a feature selection approach (63). Specifically, using a model that considers the addition or removal of shape features one nucleotide position at a time, we identified regions of the binding site where shape features contribute more substantially to model performance. To investigate degeneracy of the central region, we allowed up to two mismatches at the peripheral half-sites and considered any sequence composition within the central N4 region of the core motif (we used YTAN4TAR for filtering, and allowed up to two mismatches at the YTA or TAR half-sites combined).
Using this model, shape had the greatest contribution at the center of the binding site (nucleotide positions −1/+1) (Figure 4C and Supplementary Figure S7). Adding (Figure 4C, top panel) or removing (Figure 4C, bottom panel) shape features at these positions had the largest impact on model performance. However, when we used the stringent filtering approach based on motif YTAW4TAR and allowed only one mismatch, we found that shape features at positions −2/+2 of the central 4-bp region had the largest contributions to model performance (Supplementary Figure S7). This outcome might be a result of the sequence preference already introduced by the filtering method because AT-rich regions share intrinsic structural characteristics. Although none of these models fully captured the large variability of sequences bound by MEF2B, there is a trade-off between the aforementioned sequence selection (and, therefore, the number of sequences present) and the model performance. Nonetheless, considering distinct motifs allowed us to discern possible sequence context-dependent readout mechanisms that we would not otherwise reveal.
Our results indicate that shape parameters and interdependencies between nucleotide positions are key features for MEF2B–DNA recognition. Other studies aimed at modeling binding affinities for a wide range of TFs have shown that shape features, in addition to sequence, can increase model performance for some TF families more than for others (63,76). Previous studies have identified DNA structural features that are recognized by MADS-box TFs in plants (77,78), and have shown that in vivo binding predictions for human MADS-domain TFs benefit from the inclusion of DNA shape (79). Using a feature selection model that evaluated shape contributions to model performance one nucleotide position at a time, we identified positions where shape features were most important for binding specificity. Pre-processing of SELEX-seq data was a critical factor in the analysis, and its effect should be considered (77). Alignment bias was noticeable from our results (80), highlighting the need to develop alignment-free methods (81). At the same time, variations in data pre-processing emphasize the promiscuous binding of MEF2 TFs. These observations are consistent with the structural analysis, from which we inferred that DNA shape and minor groove interactions are important for MEF2 recognition. These findings prompted us to investigate the structural components that could be involved in DNA shape recognition.
Mechanistic insights into sources of DNA recognition by MEF2B
To interpret the readout mechanisms inferred from our high-throughput selection studies, we analyzed available crystal structures (27) and performed MD simulations for complexes for which no co-crystal structure has been solved yet.
We specifically focused on interactions between MEF2B and DNA, which are pronounced at the N-terminal tail and helix H1 (Figure 1A). Basic residues at these sites interacted with DNA in a region of narrow minor groove and enhanced negative electrostatic potential (Figure 5A). For example, K31 inserted into the minor groove of the central AT-rich region, and R3 inserted deeply into the minor groove in the region of the peripheral half-sites (Figure 5A and B). Other basic residues (K4, K5 and R24) were located near the minor groove, where they interacted with the phosphodiester backbone (Figure 5A and B, Supplementary Figure S2). The narrowest region of the minor groove measured a width of 2.7 Å (Figure 5A, blue line), much smaller than the typical value of 5.8 Å for standard B-form DNA. Electrostatic potential was most negative in the central region of the binding site, where the minor groove was narrowest, and less negative moving outward toward the peripheral half-sites, with a variation of about 7 kT/e across the binding site (Figure 5A, red line). Co-crystal structures of other MEF2 family members in complex with DNA showed similar features of enhanced negative electrostatic potential coinciding with minor groove narrowing (Supplementary Figure S8). The predicted minor groove of unbound DNA was also narrow in the central region of the binding site (Supplementary Figure S8), and this narrowing is likely an intrinsic feature of the DNA target. Thus, our structural analysis showed that recognition of DNA binding sites by MEF2 TFs, including MEF2B, exhibits characteristics of shape readout mechanisms (39,82); arginine and lysine residues insert into the minor groove in a region of enhanced negative electrostatic potential, as was previously described for other TF families (83).
Figure 5.

MEF2B residues recognize DNA shape. (A) Minor groove width and electrostatic potential plotted as function of sequence, based on analysis of co-crystal structures of MEF2B with DNA from PDB ID 1N6J (27). Supplementary Figure S8 shows consistency with co-crystal structures, including PDB ID 1TQE used as starting configuration for MD simulations. (B) Electrostatic potential of DNA shown on molecular surface of DNA. Red mesh represents regions with isopotential of −5 kT/e. Positively charged MEF2B residues near minor groove are shown in stick representation. (C) Snapshot of interactions between K23 and DNA. Bases interacting with K23 are underlined. Panels display interactions between K23 and regions 3′ (left panel) or 5′ (right panel) of the A-tract. Snapshot was obtained from the most representative structure of MD simulations based on the main cluster. (D) Difference contact map between MEF2B−DNA complex with DNA substitutions and co-crystal structure with PDB ID 1TQE shows changes in distance matrices induced by DNA substitutions. Negative differences represent regions that are closer in contact. Positive differences represent regions that are further away. Map edges are color-coded to represent specific regions of the protein as described in the figure.
Given our observations of A-tract polarity and sequence preferences at the peripheral half-sites, we further investigated conformational dynamics of the MEF2B–DNA complex using MD simulations (Figure 5C-D). Starting from an input complex previously solved by X-ray crystallography (PDB ID: 1TQE), we performed MD simulations with the original complex or introduced mutations in the DNA to investigate the effect on protein–DNA interaction dynamics. Our results revealed that the two monomers displayed variable interactions with the DNA. Hydrogen bonds (determined at a 3.5 Å distance cutoff) were more predominant in the region 3′ of the central A-tract (Figure 5C). Difference contact maps from MD simulations showed that compared to the original structure, mutating the 3′ peripheral triplet and introducing palindromic half-sites resulted in closer proximity of the N-terminal tails and helix H1 to the DNA (Figure 5D, top panel). When considering a full palindromic core motif for the 10 bp of the binding site, this same effect was accompanied by closer interactions of certain residues at the N-terminal and MEF2 domain with the DNA (Figure 5D, bottom panel). Minor groove narrowing is one of the mechanisms through which such effects could be modulated as observed by introduction of palindromic DNA regions (Supplementary Figure S9A).
Our studies strongly indicate that DNA shape and structural conformation play a major role in DNA recognition by MEF2B. Moreover, our data are consistent with early crystallographic studies, which demonstrate a narrow minor groove when MEF2 is bound to DNA (24). MEF2B engages in a unique DNA shape recognition mode that involves the center of the binding site and MEF2B residues at the main recognition helix H1 and N-terminal tail. For other proteins, including bHLH and Hox TFs, as well as GR (30,37,84), shape readout is achieved at either flanking regions outside the core motif or at the spacer between the TF and cofactor binding sites, and the main DNA recognition components interact with the major groove. These DNA sequence and shape readout contributions are intrinsically intertwined because DNA shape is a result of the interdependency between nucleotides, dominated by stacking interactions between adjacent bp (66).
MEF2B protein mutants modulate changes in DNA sequence and shape preferences
To analyze the effects of certain protein mutations on DNA binding, we performed SELEX-seq experiments on four single amino-acid mutations of MEF2B associated with cancer (K4E, K5E, R15G or K23R). DNA binding preferences from SELEX-seq data were mainly determined by variations at specific protein regions (Figure 6A). PWMs for high-affinity k-mers display contributions of individual positions to protein binding (Supplementary Figure S10). In addition, position frequency matrices (PFMs) generated from the most enriched sequences based on the consensus motif shared by the MEF2 family revealed that binding preferences were most correlated between wild-type MEF2B and the two N-terminal mutants, K4E and K5E (Figure 6B). These standard approaches suggest contributions of individual nucleotide positions to binding whereas interdependencies between positions and A-tract polarity relate to structural features that are important for MEF2B binding. On the other hand, the two helix-H1 mutants, R15G and K23R, showed the largest dissimilarities in DNA shape preferences compared to wild-type as measured by Euclidean distances of DNA shape features (Figure 6C). Trinucleotide preferences at the peripheral half-sites were mostly conserved, except for the K23R mutation, which is known to make major groove contacts in this region (Figure 6D). At the 5′ end, every triplet with a TpA step starting at position −4 was favored. Similarly, every triplet with a TpA step starting at position +3 was also favored. Tolerance for 5′ CpC and 3′ GpG dinucleotides was diminished for the K23R mutant (Figure 6D).
Figure 6.

DNA binding preferences of MEF2B protein mutants. (A) Single point mutation locations are indicated as magenta stick representations of individual residues. Residues in one MEF2B subunit of the homodimer are labeled. (B) Position frequency matrix (PFM) correlations (purple) and (C) DNA shape dissimilarity based on Euclidean distance (blue) between DNA binding sites of MEF2B proteins. (D) Trinucleotide frequency at 5′ and 3′ regions of DNA binding sites. Sequences used in (B–D) represent top k-mers (n = 100) based on MEF2 consensus site. (E) Eigenvectors obtained from MD simulations of wild-type and protein mutants. Colors represent projections of values along Eigenvector 1 obtained from MD trajectories of wild-type MEF2B and mutant proteins.
Since co-crystal structures for MEF2B mutants were unavailable, we reverted to MD simulations to study structural readout modes of MEF2B mutant–DNA complexes. MD simulations were generated for each of the four mutants based on a starting complex of MEF2B bound to DNA, from PDB ID 1TQE (58). To better understand how these mutants affect binding mechanisms, we visualized the most flexible parts of each complex, as reflected by extremes on the projection of each trajectory (wild-type MEF2B and four mutant proteins) along the first principal eigenvector (Figure 6E). The K4E mutant exhibited the greatest movement, found within the N-terminal tail, suggesting disruption of stabilizing contacts in this region. Interestingly, this mutant is also reported as most strongly linked to non-Hodgkin's lymphoma (85). To a lesser extent, the K5E mutant also exhibited elevated conformational flexibility in the region, with the most dramatic effect occurring at the 3′ end of the A-tract (Figure 6D). The K23R mutant exhibited only slightly increased flexibility of helix H1 compared to the rest of the structure. As arginine (like lysine) is a positively charged residue, the contacts are likely slightly shifted to accommodate the geometric change. The most distinct conformational variations of mutants with respect to the wild-type trajectory were mainly due to DNA oscillations in regions not stabilized by protein contacts. As expected, conformational variations within the rest of the protein were uniform and limited. Minor groove width amongst the different complexes further indicates how each mutant protein is exerting a distinct effect on DNA conformation upon binding (Supplementary Figure S9B). Our results suggest that dynamic changes occur at one of the peripheral half-sites, based on the A-tract polarity. Mutations at the N-terminal tail (K4E and K5E) might destabilize interactions at this half-site (Figure 6E). The binding preferences of the K4E and K5E mutants are most closely related to wild-type MEF2B. This is likely a result of the K4E and K5E mutants’ lower preference for sequences that deviate from the consensus motif as indicated by the PWMs (Supplementary Figure S10). Conformational flexibility at one half-site is common between the wild-type and the K4E and K5E mutants (Figure 6E). Our results indicate a mode of protein–DNA binding where A-tract polarity modulates binding at the peripheral half-sites. Protein mutations can destabilize these interactions and our data indicate an intricate balance of complex binding mechanisms.
CONCLUSIONS
Recognition of DNA target sites by protein factors is a critical step in gene regulation. Our high-throughput binding assays coupled with analyses of co-crystal structures of MEF2–DNA complexes revealed that members of this TF family use a combination of base and shape readout mechanisms to achieve DNA binding specificity.
We performed sequence and shape analyses of a large number of MEF2B binding sites derived from SELEX-seq experiments. MEF2B is unique with respect to other TFs whose mechanisms of recognition rely mainly on base readout within their central core motif (Figure 7). Our approach allowed us to probe how variations within the core motif affect binding specificity in a sequence context-dependent manner. Our results are consistent with DNA characteristics of MEF2 target sites described by DNA selection studies and X-ray crystallography (14,24,27). Previous structural studies revealed MEF2 consensus sequence preferences and demonstrated that MEF2 TFs bind with few specific contacts to their DNA binding sites and recognize a region of narrow minor groove (24). Furthermore, the AT-rich regions of higher-affinity sites exhibit an overall increase in helix twist and more negative propeller twist. Certain features of the protein–DNA interface, such as the insertion of basic residues into the minor groove, indicate the use of DNA shape readout in that region (39,82). By modeling relative affinities using L2-regularized MLR considering DNA sequence and shape features, we predicted binding affinities with high accuracy and showed that DNA shape or the interdependency between nucleotide positions is an important feature that improves model performance.
Figure 7.
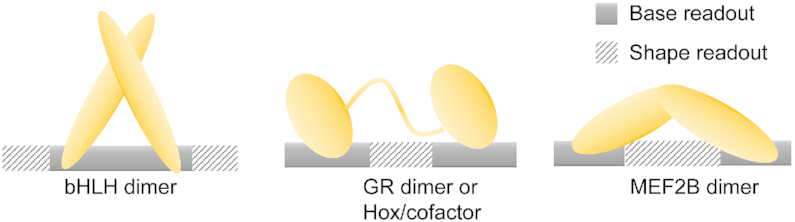
Different forms of cooperating base and shape readout. Mechanisms of DNA recognition are indicated as regions of either primarily base or shape readout. Schematic representations are shown for basic helix-loop-helix (bHLH) dimers, glucocorticoid receptor (GR) homodimers or Hox homedomain TF/cofactor complexes, and MEF2B homodimers.
The recognition mode described for MEF2 is intrinsically distinct from that of other TFs. MEF2 is a specific transcriptional regulator; as such, its main DNA binding elements display a small number of base-specific contacts compared to the extensive interactions with the phosphodiester backbone and minor groove. By contrast, the main recognition mode of other TFs, including bHLH TFs, Hox TFs and GR (Figure 7), is base readout. Nevertheless, the contributions of base and shape readout cannot be entirely disentangled. Our observations support the notion that the two readout modes coexist to different extent.
In summary, using SELEX-seq experiments and computational methods, we demonstrated how variations in DNA features can affect the relative binding affinities of MEF2B TFs. Variations in TF binding sites in regulatory regions have emerged as a major source of diversity (86) and can contribute to pathological differences in gene regulation (4). For example, mutations in gene regulatory regions of MEF2 family members were found to disrupt DNA binding and were associated with cardiac disease (87). Coding missense mutations disrupted binding (88) and were linked to pathological outcomes (21,85). Furthermore, MEF2B is highly expressed in lymph nodes and across multiple other tissues (34). Questions remain as to how individual MEF2 residues contribute to binding specificity. A systematic analysis—such as the approach presented here to elucidate how MEF2B binds to its target sites in vitro—could be used to uncover the fundamental mechanisms of gene regulation.
DATA AVAILABILITY
SELEX-seq sequencing data for MEF2B wild-type and mutants were submitted to the Gene Omnibus (GEO) at https://www.ncbi.nlm.nih.gov/geo/ and are available under accession number GSE116401.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Harmen Bussemaker, Richard Mann, Judith Kribelbauer and Namiko Abe for helpful discussions and advice for setting up the experimental SELEX-seq system in the Rohs laboratory; Lin Yang for help with the feature selection approach; Angana Ray for help with setting up MD simulations and Jared Peace for helpful discussions and comments on the manuscript.
Notes
Present address: Ana Carolina Dantas Machado, Department of Medicine, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Present address: Xiao Lei, The Rockefeller University, 1230 York Avenue, New York, NY 10065, USA.
Contributor Information
Ana Carolina Dantas Machado, Quantitative and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Brendon H Cooper, Quantitative and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Xiao Lei, Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Rosa Di Felice, Quantitative and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA; Department of Physics & Astronomy, University of Southern California, Los Angeles, CA 90089, USA.
Lin Chen, Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA; Department of Chemistry, University of Southern California, Los Angeles, CA 90089, USA; Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, CA 90033, USA.
Remo Rohs, Quantitative and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA; Department of Physics & Astronomy, University of Southern California, Los Angeles, CA 90089, USA; Department of Chemistry, University of Southern California, Los Angeles, CA 90089, USA; Norris Comprehensive Cancer Center, University of Southern California, Los Angeles, CA 90033, USA; Department of Computer Science, University of Southern California, Los Angeles, CA 90089, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01GM106056, R35GM130376 to R.R., R01HG003008 in part to R.R., R01AI113009, R21HG010528 to L.C.]; Human Frontier Science Program [RGP0021/2018 to R.R.]; USC Dornsife Bridge Catalyst grant [to R.R.]; DNA sequencing was performed in the USC Norris Cancer Center which is supported by the National Cancer Institute [P30CA014089]. Funding for open access charge: National Institutes of Health [R35GM130376 to R.R.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Slattery M., Zhou T., Yang L., Dantas Machado A.C., Gordân R., Rohs R.. Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 2014; 39:381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xin B., Rohs R.. Relationship between histone modifications and transcription factor binding is protein family specific. Genome Res. 2018; 28:321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang X., Zhou T., Wunderlich Z., Maurano M.T., DePace A.H., Nuzhdin S.V., Rohs R.. Analysis of genetic variation indicates DNA shape involvement in purifying selection. Mol. Biol. Evol. 2018; 35:1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khurana E., Fu Y., Chakravarty D., Demichelis F., Rubin M.A., Gerstein M.. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016; 17:93–108. [DOI] [PubMed] [Google Scholar]
- 5. Harrison S.C., Aggarwal A.K.. DNA recognition by proteins with the helix-turn-helix motif. Annu. Rev. Biochem. 1990; 59:933–969. [DOI] [PubMed] [Google Scholar]
- 6. Lawson C.L., Berman H.M.. Chapter 4: Indirect Readout of DNA Sequence by Proteins. Protein-Nucleic Acid Interactions: Structural Biology. 2008; Cambridge: The Royal Society of Chemistry; 66–90. [Google Scholar]
- 7. Seeman N.C., Rosenberg J.M., Rich A.. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. U.S.A. 1976; 73:804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pabo C.O., Lewis M.. The operator-binding domain of lambda repressor: structure and DNA recognition. Nature. 1982; 298:443–447. [DOI] [PubMed] [Google Scholar]
- 9. Garvie C.W., Wolberger C.. Recognition of specific DNA sequences. Mol. Cell. 2001; 8:937–946. [DOI] [PubMed] [Google Scholar]
- 10. Hong M., Marmorstein R.. Chapter 3: Structural basis for sequence-specific DNA recognition by transcription factors and their complexes. Protein-Nucleic Acid Interactions: Structural Biology. 2008; 47–65. [Google Scholar]
- 11. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Potthoff M.J., Olson E.N.. MEF2: a central regulator of diverse developmental programs. Development. 2007; 134:4131–4140. [DOI] [PubMed] [Google Scholar]
- 13. Pon J.R., Marra M.A.. MEF2 transcription factors: developmental regulators and emerging cancer genes. Oncotarget. 2016; 7:2297–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pollock R., Treisman R.. Human SRF-related proteins: DNA-binding properties and potential regulatory targets. Genes Dev. 1991; 5:2327–2341. [DOI] [PubMed] [Google Scholar]
- 15. Andrés V., Cervera M., Mahdavi V.. Determination of the consensus binding site for MEF2 expressed in muscle and brain reveals tissue-specific sequence constraints. J. Biol. Chem. 1995; 270:23246–23249. [DOI] [PubMed] [Google Scholar]
- 16. Mao Z., Bonni A., Xia F., Nadal-Vicens M., Greenberg M.E.. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999; 286:785–790. [DOI] [PubMed] [Google Scholar]
- 17. Han J., Jiang Y., Li Z., Kravchenko V.V., Ulevitch R.J.. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997; 386:296–299. [DOI] [PubMed] [Google Scholar]
- 18. Molkentin J.D., Firulli A.B., Black B.L., Martin J.F., Hustad C.M., Copeland N., Jenkins N., Lyons G., Olson E.N.. MEF2B is a potent transactivator expressed in early myogenic lineages. Mol. Cell. Biol. 1996; 16:3814–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhagavatula M.R.K., Fan C., Shen G.-Q., Cassano J., Plow E.F., Topol E.J., Wang Q.. Transcription factor MEF2A mutations in patients with coronary artery disease. Hum. Mol. Genet. 2004; 13:3181–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zweier M., Gregor A., Zweier C., Engels H., Sticht H., Wohlleber E., Bijlsma E.K., Holder S.E., Zenker M., Rossier E. et al.. Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum. Mutat. 2010; 31:722–733. [DOI] [PubMed] [Google Scholar]
- 21. Morin R.D., Mendez-Lago M., Mungall A.J., Goya R., Mungall K.L., Corbett R.D., Johnson N.A., Severson T.M., Chiu R., Field M. et al.. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011; 476:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morin R.D., Assouline S., Alcaide M., Mohajeri A., Johnston R.L., Chong L., Grewal J., Yu S., Fornika D., Bushell K. et al.. Genetic landscapes of relapsed and refractory diffuse large B-Cell lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016; 22:2290–2300. [DOI] [PubMed] [Google Scholar]
- 23. Jayathilaka N., Han A., Gaffney K.J., Dey R., Jarusiewicz J.A., Noridomi K., Philips M.A., Lei X., He J., Ye J. et al.. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res. 2012; 40:5378–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santelli E., Richmond T.J.. Crystal structure of MEF2A core bound to DNA at 1.5 Å resolution. J. Mol. Biol. 2000; 297:437–449. [DOI] [PubMed] [Google Scholar]
- 25. de Folter S., Angenent G.C.. trans meets cis in MADS science. Trends Plant Sci. 2006; 11:224–231. [DOI] [PubMed] [Google Scholar]
- 26. Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G. et al.. DNA-binding specificities of human transcription factors. Cell. 2013; 152:327–339. [DOI] [PubMed] [Google Scholar]
- 27. Han A., Pan F., Stroud J.C., Youn H.-D., Liu J.O., Chen L.. Sequence-specific recruitment of transcriptional co-repressor Cabin1 by myocyte enhancer factor-2. Nature. 2003; 422:730–734. [DOI] [PubMed] [Google Scholar]
- 28. Ferré-D’Amaré A.R., Pognonec P., Roeder R.G., Burley S.K.. Structure and function of the b/HLH/Z domain of USF. EMBO J. 1994; 13:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joshi R., Passner J.M., Rohs R., Jain R., Sosinsky A., Crickmore M.A., Jacob V., Aggarwal A.K., Honig B., Mann R.S.. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007; 131:530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luisi B.F., Xu W.X., Otwinowski Z., Freedman L.P., Yamamoto K.R., Sigler P.B.. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. 1991; 352:497–505. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y., Dey R., Han A., Jayathilaka N., Philips M., Ye J., Chen L.. Structure of the MADS-box/MEF2 domain of MEF2A bound to DNA and its implication for myocardin recruitment. J. Mol. Biol. 2010; 397:520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaufmann K., Muiño J.M., Jauregui R., Airoldi C.A., Smaczniak C., Krajewski P., Angenent G.C.. Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol. 2009; 7:e1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gramzow L., Theissen G.. A hitchhiker's guide to the MADS world of plants. Genome Biol. 2010; 11:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fagerberg L., Hallström B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K. et al.. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics MCP. 2014; 13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pasqualucci L., Trifonov V., Fabbri G., Ma J., Rossi D., Chiarenza A., Wells V.A., Grunn A., Messina M., Elliot O. et al.. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011; 43:830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Riley T.R., Slattery M., Abe N., Rastogi C., Liu D., Mann R.S., Bussemaker H.J.. SELEX-seq: a method for characterizing the complete repertoire of binding site preferences for transcription factor complexes. Methods Mol. Biol. 2014; 1196:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Slattery M., Riley T., Liu P., Abe N., Gomez-Alcala P., Dror I., Zhou T., Rohs R., Honig B., Bussemaker H.J. et al.. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011; 147:1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lavery R., Sklenar H.. Defining the structure of irregular nucleic acids: conventions and principles. J. Biomol. Struct. Dyn. 1989; 6:655–667. [DOI] [PubMed] [Google Scholar]
- 39. Rohs R., West S.M., Sosinsky A., Liu P., Mann R.S., Honig B.. The role of DNA shape in protein–DNA recognition. Nature. 2009; 461:1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Honig B., Nicholls A.. Classical electrostatics in biology and chemistry. Science. 1995; 268:1144–1149. [DOI] [PubMed] [Google Scholar]
- 41. Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A.. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995; 117:5179–5197. [Google Scholar]
- 42. Sagendorf J.M., Berman H.M., Rohs R.. DNAproDB: an interactive tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2017; 45:W89–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sagendorf J.M., Markarian N., Berman H.M., Rohs R.. DNAproDB: an expanded database and web-based tool for structural analysis of DNA-protein complexes. Nucleic Acids Res. 2020; 48:D277–D287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chiu T.P., Xin B., Markarian N., Wang Y., Rohs R.. TFBSshape: an expanded motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2020; 48:D246–D255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou T., Yang L., Lu Y., Dror I., Dantas Machado A.C., Ghane T., Felice R.D., Rohs R.. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013; 41:W56–W62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Foat B.C., Morozov A.V., Bussemaker H.J.. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006; 22:e141–e149. [DOI] [PubMed] [Google Scholar]
- 48. Pon J.R., Wong J., Saberi S., Alder O., Moksa M., Grace Cheng S.-W., Morin G.B., Hoodless P.A., Hirst M., Marra M.A.. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat. Commun. 2015; 6:7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013,preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 50. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chiu T.P., Comoglio F., Zhou T., Yang L., Paro R., Rohs R.. DNAshapeR: an R/Bioconductor package for DNA shape prediction and feature encoding. Bioinforma. Oxf. Engl. 2016; 32:1211–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li J., Sagendorf J.M., Chiu T.P., Pasi M., Perez A., Rohs R.. Expanding the repertoire of DNA shape features for genome-scale studies of transcription factor binding. Nucleic Acids Res. 2017; 45:12877–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pasi M., Maddocks J.H., Beveridge D., Bishop T.C., Case D.A., Cheatham T., Dans P.D., Jayaram B., Lankas F., Laughton C. et al.. μABC: a systematic microsecond molecular dynamics study of tetranucleotide sequence effects in B-DNA. Nucleic Acids Res. 2014; 42:12272–12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Berman H.M., Olson W.K., Beveridge D.L., Westbrook J., Gelbin A., Demeny T., Hsieh S.H., Srinivasan A.R., Schneider B.. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992; 63:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Azad R.N., Zafiropoulos D., Ober D., Jiang Y., Chiu T.P., Sagendorf J.M., Rohs R., Tullius T.D.. Experimental maps of DNA structure at nucleotide resolution distinguish intrinsic from protein-induced DNA deformations. Nucleic Acids Res. 2018; 46:2636–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E.. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1–2:19–25. [Google Scholar]
- 58. Han A., He J., Wu Y., Liu J.O., Chen L.. Mechanism of recruitment of class II histone deacetylases by myocyte enhancer Factor-2. J. Mol. Biol. 2005; 345:91–102. [DOI] [PubMed] [Google Scholar]
- 59. Ivani I., Dans P.D., Noy A., Pérez A., Faustino I., Hospital A., Walther J., Andrio P., Goñi R., Balaceanu A. et al.. Parmbsc1: a refined force field for DNA simulations. Nat. Methods. 2016; 13:55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pérez A., Marchán I., Svozil D., Sponer J., Cheatham T.E., Laughton C.A., Orozco M.. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abe N., Dror I., Yang L., Slattery M., Zhou T., Bussemaker H.J., Rohs R., Mann R.S.. Deconvolving the recognition of DNA shape from sequence. Cell. 2015; 161:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang L., Zhou T., Dror I., Mathelier A., Wasserman W.W., Gordân R., Rohs R.. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014; 42:D148–D155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang L., Orenstein Y., Jolma A., Yin Y., Taipale J., Shamir R., Rohs R.. Transcription factor family-specific DNA shape readout revealed by quantitative specificity models. Mol. Syst. Biol. 2017; 13:910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Treisman R. Identification and purification of a polypeptide that binds to the c-fos serum response element. EMBO J. 1987; 6:2711–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mathelier A., Fornes O., Arenillas D.J., Chen C.Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. et al.. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016; 44:D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shakked Z., Crothers D.M.. Neidle S. DNA bending by adenine-thymine tracts. Oxford Handbook of Nucleic Acid Structure. 1999; London: Oxford University Press; 455–470. [Google Scholar]
- 67. Ogawa M., Sakakibara Y., Kamemura K.. Requirement of decreased O-GlcNAc glycosylation of Mef2D for its recruitment to the myogenin promoter. Biochem. Biophys. Res. Commun. 2013; 433:558–562. [DOI] [PubMed] [Google Scholar]
- 68. Black B.L., Ligon K.L., Zhang Y., Olson E.N.. Cooperative transcriptional activation by the neurogenic basic helix-loop-helix protein MASH1 and members of the myocyte enhancer factor-2 (MEF2) family. J. Biol. Chem. 1996; 271:26659–26663. [DOI] [PubMed] [Google Scholar]
- 69. Haran T.E., Mohanty U.. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009; 42:41–81. [DOI] [PubMed] [Google Scholar]
- 70. Merling A., Sagaydakova N., Haran T.E.. A-tract polarity dominate the curvature in flanking sequences. Biochemistry. 2003; 42:4978–4984. [DOI] [PubMed] [Google Scholar]
- 71. Katoh Y., Molkentin J.D., Dave V., Olson E.N., Periasamy M.. MEF2B is a component of a smooth muscle-specific complex that binds an A/T-rich element important for smooth muscle myosin heavy chain gene expression. J. Biol. Chem. 1998; 273:1511–1518. [DOI] [PubMed] [Google Scholar]
- 72. Cserjesi P., Olson E.N.. Myogenin induces the myocyte-specific enhancer binding factor MEF-2 independently of other muscle-specific gene products. Mol. Cell. Biol. 1991; 11:4854–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Meierhans D., Sieber M., Allemann R.K.. High affinity binding of MEF-2C correlates with DNA bending. Nucleic Acids Res. 1997; 25:4537–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. West A.G., Shore P., Sharrocks A.D.. DNA binding by MADS-box transcription factors: a molecular mechanism for differential DNA bending. Mol. Cell. Biol. 1997; 17:2876–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stella S., Cascio D., Johnson R.C.. The shape of the DNA minor groove directs binding by the DNA-bending protein Fis. Genes Dev. 2010; 24:814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhou T., Shen N., Yang L., Abe N., Horton J., Mann R.S., Bussemaker H.J., Gordân R., Rohs R.. Quantitative modeling of transcription factor binding specificities using DNA shape. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:4654–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Muiño J.M., Smaczniak C., Angenent G.C., Kaufmann K., Dijk A.D.J. van. Structural determinants of DNA recognition by plant MADS-domain transcription factors. Nucleic Acids Res. 2014; 42:2138–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Käppel S., Melzer R., Rümpler F., Gafert C., Theißen G.. The floral homeotic protein SEPALLATA3 recognizes target DNA sequences by shape readout involving a conserved arginine residue in the MADS-domain. Plant J. Cell Mol. Biol. 2018; 95:341–357. [DOI] [PubMed] [Google Scholar]
- 79. Mathelier A., Xin B., Chiu T.P., Yang L., Rohs R., Wasserman W.W.. DNA shape features improve transcription factor binding site predictions in vivo. Cell Syst. 2016; 3:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Orenstein Y., Shamir R.. Modeling protein-DNA binding via high-throughput in vitro technologies. Brief. Funct. Genomics. 2017; 16:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ma W., Yang L., Rohs R., Noble W.S.. DNA sequence+shape kernel enables alignment-free modeling of transcription factor binding. Bioinformatics. 2017; 33:3003–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Deng Z., Wang Q., Liu Z., Zhang M., Dantas Machado A.C., Chiu T.P., Feng C., Zhang Q., Yu L., Qi L. et al.. Mechanistic insights into metal ion activation and operator recognition by the ferric uptake regulator. Nat. Commun. 2015; 6:7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chiu T.P., Rao S., Mann R.S., Honig B., Rohs R.. Genome-wide prediction of minor-groove electrostatic potential enables biophysical modeling of protein-DNA binding. Nucleic Acids Res. 2017; 45:12565–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gordân R., Shen N., Dror I., Zhou T., Horton J., Rohs R., Bulyk M.L.. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013; 3:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ying C.Y., Dominguez-Sola D., Fabi M., Lorenz I.C., Hussein S., Bansal M., Califano A., Pasqualucci L., Basso K., Dalla-Favera R.. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat. Immunol. 2013; 14:1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. GTEx Consortium Genetic effects on gene expression across human tissues. Nature. 2017; 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Oishi Y., Manabe I., Imai Y., Hara K., Horikoshi M., Fujiu K., Tanaka T., Aizawa T., Kadowaki T., Nagai R.. Regulatory polymorphism in transcription factor KLF5 at the MEF2 element alters the response to angiotensin II and is associated with human hypertension. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010; 24:1780–1788. [DOI] [PubMed] [Google Scholar]
- 88. Molkentin J.D., Black B.L., Martin J.F., Olson E.N.. Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol. Cell. Biol. 1996; 16:2627–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SELEX-seq sequencing data for MEF2B wild-type and mutants were submitted to the Gene Omnibus (GEO) at https://www.ncbi.nlm.nih.gov/geo/ and are available under accession number GSE116401.